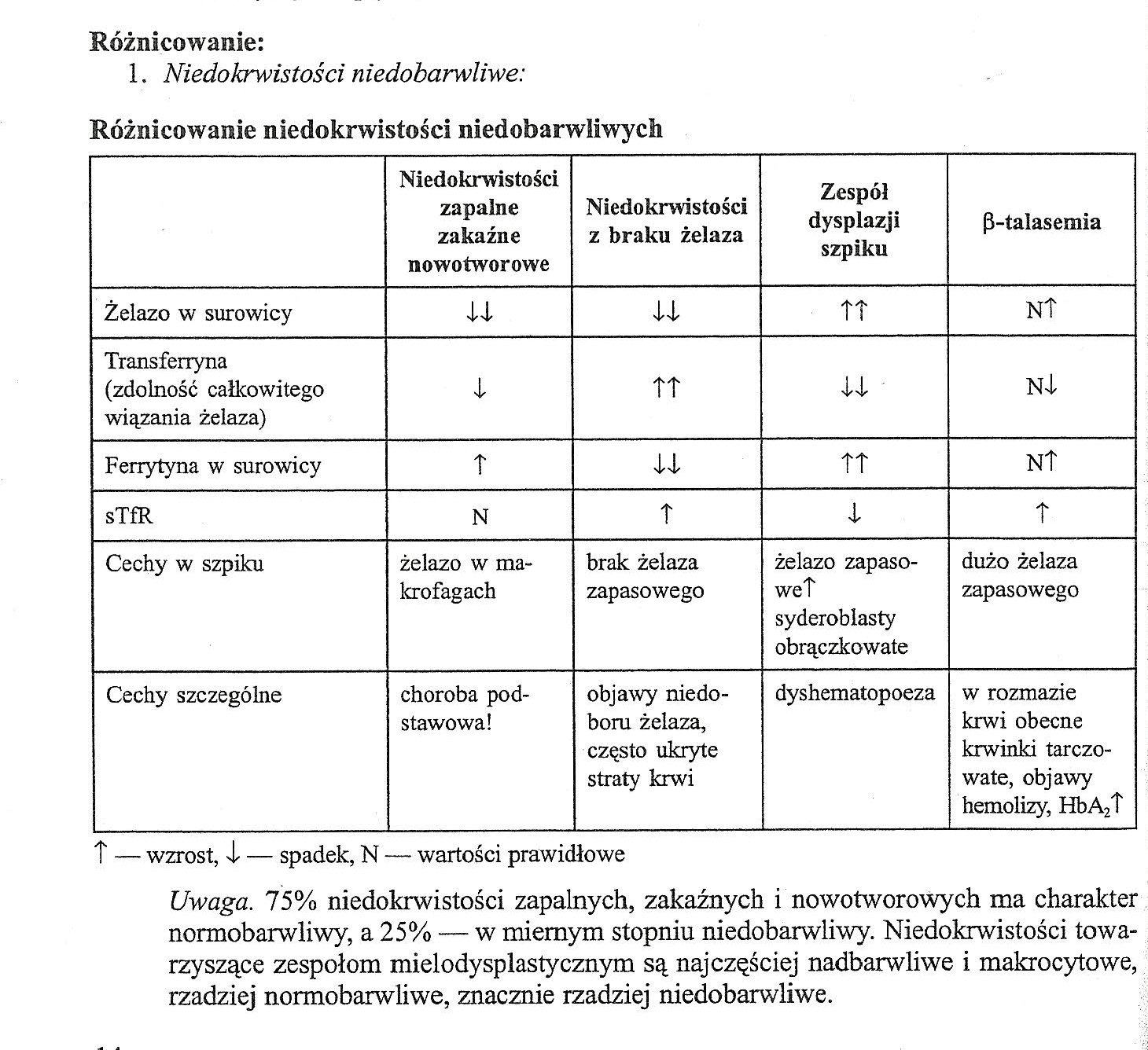
Niedokrwistości - definicja.
Niedokrwistość jest to zmniejszenie stężenia hemoglobiny, hematokrytu lub liczby erytrocytów poniżej wartości prawidłowych:
Hb < 13,5 g/dl (< 8,3 mmol/l) (M)
< 12,0 g/dl (<7,4 mmol/l) (K)
Ht < 0,40 (40%) M
< 0,37 (37%) K
Niedokrwistości spowodowane niedoborem żelaza.
Występowanie:
w Europie występuje u ok.10% ,a w krajach rozwijających się u ponad 50% kobiet w wieku rozrodczym
stanowi 80% wszystkich niedokrwistości
Wśród wszystkich chorych na niedokrwistość z niedoboru żelaza 80% stanowią kobiety (większe zapotrzebowanie na żelazo spowodowane menstruacją, ciążą i laktacją)
Etiologia.
niewystarczająca podaż (u niemowląt, małych dzieci, wegetarian),
upośledzone wchłanianie: stan po resekcji żołądka, zespół złego wchłaniania,
wzmożone zapotrzebowanie (okres wzrostu, ciąża, okres karmienia piersią itp.),
najczęstsza przyczyna - utrata żelaza wskutek przewlekłych krwawień (80%).
Klinika:
Występowanie objawów niedoboru żelaza jeszcze przed pojawieniem się klinicznie jawnej niedokrwistości określa się mianem syderopenii lub utajonego niedoboru żelaza.
Objawy na skórze i błonach śluzowych (zajady w kącikach ust, łamliwość włosów i paznokci, zespół Plummera-Vinsona - dysfagia).
Nieswoiste zaburzenia psychiczne lub neurologiczne.
Ogólne objawy niedokrwistości (bladość skóry i błon śluzowych, osłabienie lub duszność wysiłkowa, szmer skurczowy nad sercem, osłabienie koncentracji, bóle głowy).
Badania laboratoryjne.
a) przedutajony niedobór żelaza:
zmniejszenie stężenia ferrytyny w surowicy i żelaza w szpiku kostnym, niedobór żelaza
Zmniejszone stężenie w surowicy rozpuszczalnego receptora dla transferyny
b) czynnościowy niedobór żelaza:
dodatkowo:
Wysycenie transferyny < 20%, zwiększenie stężenia transferyny
Zmniejszona liczba syderoblastów w szpiku kostnym
Zmniejszone stężenie żelaza w surowicy
c) Klinicznie jawny niedobór żelaza:
Zmniejszone stężenie hemoglobiny, mniejsza liczba erytrocytów i wartość hematokrytu
Niedokrwistości z niedoboru żelaza powodują pierwotnie zahamowanie wytwarzania hemoglobiny, natomiast w mniejszym stopniu wytwarzania erytrocytów.
Niedobarwliwość - blade erytrocyty, zmniejszenie średniej zawartości hemoglobiny w erytrocycie (MCH) poniżej 28 pg.
Objawem niedoboru żelaza jest zwiększanie odsetka niedobarwliwych erytrocytów.
W niektórych przypadkach niedokrwistości spowodowanej niedoborem żelaza towarzyszy nadpłytkowość.
Rozpoznanie.
niedokrwistości z niedoboru żelaza (wywiady i obraz kliniczny, wyniki badań laboratoryjnych)
ustalenie przyczyn (najczęstsze przyczyny to krwawienia)
Leczenie.
przyczynowe
objawowe (uzupełnianie niedoborów żelaza)
Niedokrwistości megaloblastyczne.
Jest to niedobór witaminy B12 i/lub kwasu foliowego powodujący zaburzenia syntezy DNA, a w zakresie mielopoezy upośledzenie dojrzewania jąder komórkowych, będące przyczyną występowania megaloblastów.
Objawem wiodącym jest niedokrwistość megaloblastyczna (w przypadku niedoboru witaminy B12 występują dodatkowo objawy ze strony układu nerwowego i pokarmowego).
Epidemiologia: najczęściej powodowane są niedoborem kobalaminy (wit. B12)
Niedobór witaminy B12:
niewystarczająca podaż w diecie,
niedobór czynnika wewnętrznego (np. stan po resekcji żołądka),
zespół złego wchłaniania,
zwiększone zużycie (tasiemiec bruzdogłowy szeroki),
nadmierny rozwój bakterii
2. Niedobór kwasu foliowego:
niedobory w pożywieniu,
zwiększone zapotrzebowanie (hemoliza, ciąża),
zespół upośledzonego wchłaniania,
leczenie antagonistami kwasu foliowego.
Klinika niedoboru wit. B12:
W pełni wyrażony niedobór witaminy B12 charakteryzuje się triadą objawów - zaburzenia hematologiczne + neurologiczne + gastroenterologiczne:
zespół hematologiczny - ogólne objawy niedokrwistości,
zespół żołądkowo - jelitowy (zanikowe autoimmunologiczne zapalenie żołądka typu A, troficzne zmiany błon śluzowych, zanikowe zapalenie języka),
zespół neurologiczny - choroba sznurów rdzenia ze zmianami zanikowymi osłonek rdzenia.
Klinika niedoboru kwasu foliowego:
niedobór kwasu foliowego powoduje niedokrwistość megaloblastyczną bez objawów neurologicznych,
niedobór u kobiety ciężarnej zwiększa ryzyko uszkodzenia układu nerwowego u płodu
Badania laboratoryjne.
rozmaz krwi obwodowej: niedokrwistość megalocytowa (megalocyty mają zwiększoną objętość krwinki i są nadbarwliwe, jednak stężenie hemoglobiny nie jest bezwzględnie podwyższone
szpik kostny: zaburzone dojrzewanie i eliminacja we wszystkich 3 układach komórkowych - nieefektywna erytro-, granulo- i trombocytopoeza
objawy nieefektywnej erytropoezy z towarzyszącą hemolizą (Ⴍ stężęnie żelaza, Ⴍaktywności LDH, Ⴍ stężenie bilirubiny niezwiązanej)
pomiar stężenia witaminy B12 i kwasu foliowego - stwierdzenie niedoborów tych substancji
badanie zaburzeń wchłaniania witaminy B12 testem Schillinga
Rozpoznanie:
wywiad, objawy kliniczne,
badania laboratoryjne: pełny obraz krwi
oznaczanie witaminy B12 i kwasu foliowego,
badanie szpiku kostnego.
Leczenie:
niedobór wit. B12
leczenie przyczynowe,
podaż wit. B12 (pozajelitowo, doustnie),
w przewlekłym zanikowym zapaleniu żołądka typu A - kontrolne gastroskopie,
niedobór kwasu foliowego:
leczenie przyczynowe (abstynencja alkoholowa, odpowiednia dieta),
substytucja kwasu foliowego.
Niedokrwistości hemolityczne- typy hemolizy, podział niedokrwistości, ogólna diagnostyka laboratoryjna.
Definicja:
Niedokrwistości hemolityczne są spowodowane szybszym rozpadem krwinek czerwonych z powodu ich błędnej budowy bądź zewnętrznych czynników. Nasilenie hemolizy jest wtedy większe niż zdolność odnawiania szpiku.
Okres życia erytrocytu podczas tej choroby jest krótszy niż 50 dni.
Jeżeli w następstwie zwiększonej erytropoezy zawartość hemoglobiny we krwi pozostaje prawidłowa, hemolizę określa się jako wyrównaną, w przeciwnym razie rozwija się niedokrwistość hemolityczna.
Typy hemolizy.
Do hemolizy erytrocytów może dojść na zewnątrz i wewnątrz naczyń krwionośnych. Najczęściej hemoliza zachodzi pozanaczyniowo w komórkach fagocytujących śledziony (głównie), wątroby lub szpiku kostnego.
Hemoliza zewnątrznaczyniowa.
Do stanów w których występuje hemoliza zewnątrznaczyniowa należy zaliczyć:
późne reakcje poprzetoczeniowe,
hemoglobinopatie,
choroby autoimmunohemolityczne,
sferocytozę,
hipersplenizm,
choroby wątroby.
W razie wyczerpania się możliwości degradacyjnych układu siateczkowo - śródbłonkowego lub nagłej hemolizy dochodzi do hemolizy wewnątrznaczyniowej.
W takim przypadku hemoglobina zostaje związana przez haptoglobinę. Gdy wskutek hemolizy wyczerpana zostaje możliwość wiązania hemoglobiny przez haptoglobinę, w surowicy pojawia się wolna hemoglobina, która jest metabolizowana do hematyny i jej pochodnych. Te ostatnie przenoszone są za pomocą hematopeksyny do układu siateczkowo - śródbłonkowego.
Najbardziej czułym parametrem hemolizy wewnątrznaczyniowej jest zmniejszenie stężenia haptoglobiny. Dodatkowe oznaczenie hemopeksyny pozwala na określenie nasilenie hemolizy.
Hemoliza wewnątrznaczyniowa.
Hemoliza wewnątrznaczyniowa może zachodzić:
po wymianie zastawek serca,
po przetoczeniu krwi niezgodnej grupowo,
w zimnicy.
Podział niedokrwistości hemolitycznych.
Niedokrwistości hemolityczne spowodowane czynnikami wewnątrzkrwinkowymi
(tzw. korpuskularne).
Niedokrwistości hemolityczne spowodowane czynnikami pozakrwinkowymi (tzw.pozakorpuskularne).
Niedokrwistości hemolityczne spowodowane czynnikami wewnątrzkrwinkowymi.
Wrodzone defekty błony erytrocytów, np. sferocytoza i owalocytoza.
2. Wrodzone defekty enzymatyczne erytrocytów (niedokrwistości hemolityczne z niedoboru enzymów)
defekty cyklu heksozomonofosforanów (np. niedobór G-6-PD)
defekty glikolizy (np. niedobór kinazy pirogronianowej).
3. Wrodzone zaburzenia syntezy hemoglobiny (hemoglobinopatie)
nieprawidłowe hemoglobiny (warianty nieprawidłowej struktury Hb)
talasemie (warianty, w których tworzy się jeden lub więcej nieprawidłowych łańcuchów polipeptydowych Hb)
4. Nabyte defekty błony krwinki czerwonej: napadowa nocna hemoglobinuria.
Niedokrwistości hemolityczne spowodowane czynnikami pozakrwinkowymi.
Niedokrwistości izoimmunohemolityczne wywołane przez izoprzeciwciała (alloprzeciwciała)
konflikt Rh noworodka,
błędy w przetaczaniu.
Niedokrwistości autoimmunohemolityczne (z ang. autoimmune hemolytic anemia - AIHA)
AIHA z przeciwciałami ciepłymi,
AIHA z przeciwciałami zimnymi,
napadowa zimna hemoglobinuria.
Niedokrwistości immunohemolityczne indukowane lekami, wyróżnia się 3 mechanizmy patofizjologiczne:
typ fanacetynowy (lek działa jak hapten),
typ penicylinowy (lek wiąże się z błoną krwinki czerwonej),
typ ၡ-metylodopa (lek indukuje wytwarzanie autoprzeciwciał).
Hemoliza towarzysząca chorobom zakaźnym (np. malarii).
Niedokrwistości hemolityczne wywołane czynnikami fizycznymi i chemicznymi (uszkodzenia mechaniczne erytrocytów, termiczne uszkodzenie erytrocytów, trucizny chemiczne - jad żmii).
Niedokrwistości hemolityczne u chorych z zaburzeniami przemiany materii: zespół Zievego (poalkoholowe uszkodzenie wątroby, niedokrwistość hemolityczna i hiperlipidemia).
Mikroangioplastyczne niedokrwistości hemolityczne (MHA):
zespół hemolityczno - mocznicowy (HUS) = zespół Gassera
plamica zakrzepowo - małopłytkowa = zespół Moschcowitza
mikroangioplastyczne niedokrwistości hemolityczne (MHA) indukowane lekami (np. mitomycyną C)
MHA towarzyszące przerzutom nowotworowym
Wrodzone defekty błony erytrocytów.
Sferocytoza (mikrosferocytoza wrodzona).
Występowanie : najczęstsza wrodzona niedokrwistość hemolityczna w Europie Północnej.
Etiologia: ilościowy defekt dwóch głównych składników błony erytrocytu (spektryny i ankyryny).
Patogeneza: defekt błony erytrocytu zaburzenia przepuszczalności jonów, powodujące nagromadzenie wody i sodu wewnątrz krwinki zmiana dwuwklęsłego na kulisty kształt krwinki fagocytoza sferocytów w śledzionie, skracająca okres przeżycia erytrocytów
Klinika:
niedokrwistość i/lub żółtaczka w wieku dziecięcym
w 95% przypadków wywiad rodzinny jest obciążający
rzadko przełomy hemolityczne
powiększenie śledziony (splenomegalia), kamienie żółciowe zawierające bilirubinę
Powikłania: rzadko przełomy hemolityczne, często kamica żółciowa
Badania laboratoryjne:
niedokrwistość normobarwliwa + objawy hemolizy (Ⴍ stężenie bilirubiny pośredniej, Ⴍ stężenie LDH, Ⴏ stężenie haptoglobiny - wychwyt hemoglobiny)
obecność mikrosferocytów (bez centralnego przejaśnienia) i zmniejszona oporność osmotyczna)
Leczenie:
w niektórych przypadkach - splenektomia.
Wrodzone defekty enzymatyczne erytrocytów.
Niedobór dehydrogenazy glukozo-6-fosforanowej (G-6-PD)
- najczęściej występująca po cukrzycy choroba dziedziczna
- dziedziczenie związane jest z chromosomem X
Patofizjologia: niedobór G-6-PD prowadzi do zmniejszenia wytwarzania zredukowanego glutationu, ochraniającego erytrocyty przed uszkodzeniami spowodowanymi substancjami utleniającymi
Klinika: przełomy hemolityczne wywołane przez stres oksydacyjny, spożycie bobu, leki (powstawanie nadtlenków i uszkadzanie erytrocytów), obecność ciałek Heinza wewnątrz erytrocytów (produkty degradacji hemoglobiny).
Rozpoznanie: wywiad i badanie przedmiotowe, wykazanie zmniejszonej aktywności G-6-PD w erytrocytach.
Niedobór kinazy pirogronianowej (PK).
najczęstszy wrodzony defekt glikolizy,
dziedziczy się autosomalnie, recesywnie.
Patogeneza: dojrzały erytrocyt nie zawiera mitochondriów i dlatego źródłem energii jest glikoliza, gradient sodowo - potasowy błony jest utrzymywany przez ATP, a więc defekt PK powoduje Ⴏ ilość ATP co w efekcie daje niedokrwistość hemolityczną
Klinika: powiększenie śledziony, w rozmazie krwi - akantocyty (obkurczone erytrocyty z wypustkami)
Rozpoznanie: wywiad i badanie przedmiotowe, zmniejszona aktywność PK erytrocytów
Leczenie: splenektomia - w razie nasilenie hemolizy w śledzionie
Hemoglobinopatie.
Niedokrwistość sierpowatokrwinkowa.
najczęstsza hemoglobinopatia
choroba dziedziczona autosomalnie dominująco
Patogeneza: u homozygotycznych nosicieli HbS całkowita Hb składa się w 80% z HbS i w 20% z HbF. W stanie zredukowanym HbS ulega precypitacji. Erytrocyty przybierają postać sierpowatą, traca swą prawidłową elastyczność i zatykają naczynia w mikrokrążeniu, co powoduje zawały narządowe.
Klinika: w chorych homozygotycznych - niedokrwistość hemolityczna oraz przełomy choroby wywołane zatorami naczyniowymi, zawały narządowe, splenomegalia
Rozpoznanie: badanie mikroskopowe - test na obecność krwinek sierpowatych oraz elektroforeza hemoglobiny
Leczenie: przyczynowe (allogeniczny przeszczep szpiku), objawowe (unikanie niedoborów tlenu i odwodnienia).
Talasemia (niedokrwistość śródziemnomorska, niedokrwistość tarczowatokrwinkowa).
Jest to ilościowe, a nie jakościowe zaburzenie syntezy Hb; genetycznie uwarunkowana błędna regulacja syntezy łańcuchów globinowych.
ၢ-talasemia - zmniejszona synteza łańcuchów ၢ
ၡ-talasemia - zmniejszona synteza łańcuchów ၡ
ၡ-talasemia
upośledzenie wytwarzania łańcuchów ၡ hemoglobiny,
rozpoznanie: elektroforeza hemoglobiny, badania genetyczne,
leczenie: zależy od natężenia objawów
ၢ-talasemia
najczęstsza talasemia
niedokrwistość niedobarwliwa
patogeneza: mutacja genu globiny ၢ powodująca zmniejszone wytwarzanie łańcuchów ၢ z kompensacyjnym wytwarzaniem łańcuchów δ lub γ
rozpoznanie: wywiad (miejsce urodzenia), obraz kliniczny i badanie elektroforetyczne hemoglobiny
Nabyte defekty błony erytrocytów.
Napadowa nocna hemoglobinuria (PHN, choroba Marchiafavy).
Jest to nabyta choroba klonu komórek pnia szpiku, polegająca na braku białka sprzężonego z glikanem fosfatydyloinozytolu (GPI).
Patogeneza: czynnikiem nasilającym hemolizę jest zmniejszenie wartości pH krwi występujące w nocy, przyczyną hemolizy są nieprawidłowości w obrębie struktur błonowych regulujących czynność dopełniacza (DAF, MIRL). Zakotiwczenie wymienionych białek w błonie komórkowej wymaga GPI - u tych chorych produkcja GPI w komórkach szpiku kostnego jest upośledzona lub zniesiona.
Następstwo:
aktywacja dopełniacza nasilona hemoliza aktywacja płytek krwi (tworzenie zakrzepów)
Klinika:
- triada objawów: hemoliza, cytopenia, zakrzepica (zmienne natężenie).
napadowy przebieg choroby (zakażenie, stres, leki)
hepatosplenomegalia
może prowadzić do niedokrwistości powodowanych niedoborem żelaza
Badania laboratoryjne:
objawy hemolizy wewnątrznaczyniowej, hemoglobinuria, hemosyderynuria, w niektórych przypadkach pancytopenia, badanie szpiku kostnego, obraz szpiku nieswoisty, zwykle występuje przerost komórek erytropoezy
Powikłania:
zakrzepy w obszarze krążenie wrotnego żył wątrobowych
zwiększenie ryzyka rozwoju zespołu mielodysplastycznego lub ostrej białaczki szpikowej.
Niedokrwistości hemolityczne spowodowane czynnikami pozakrwinkowymi.
Niedokrwistości hemolityczne wywołane przeciwciałami.
Przeciwciała przeciw erytrocytom:
w klasie IgM - dzięki większemu wymiarowi średnicy mogą wypełnić odstęp pomiędzy 2 erytrocytami stąd są to „przeciwciała kompletne”, typu zimnego,
w klasie IgG - nie wypełniają swą średnicą odstępu między 2 erytrocytami, „przeciwciała niekompletne”.
Hemolityczne odczyny poprzetoczeniowe.
Etiologia: najczęściej następstwo pomyłkowego przetoczenia krwi niezgodnej grupowo w układzie AB0
Klinika:
natychmiastowe hemolityczne odczyny poprzetoczeniowe - przebieg ostry, ciężki, hemoglobinuria, żółtaczka, pokrzywka itp.
późne hemolityczne odczyny poprzetoczeniowe - łagodna żółtaczka, gorączka, zmniejszenie stężenia hemoglobiny
Badania laboratoryjne:
Wolna Hb w surowicy i w moczu (czerwone zabarwienie)
Zmniejszone stężenie haptoglobiny, zwiększenie aktywności LDH i stężenie bilirubiny pośredniej
Zbyt małe zwiększenie stężenia Hb, liczby erytrocytów i wartości hematokrytu po przetoczeniu krwi
Rozpoznanie: wywiad + badanie przedmiotowe + diagnostyka serologiczna.
Niedokrwistość hemolityczna noworodków.
Choroba hemolityczna noworodków, konflikt serologiczny, ujawnia się w późnym okresie życia płodowego lub we wczesnym okresie noworodkowym jako wynik przedostania się przez łożysko przeciwciał anty-Rh (D) od matki mającej czynnik krwi Rh-ujemny do płodu mającego Rh-dodatni (rzadziej przyczyną jest konflikt w zakresie grup głównych - ABO).
Objawy: niedokrwistość (anemia), żółtaczka (nadmiar bilirubiny pośredniej), pojawienie się we krwi obwodowej licznych erytroblastów, wynikające z rozpadu (hemolizy) krwinek czerwonych płodu po reakcji z przeciwciałami matki.
Niedokrwistości autoimmunohemolityczne (autoimmune hemolytic anemia - AIHA).
1. AIHA wywołane niekompletnymi przeciwciałami ciepłymi z klasy IgG.
2. AIHA spowodowana przez zimne aglutyniny typu IgM.
AIHA wywołane niekompletnymi przeciwciałami ciepłymi z klasy IgG.
Patogeneza: autoprzeciwciała ciepłe typu IgG wiążą się w temperaturze ciała z erytrocytami, nie powodując ich hemolizy. Erytrocyty obładowane przeciwciałami niszczone są na drodze fagocytozy w wątrobie i śledzionie. W przewlekłym przebiegu niedokrwistość ujawnia się wówczas, gdy erytropoeza, która może wzrosnąć nawet 10-krotnie w stosunku do prawidłowej, nie może już skompensować utraty erytrocytów.
Klinika:
niedokrwistość hemolityczna,
możliwość wystąpienie przełomu hemolitycznego.
Rozpoznanie: zmniejszone stężenie Hb, objawy niedokrwistości hemolitycznej, istnienie przeciwciał ciepłych
AIHA spowodowana przez zimne aglutyniny typu IgM.
Patogeneza: zimne aglutyniny ulegają aktywacji przy udziale układu dopełniacza w temperaturze 0 - 5ႰC, stając się autoprzeciwciałami wywołującymi nasiloną aglutynację. W warunkach prawidłowych miano zimnych aglutynin jest bardzo małe. Kliniczne znaczenie nie zależy od tego parametru, lecz od zdolności wiązania przeciwciała z antygenem w temperaturze 30ႰC (czyli w temperaturze dystalnych części kończyn w zwykłej temperaturze zewnętrznej)
Rozpoznanie: wskazówki pośrednie: trudności w pobieraniu krwi do badania (spowodowane aglutynacją erytrocytów w igle), trudność w liczeniu erytrocytów, wykonaniu rozmazu krwi obwodowej i próby krzyżowej, OB. w temp. pokojowej jest podwyższone, oznaczenie miana zimnych aglutynin
AIHA spowodowana hemolizynami bitermicznymi typu IgG.
jest to najczęściej ostry odczyn po zakażeniu wirusowym występujący u dzieci (najczęstsza AIHA wieku dziecięcego),
choroba polega na gwałtownym rozwoju hemolizy wewnątrznaczyniowej, stężenie hemoglobiny jest mniejsze niż 5 g/dl,
objawy mają charakter przejściowy, wyzdrowienie następuje po ustąpieniu zakażenia,
najczęściej stwierdza się obecność przeciwciał poliklonalnych klasy IgG.
Rozpoznanie: odczyn Donatha-Landsteinera - bitermiczne hemolizyny przyłączają się w niskich temperaturach z dopełniaczem do erytrocytów i po ogrzaniu prowadzą do hemolizy, są to tzw. przeciwciała Donatha-Landsteinera.
Niedokrwistość aplastyczna- patogeneza, diagnostyka laboratoryjna.
Definicja:
Niedokrwistość aplastyczna jest to niewydolność szpiku powstająca w następstwie jego aplazji lub hipoplazji, powodująca pancytopenię. Rozpoznanie można postawić, gdy spełnione są przynajmniej 2 spośród 3 wymienionych niżej kryteriów:
3 stopnie ciężkości niedokrwistości aplastycznej |
Liczba granulocytów |
Liczba trombocytów |
Liczba retikulocytów |
Umiarkowana (nSAA) |
< 1500 / µl |
< 50 000 / µl |
< 60 000 / µl |
Ciężka (SAA) |
< 500 / µl |
< 20 000 / µl |
< 20 000 / µl |
Bardzo ciężka (vSAA) |
< 200 / µl |
< 20 000 / µl |
< 20 000 / µl |
Niedokrwistość aplastyczna charakteryzuje się zaburzeniem wytwarzania komórek krwi w szpiku kostnym. Następuje zastępowanie tkanki krwiotwórczej tkanką tłuszczową (hipoplazja lub aplazja). Aplazja prowadzi do zmniejszenia liczby wszystkich elementów morfotycznych we krwi obwodowej, czyli do pancytopenii.
Przyczyny wrodzone:
Aplazja wywołana czynnikami wrodzonymi jest niezwykle rzadka. Do wrodzonych niedokrwistości aplastycznych należy np. zespół Fanconiego.
Przyczyny nabyte:
Niedokrwistość aplastyczna samoistna stanowi około 70% wszystkich przypadków, jednak jej przyczyna nie jest znana.
Wtórna niedokrwistość aplastyczna, wywoływana przez różnego rodzaju czynniki.
- Leki - chloramfenikol, fenylbutazon, inne niesteroidowe leki przeciwzapalne, preparaty złota, kolchicyna, fenytoina, powodują ok. 10% przypadków
Substancje toksyczne - benzen, związane głównie z narażeniem na ich działanie w miejscu pracy, powodują ok. 10% przypadków
Promieniowanie jonizujące
Zakażenia wirusowe i bakteryjne np. wirusami zapalenia wątroby
Patogeneza:
Czynniki egzogenne takie jak leki lub zakażenia wirusowe działające na osoby chore, posiadające predyspozycje genetyczne, wyzwalają reakcję autoimmunologiczną skierowaną przeciwko tkance hematopoetycznej. U części chorych pojawiają się autoagresywne limfocyty T, skierowane przeciwko komórkom pnia.
Obraz kliniczny:
Niedokrwistość |
Granulocytopenia |
Trombocytopenia |
Bladość powiek |
Zakażenie, gorączka |
wybroczyny |
Duszność |
Martwica tkanek |
Krwawienia z dziąseł, nosa i innych |
Zmęczenie |
grzybica |
|
Przyspieszone opadanie krwinek czerwonych (OB) spowodowane jest samym zmniejszeniem ich liczby, niezależnie od przyczyny niedokrwistości.
Różnicowanie niedokrwistości aplastycznej:
Pancytopenia u chorych ze szpikiem normo lub bogatokomórkowym: zespół mielodysplastyczny, hiperslenizm (gromadzenie komórek krwi w śledzionie), niedobór witaminy B12 lub kwasu foliowego
Napadowa nocna hemoglobinuria
Układowy toczeń rumieniowaty
Nacieczenie szpiku spowodowane białaczką, chłoniakiem złośliwym lub innym nowotworem
Zwłóknienie szpiku
Aplazja wstępująca po intensywnej chemioterapii
Rozpoznanie:
Poprzez obraz krwi obwodowej oraz badanie cytologiczne/histologiczne szpiku. Obserwowana pancytopenia oraz aplazja szpiku, w którym stwierdza się wzrost tkanki tłuszczowej oraz zwiększenie liczby komórek limfoidalnych i plazmatycznych.
Dodatkowo wykonywane są badania mające na celu wykluczenie innych przyczyn
Leczenie:
Leczenie objawowe - polega na przetaczaniu krwinek czerwonych i płytek krwi oraz zapobieganiu i leczeniu zakażeń.
Leczenie przyczynowe - u chorych na ciężką postać niedokrwistości aplastycznej.
Leczenie przyczynowe:
Alogeniczny przeszczep szpiku kostnego lub komórek pnia histokompatybilnego dawcy rodzinnego, wskazany przy ciężkiej i bardzo ciężkiej niedokrwistości aplastycznej. Może wywoływać niepożądane powikłania takie jak działanie toksyczne, zakażenia, reakcja „przeszczep przeciwko gospodarzowi” lub odrzucenie przeszczepu.
Leczenie immunosupresyjne wskazane przy umiarkowanej niedokrwistości aplastycznej, u chorych na cięższe postaci u których brak zgodnego dawcy rodzinnego do przeszczepu lub u osób z przeciwwskazaniami do przeszczepu.
Inne metody leczenia takie jak preparaty immunoglobulin lub cytokiny.
Rokowanie:
Śmiertelność u nieleczonych dorosłych wynosi około 70%. Najważniejszym czynnikiem prognostycznym jest liczba granulocytów w momencie rozpoznania choroby. Po przeszczepie rodzinnym u 75% chorych remisja utrzymuje się przez około 10 lat. Po 10 latach terapii immunosupresyjnej żyje około 50% chorych.
Pancytopenia będąca objawem najcięższego uszkodzenia szpiku może powodować:
Izolowaną aplazję granulocytarną (występują ciężkie zakażenia bakteryjne)
Izolowaną aplazję trombocytarną (skaza krwotoczna z trombocytopenią)
Izolowaną aplazję erytrocytarną (objawy związane z niedokrwistością)
Niedokrwistość aplastyczna czysto czerwonokrwinkowa:
Charakteryzuje się wybiórczą aplazją szpiku kostnego dotyczącą układu czerwono-krwinkowego. Zaburzenie wytwarzania komórek tego układu w szpiku kostnym może wynikać z zahamowania dojrzewania erytroblastów lub pośrednio z hamowania wydzielania erytropoetyny. Niedokrwistość ta może mieć charakter pierwotny lub wystąpić wtórnie w niedokrwistościach hemolitycznych, chłoniakach lub infekcjach wirusowych.
15
Wyszukiwarka
Podobne podstrony:
Choroba niedokrwienna serca
(33) Leki stosowane w niedokrwistościach megaloblastycznych oraz aplastycznych
Zajecia 6 7 Test Niedokonczonych Zdan
09 Choroba niedokrwienna sercaid 7754 ppt
Mukowiscydoza, Niedokrwistość sierpowao krwinkowa
Niedokrwistosci
49 CHOROBA NIEDOKRWIENNA SERCA
NIEDOKRWISTOŚCI
NIEDOKRWISTOŚCI SEM 2011 2012
wykład choroba niedokrwienna serca
Kryterium niedokrwienia mięśnia sercowego w elektrokardiografie wysiłkowym
Czynniki ryzyka choroby niedokrwiennej serca cz
Stres a rozwój choroby niedokrwiennej serca
Choroba niedokrwienna, ostre zespoły wieńcowe
więcej podobnych podstron