Choroba syropu klonowego (ang: maple syrup urine disease, MSUD) jest wrodzoną wadą metabolizmu aminokwasów o łańcuchach rozgałęzionych czyli leucyny, izoleucyny i waliny. W pierwszym etapie ich metabolizmu (ryc.1) powstają związki tzw. α - ketokwasy, które nie podlegają dalszemu procesowi przemiany na skutek całkowitego lub częściowego braku wspólnego enzymu dehydrogenazy α - ketokwasów aminokwasów rozgałęzionych (BCKAD). Taka sytuacja powoduje znaczne podwyższenie stężenia zarówno leucyny, izoleucyny i waliny, jak i powstających z nich odpowiednich α -ketokwasów we krwi, erytrocytach, płynie mózgowo-rdzeniowym i moczu.
Nazwa choroby pochodzi od charakterystycznego zapachu moczu, przypominającego zapach syropu otrzymywanego z klonu kanadyjskiego, w Polsce kojarzy się najczęściej z zapachem maggi.
Ryc. 1 Metabolizm aminokwasów rozgałęzionych i miejsce bloku enzymatycznego
w chorobie syropu klonowego
Dziedziczenie MSUD.
Choroba syropu klonowego dziedziczy się w sposób autosomalny recesywny tzn., że aby pojawiło się ryzyko wystąpienia tej choroby u potomstwa, oboje rodziców muszą być nosicielami wadliwego genu. Oznacza to, że sami rodzice nie chorują, natomiast prawdopodobieństwo połączenia genów rodziców rozkłada się następująco (ryc. 2): 25% dla urodzenia się potomstwa zdrowego, 50% ryzyka, że potomstwo będzie nosicielem wadliwego genu MSUD i 25% ryzyko urodzenia dziecka z chorobą syropu klonowego. 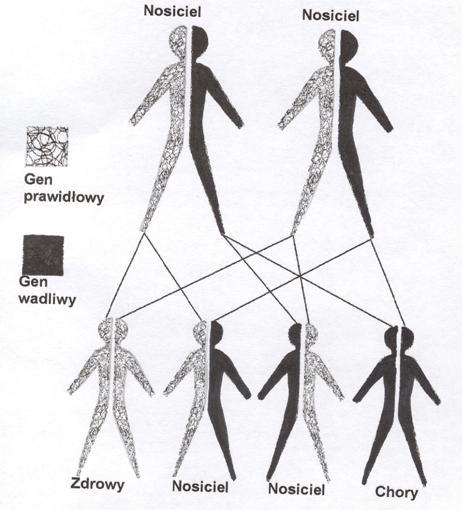
Ryc. 2 Dziedziczenie autosomalne recesywne
Diagnostyka
MSUD występuje we wszystkich grupach etnicznych ze światową częstością szacowaną na 1 : 185 000 żywych urodzeń. W Polsce choroba ta wykrywana jest w badaniu przesiewowym tzw. skriningu selektywnym obejmującym tylko noworodki lub niemowlęta z grupy ryzyka.
Częstość występowania MSUD w polskiej populacji określana jest na 1 : 250 000, co oznacza 1 do 2 nowych rozpoznań rocznie. Podstawą rozpoznania choroby jest stwierdzenie w surowicy i moczu metodą GC-MS obecności kwasów organicznych: kwasu α - keto-izokapronowego (KIC), kwasu α - keto-metylowalerianowego (KMV) i kwasu α - keto-izowalerianowego (KIV) oraz wysokiego stężenia w surowicy aminokwasów leucyny, izoleucyny i waliny metodą np. tandemowej spektrometrii masowej.
Diagnozę MSUD można potwierdzić analizą aktywności enzymu BCKAD w leukocytach, a także w hodowli fibroblastów skóry dziecka.
Postaci MSUD
W zależności od czasu wystąpienia ostrej fazy choroby, wartości stężenia aminokwasów rozgałęzionych w surowicy oraz szczątkowej aktywności kompleksu enzymatycznego BCKAD wyróżnia się 5 klinicznych i biochemicznych fenotypów:
* klasyczna - początkowo u noworodka zazwyczaj urodzonego w dobrym stanie, w ciągu pierwszych kilku dni narastają objawy niechęci do jedzenia, słabszego ssania, apatii i wiotkości. Prowadzi to do spadku masy ciała, szybko narastających objawów zatrucia i zagrożenia życia. Pojawiają się bezdechy, mogą wystąpić drgawki a nawet śpiączka. Stężenie aminokwasów rozgałęzionych, a zwłaszcza leucyny wzrasta we krwi do wartości 1000 - 5000 μmol/L (wartości fizjologiczne 65-220 μmolL). Aktywność enzymu BCKAD wynosi od 0 do 2% normy.
* przerywana - wczesny rozwój dziecka jest prawidłowy, pierwsze objawy kliniczne ujawniają się w tej postaci MSUD w wieku od 5 miesięcy do 2 lat, lub później, zwykle w czasie infekcji, po szczepieniu lub nagłym zwiększeniu zawartości pełnowartościowego białka w pożywieniu dziecka. Aktywność enzymu BCKAD ocenia się na 5 -20% normy. Tylko w czasie ostrej dekompensacji wzrasta stężenie leucyny do wartości 400-2000 μmol/L. Zazwyczaj rozwój psychoruchowy jest prawidłowy, jednakże sytuacje stresowe zawsze stwarzają ryzyko wystąpienia dekompensacji metabolicznej ze wszystkimi jej konsekwencjami.
* pośrednia - ta rzadka postać choroby może być wykrywana przypadkowo u dzieci bez objawów klinicznych, z nieznacznie ale przewlekle podwyższonymi stężeniami leucyny w granicach 50-1000 μmol/L i opóźnieniem rozwoju. Aktywność BCKAD mieści się w granicach 3-30% wartości prawidłowej.
* tiamino-zależna - postać MSUD o klinicznych cechach podobnych jak w postaci pośredniej. Leczenie witaminą B1 prowadzi do wyraźnego obniżenia stężenia leucyny.
* z deficytem podjednostki E3 (niedobór dehydrogenazy dwuhydrolipoilowej) - jest rzadką postacią, o fenotypie zbliżonym do postaci pośredniej, której towarzyszy ciężka kwasica mleczanowa. U pacjentów występuje łączny deficyt kompleksu BCKAD, dehydrogenazy pirogronianowej i dehydrogenazy α-ketoglutaranu.
Zaburzenia neurologiczne
Neurologiczna dysfunkcja jest często obserwowana u dzieci z chorobą syropu klonowego. Stała obecność w surowicy nie fizjologicznych stężeń leucyny, a zwłaszcza powstającego z niej kwasu α - ketoizokapronowego (KIC) może zgodnie z ich potencjalnie toksycznym działaniem prowadzić do ostrej lub przewlekłej dysfunkcji mózgu. Zrozumieniu patofizjologii zaburzeń neurologicznych służą badania (in vitro) na zwierzętach, w których wykazano wpływ stężenia α- ketokwasów na parametry metabolizmu energetycznego w korze mózgowej szczurów. Stwierdzono, że w MSUD większość metabolitów powstających z aminokwasów rozgałęzionych hamuje aktywność kompleksu łańcucha oddechowego, powodując obniżenie wydolności funkcjonowania mózgu. Zmiany w obrazie MRI obserwowane u dzieci z MSUD podczas ostrej metabolicznej dekompensacji całkowicie ustępowały po leczeniu wyrównującym stężenie leucyny, izoleucyny i waliny w surowicy. Wysoką istotną korelację pomiędzy zasięgiem zmian w istocie białej mózgu widocznych w badaniu MRI a zakresem stężeń aminokwasów rozgałęzionych stwierdzono w trzyletniej obserwacji u młodzieży i dorosłych z chorobą syropu klonowego. Według autorów badania podstawowym powodem objawów dysmielinizacji była utrzymująca się nierównowaga biochemiczna z podwyższonymi przez kilka lat stężeniami aminokwasów rozgałęzionych we krwi.
Podstawy leczenia dietetycznego w MSUD
Wczesne rozpoznanie i leczenie jest niezbędne dla zminimalizowania zmian w ośrodkowym układzie nerwowym. Przez całe życie obowiązuje ograniczenie zawartości aminokwasów rozgałęzionych w diecie, dostosowane do indywidualnej tolerancji i aktualnego stanu metabolicznego organizmu.
W okresie diagnostycznym, ze względu na ciężki stan intoksykacji (zatrucia) organizmu wymagane jest odmienne postępowanie terapeutyczne polegające na całkowitym odstawieniu podaży białka i na zahamowaniu katabolizmu białek ustrojowych poprzez znacznie zwiększony „dowóz” energii w diecie do wartości 130 - 140 kcal/ kg masy ciała. Początkowo konieczna jest zwykle dożylna podaż stężonych roztworów glukozy oraz lipidów do naczyń centralnych. Obniżanie stężenia toksycznych metabolitów w sposób fizjologiczny jest często zbyt wolne i nieefektywne, wtedy niezbędna bywa pozaustrojowa metoda eliminacji np. dializa otrzewnowa.
Po osiągnięciu obniżenia leucyny w surowicy do wartości ok. 1000umol/L dializa powinna być zakończona, a stężenie leucyny w dalszym ciągu powinno być obniżane poprzez wyłączne żywienie pozajelitowe lub w połączeniu z podażą wysokoenergetycznej diety z zastosowaniem specjalnego preparatu wolnego od aminokwasów rozgałęzionych, a dostarczających pozostałych aminokwasów niezbędnych do syntezy białka. Po osiągnięciu wyrównania biochemicznego, stopniowo można wprowadzać do diety białko naturalne w postaci pokarmu matki lub mleka modyfikowanego, po wyliczeniu zawartości aminokwasów rozgałęzionych zgodnie z normami.
Okresowo występujące infekcje, podwyższona temperatura ciała, obniżenie łaknienia przy ząbkowaniu lub luźne stolce zwłaszcza w pierwszych latach życia także zwiększają ryzyko metabolicznej dekompensacji. Często w czasie poprzedzającym ujawnienie się infekcji zmniejsza się tolerancja białka co prowadzi do wzrostu stężenia leucyny, izoleucyny i waliny w surowicy, pochodzących z katabolizmu białka endogennego, czyli organizmu ( białko mięśni). Dlatego każdy pierwszy sygnał jakiejkolwiek rozwijającej się infekcji upoważnia do rozpoczęcia postępowania zapobiegającego wzrostowi stężenia leucyny i jej toksycznego metabolitu kwasu KIC w surowicy, który mógłby spowodować nagłe neurologiczne pogorszenie. W warunkach domowych wczesne tymczasowe wycofanie źródeł białka naturalnego lub obniżenie spożycia do 25% dotychczasowej ilości w diecie, połączone z obfitym pojeniem co 1-2 godziny wysokoenergetycznymi roztworami maltodekstryny lub polimerów glukozy może być pomocne w ograniczeniu akumulacji niepożądanych związków. Podawanie w tym czasie preparatów nie zawierających aminokwasów rozgałęzionych jest niezbędne dla promowania syntezy białka, która jest jedyną drogą obniżania stężenia aminokwasów. Powrót do diety wyjściowej zwykle jest możliwy w ciągu kilku następnych dni.
Natomiast w sytuacji, gdy dziecko odmawia przyjmowania płynów już na początku rozwijającej się infekcji należy niezwłocznie zgłosić się do szpitala.
Postępowanie dietetyczne w stanie wyrównania metabolicznego ma na celu utrzymanie stężeń aminokwasów rozgałęzionych w zakresie najbardziej zbliżonym do wartości fizjologicznych oraz dostarczenie wszystkich składników odżywczych niezbędnych do utrzymania prawidłowego rozwoju fizycznego i umysłowego dziecka.
Aminokwasy rozgałęzione należą do aminokwasów egzogennych, tzn. takich, których organizm nie może sam wytworzyć, w związku z czym konieczne jest dostarczenie niewielkiej ich ilości z żywnością. Głównymi źródłami leucyny w diecie dla dzieci z MSUD są produkty zawierające białko o niskiej wartości biologicznej takie jak: ziemniaki, warzywa oraz niewielka ilość kasz. Żywność, którą charakteryzuje wysoka wartość biologiczna białka jest niedozwolona ze względu na zbyt wysoki udział leucyny. Zawartość izoleucyny i waliny w produktach spożywczych jest zazwyczaj znacznie niższa od zawartości leucyny. W sytuacji długo trwającej niedostatecznej podaży izoleucyny w diecie, dochodzi niekiedy do znacznego obniżenia jej stężenia w surowicy, prowadzącego do pojawienia się zmian skórnych przypominających acrodermatitis enteropatica. Suplementacja diety L-izoleucyną powoduje szybkie i całkowite ustąpienie zmian.
Nieodzownym elementem diety o obniżonej zawartości białka naturalnego jest udział preparatów wolnych od BCAA, o różnej kompozycji składników, w zależności od wieku pacjentów. Preparaty dla niemowląt w odróżnieniu od tych przeznaczonych dla dzieci starszych, zawierają także węglowodany i tłuszcz. Zróżnicowanie ilościowe i jakościowe składników w preparatach białkozastępczych jest odpowiednie do zmieniającego się zapotrzebowania rosnącego organizmu dziecka z MSUD. Uzupełnienie diety w pozostałe aminokwasy pozwala także na zminimalizowanie konkurencyjności w przepływie aminokwasów, szczególnie leucyny przez barierę krew-mózg.
Zalecana dzienna ilość suplementów aminokwasowych powinna być podzielona na kilka dawek w ciągu dnia (3-4) i po rozpuszczeniu w wodzie lub soku, spożyta w czasie posiłku lub krótko po posiłku.
Odpowiednie zalecenia i spożycie energii dostarczonej w pożywieniu zapewnia prawidłową syntezę białka i normalny rozwój fizyczny. Stężenie BCAA będzie wzrastało w surowicy, jeżeli zalecenia lub spożycie energii z dietą będą niewystarczające dla utrzymania równowagi energetycznej co może uruchomić proces katabolizmu. Źródłem energii w diecie dzieci z MSUD są tłuszcze, cukier, niskobiałkowe produkty: chleb, ciasta, makarony itp. oraz owoce, warzywa i energetyczne suplementy np. polimery glukozy lub mieszanina węglowodanów z tłuszczami.
W sytuacji okresowych trudności z łaknieniem, zwłaszcza u niemowląt i małych dzieci zachodzi konieczność żywienia przez sondę dożołądkową, aby zapobiec wystąpieniu metabolicznej dekompensacji. Dobrym rozwiązaniem w sytuacjach nietolerancji sondy , a także nasilonego refluksu żołądkowo- przełykowego, czy problemów z założeniem i utrzymaniem wejścia centralnego, może być założenie Przezskórnej Endoskopowej Gastrostomii (PEG-u).
Monitorowanie leczenia dietetycznego opiera się na ocenie klinicznego, biochemicznego i żywieniowego stanu dziecka z MSUD, ze szczególnym kontrolowaniem niedoborów białka objawiających się zmianami skórnymi.
Pokrycie dziennego zapotrzebowania na witaminy i składniki mineralne w zależności od wieku, może wymagać okresowej suplementacji w postaci preparatów farmaceutycznych.
Kontrola biochemiczna
Stężenie w surowicy leucyny , izoleucyny i waliny powinno być kontrolowane kilka razy w tygodniu przez pierwsze tygodnie życia, później kontrole mogą być prowadzone w odstępach 2-4 tygodniowych, a nawet 8 tygodniowych w zależności od stopnia równowagi biochemicznej u dziecka z MSUD.
Rokowanie w chorobie syropu klonowego
Odległe rokowanie w klasycznej postaci MSUD pozostaje niepewne. Zaburzenia neurologiczne i upośledzenie rozwoju psychomotorycznego są częstym powikłaniem. Efekty leczenia zależą od wczesnej diagnozy i postępowania terapeutycznego. Stwierdzono, że rozpoznanie choroby w czasie dłuższym niż 10-14 dni znamiennie częściej prowadziło do upośledzenia umysłowego dzieci.
Alternatywnym rozwiązaniem dla osób z MSUD może być przeszczep wątroby lub terapia genowa komórek somatycznych, podnosząca aktywność enzymu BCKAD. Jednak te nowoczesne metody leczenia pozostają jeszcze na etapie badań eksperymentalnych.
Większość doniesień naukowych sugeruje, że normalny rozwój fizyczny i umysłowy może być osiągnięty, jeżeli oprócz wczesnego rozpoznania i leczenia, będzie zapewniona konsekwentna długofalowa opieka, z efektywną terapią stanów katabolizmu.
1
Wyszukiwarka
Podobne podstrony:
bartek buczek Witamina B6, Biochemia, prace
choroby naczyn i serca(1)
ŻYWIENIE A CHOROBY 4b
Choroby układu nerwowego ppt
Produkty przeciwwskazane w chorobach jelit II
Choroba niedokrwienna serca
CZLOWIEK I CHOROBA – PODSTAWOWE REAKCJE NA
Choroby nerwów czaszkowych
Choroby Cywilizacyjne 5
3 Seminarium Patofizjologia chorób rozrostowych
Replikacja DNA i choroby związane
Choroby ukadu pok
Żywienie a choroby
Krew i choroby układu krwionośnego
choroby przysadki stomatologia
więcej podobnych podstron