3. metody syntazy polimerów
Ogólna charakterystyka reakcji syntezy polimerów.
Związki wielkocząsteczkowe otrzymuje się następującymi metodami:
- polimeryzacji addycyjnej;
- polimeryzacji kondensacyjnej (też polikondensacji [4,6]);
- chemicznej modyfikacji polimerów naturalnych lub syntetycznych.
Polimeryzacja jest to reakcja kolejnego przyłączania cząsteczek monomeru do centrum aktywnego połączona z utworzeniem łańcucha polimerowego i odtworzeniem centrum aktywnego:
![]()
gdzie: R• - centrum aktywne (rodnik);
M - monomery - związki zawierające wiązania:
Początkiem tej reakcji jest pojawienie się w masie reakcyjnej centrum aktywnego (R•). Cząsteczka monomeru zawiera jedną lub więcej grup (wiązań) reakcyjnych. W przypadku polimeryzacji monomeru, którego cząsteczki zawierają jedno wiązanie reakcyjne, powstają polimery liniowe. Obecność dwóch lub więcej wiązań reaktywnych może doprowadzić do otrzymania usieciowanych polimerów - nietopliwych i nierozpuszczalnych. Makrocząsteczki takich polimerów występuje w postaci przestrzennej sieci.
Polimeryzacja kondensacyjna - jest to reakcja podstawienia, w której cząsteczki reagujące są wielofunkcyjne - zawierają co najmniej dwie grupy reakcyjne:

gdzie: Z - nowe wiązanie utworzone w trakcie reakcji;
XY - związek uboczny małocząsteczkowy;
A,B - podstawniki cząsteczki monomerów - alifatyczny, aromatyczny lub alicykliczny.
Podczas polikondensacji wydzielają się produkty uboczne mołocząsteczkowe (XY) - woda, chlorowodór lub inne proste związki. Monomery polikondensacyjne zawierają co najmniej dwie grupy funkcyjne (X,Y). Gdy jeden związek zawiera dwie różne grupy, reakcja może przebiegać między cząsteczkami tego samego monomeru tworząc homopolimer (homopolikondensacja):
nX−R−Y → X(−R−Z−R−)Y + (n-1)XY
Jeżeli używa się dwóch różnych monomerów - powstaje kopolimer (heteropolikondensacja) (wymieniona wyżej - 3.2).
Modyfikacja chemiczna - jest to proces chemiczny polegający na otrzymywaniu związków wielkocząsteczkowych o innych właściwościach z surowców polimerowych naturalnych lub syntetycznych. Najszęściej jest to proces chemicznej zmiany struktury polimeru wyjściowego przez reakcję podstawienia podstawników bocznych lub przyłączenia zmieniające strukturę jednostki:
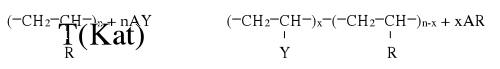
Przykładem rozpowszechnionych polimerów zmodyfikacyjnych są polimery celulozy, t.j. azotan celulozy, octan celulozy a także wielka ilość eterów celulozy termoplastycznych spośród których istnieją materiały rozpuszczalne nawet w wodzie (np. metyloceluloza).
3.1. Cechy charakterystyczne polimeryzacji addycyjnej
Do najczęściej spotykanych monomerów, ulegających reakcji polimeryzacji addycyjnej należą związki zawierające podwójne wiązania kowalencyjne (nienasycone), na przykład:
Kreska w strukturze związków chemicznych oznacza wiązanie utworzone za pomocą pary elektronów (w schemacie oznaczone punktami).
Polimeryzacja addycyjna jest reakcją łączenia i w każdym akcie łączenia monomeru zamiast wiązania π (3.6) tworzy się nowe wiązanie σ i regeneruje się centrum aktywny.
Polimeryzacja powstaje w wyniku pojawienia się w masie reakcyjnej centrów aktywnych (centrów aktywacji), którymi są rodniki lub jony. Rodzaj centrum zależy od mechanizmu rozpadu wiązania w monomerze. Rozpad wiązania może być homolityczny lub heterolityczny. Podczas rozpadu homolitycznego tworząca wiązanie π para elektronów zostaje podzielona pomiędzy sąsiednimi atomami (droga I) a w przypadku rozpadu heterolitycznego para elektronów przemieszcza się w całości do jednego atomu (droga II):
Cząsteczka zawierająca niesparowany elektron nazywa się rodnikiem. Rodnik jest związkiem zbyt krótkotrwałym - czas jego życia wynosi setne części sekundy w temperzaturze pokojowej. Wykazuje on dużą aktywność chemiczną, która zaś jest uzależniena od struktury cząsteczki. Cząsteczka zawierająca ładunek nazywa się jonem - anion o ładunku (-) i kation o ładunku (+). Jony wyróżniają się również zbyt wysoką aktywnością.
W zależności od rodzaju powstającego ośrodka aktywnego polimeryzację dzielimy na rodnikową i jonową (kationowa lub anionowa).
Polimeryzacja addycyjna jest reakcją łańcuchową w przebiegu której wyróżnia się trzy zasadnicze etapy:
- inicjowanie;
- wzrost łańcucha;
- zakończenie wzrostu łańcucha.
3.2. Polimeryzacja rodnikowa
Polimeryzacja rodnikowa jest reakcją łańcuchową o dużej szybkości. Przebiega ona w trzech etapach w wyniku powstawania centrum polimeryzacji w postaci rodnika.
3.2.1. Inicjowanie polimeryzacji rodnikowej
Inicjowanie jest początkowym etapem polimeryzacji. Inicjowanie polimeryzacji rodnikowej jest procesem tworzenia (powstawania) wolnych rodników służących do zainicjowania reakcji łączenia cząsteczek monomeru:
![]()
In _→ 2R•
gdzie: F - czynniki fizyczne;
In - czynnik chemiczny (inicjator).
Czynniki inicjowania dzialimy na fizyczne i chemiczne.
Do czynników fizycznych zaliczamy: energię cieplną, promienowanie ultrafioletowe lub radiacyjne, działanie mechaniczne i t.p.
W przypadku rozpadu wiązania-π pod działaniem temperatury mamy inicjowanie termiczne. Iniocjowanie termiczne zachodzi w podwyższonej temperaturze (120÷150°C):
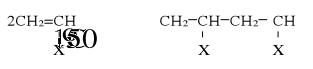
Energia aktywacji rozpadu wiązania podwójnego stanowi około 320÷350 kJ/mol. Natomist energia aktywacji tworzenia się wolnych rodników przez promienowanie ultrafioletowe lub radiacyjne jest bardzo mała i zbliża się do zera, dlatego reakcja przebiega w temperaturze pokojowej.
Do czynników chemicznych zaliczamy inicjatory - to są, najczęściej nadtlenki organiczne i nieorganiczne, wodoronadtlenki oraz niektóre związki azowe.
Inicjatory są to związki łatwo ulegające rozpadowi na wolne rodniki w temperaturze 40÷90 oC, o energii aktywacji reakcji rozpadu około 120÷130 kJ/mol. Na przykład nadtlenek di-tert-butylu (DTB) rozpada się z utworzeniem rodników w temperaturze 90 oC:
![]()
Oprócz nadtlenków alkilowych t.j. DTB stosowane są nadtlenki związków karboksylowych są to nadtlenki acylowe (np. nadtlenek benzoilu lub laurylu) i wodoronadtlenki, najczęściej spotykany z pośród których są wodoronadtlenek izopropylobenzenu, tert-butylu lub nadtlenek wodoru:
nadtlenek benzoilu wodoronadtlenek tert-butylu
Obecność jonów metali t.j. Fe2+, Mn2+, Co2+, Cr3+ lub anionu S2O3-2 znacznie przyspiesza rozpad nadtlenków:
H2O2 +Fe2+ → HO• + ![]()
+ Fe3+
![]()
+ Fe3+ → HO• + Fe2+
W wyniku małej energii aktywacji proces ten przebiega w temperaturze pokojowej. Zestaw na podstawie nadtlenku (utleniacz) i jonu redukującego (redoktor) nazywa się układem oksydacyjno-redukcyjnym lub redoks.
Szybkość inicjowania chemicznego określa się wzorem:
V=Kin⋅[In].
gdzie: Kin - stała szybkości inicjowania, s-1;
[In] - stężenie inicjatora, mol/dm3.
Ilość wprowadzonego do polimeryzacji inicjatora wynosi od 0,1 do 2 % wzgłędem do monomeru.
Porównawcze cechy energetyczne różnych sposobów inicjowania przedstawiono w tabeli 3.1.
Tabela 3.1
Energetyczne cechy różnych rodzajów inicjowania [7]
L p. |
Metoda inicjowania |
Energia aktywacji, kJ/mol |
Temperatura procesu, oC |
1. 2. 3. 4. 5. |
Inicjowanie termiczne Inicjowanie chemiczne Inicjowanie oksydacyjno-redukcyjne Fotoinicjowanie Inicjowanie radiacyjne |
352 ∼126 0÷84 13÷22 ∼0 |
120÷150 40÷120 20÷60 10÷25 0÷25 |
3.2.2. Wzrost łańcucha
Wzrost łańcucha polega na kolejnym przyłączeniu się cząsteczek monomeru do utworzonego aktywnego centrum (rodnika):
R•+M → R−M• + M → R−M−M • + . . . → R•m
gdzie R•m - rosnący makrorodnik.
Cząsteczka monomeru może polimeryzować w warunkach zmniejszenia potencjału termodynamicznego Gibbsa układu polimeryzacyjnego:
ΔG =(ΔH-TΔS)<O.
gdzie: ΔH - zmiana entalpii;
ΔS - zmiana entropii układu.
Polimeryzacja - jest procesem egzotermicznym o ujemnej ymianie entalpii ΔH=-(55÷80) kJ/mol. Wartość ΔG<0 będzie wówczas gdy TΔS będzie mniejsze od wartości (55÷80) kJ/mol [8]. Warunki te możliwe są przy zmniejszeniu entropii ΔS.
Szybkość wzrostu łańcucha jest bardzo duża i równa się 104÷106 mol/(dm3⋅s). Makrołańcuch tworzy się w czasie od setnych do dziesiątych części sekundy. Dlatego w każdym momencie czasu obserwowanego ciężar cząsteczkowy polimeru jest wartością stałą, gdy ż ilość tworzonego polimeru podczas polimeryzacji zwiększa się wraz z postępem reakcji. Przetworzenie monomeru w polimer jest konwersią.
Podczas przyłączenia monomeru do rodnika rosnącego mogą powstawać trzy konfiguracje w zależności od rozmieszczenia grupy bocznej:
głowa do ogona głowa do głowy ogon do ogona
Bardziej uprzewilejowane jest powstawanie konfiguracji typu (a). Powstająca konfiguracja może okazywać wpływ na powstawanie stereoizomerii makrocząsteczek zależnie od regularności przyłączenia. Makrocząsteczki o regularnej budowie powstają gdy przyłączenie odbywa się stale o jednakowym schemacie. W przypadku przylączenia sporadycznego o różnym schemacie powstają makrocząsteczki o budowie nieregularnej.
Szybkość reakcji wzrostu łańcucha okresla się wzorem:
VW=[R•][M]
gdzie: [R•] i [M] - stężenie rodników i monomeru.
3.2.3. Zakończenie wzrostu łańcucha
Zakończenie wzrostu łańcucha jest ostatnim etapem polimeryzacji, która polega na zaniku makrorodników na końcu rosnącej makrocząsteczki. Może nastąpić według jednej z następujących reakcji:
1) rekombinacja (łączenie się rodników):
R•m + R•m → Rm−Rm
(makrocząsteczka)
m + m = n; L = 2γ - długość łańcucha równa się dwom długościom makrorodnika (stopień polimeryzacji jest sumą liczby merów w obu rodnikach).
2) dysproporcjonowanie - przebiega przez przeniesienie atomu wodoru z jednego makrorodnika na drugi:
HR•m + HR•m → Rm + H2Rm
M = n i L = γ.
Określrenie sybkości reakcji zakonczenia łańcucha prowadzi się przez wzór:
Vz = Kz ∙ [R•m]
Dysproporcjonowanie przebiega zwłaszcza w temperaturze podwyższonej.
Reakcja zakończenia wzrostu łańcucha charakteryzuje się małą energią aktywacji - od 8 do 22 kJ/mol. Mechanizm reakcji zakończenia łańcucha wykazuje wpływ na długość łańcucha polimeru oraz na strukturę makrocząsteczki. Na ogół zgodnie z wymienionym wyżej schematem, podczas polimeryzacji powstają makrocząsteczki o liniowej strukturze łańcucha. Natomiast kiedy jest możliwość przebiegu reakcji przeniesienia łańcucha w wyniku zderzenia się makrorodnika z obojętną cząsteczką polimeru powstaje rodnik w środku makrocząsteczki:
Makrorodnik powstający przy przeniesieniu łańcucha (R•1m) jest odpowiedzialny za utworzenie się makrocząsteczki rozgałęzionej.
Prawdopodobieństwo utworzenia cząsteczek rozgałężonych wzrasta z podwyższeniem temperatury reakcji.
W zależności od sposobu i warunków prowadzenia polimeryzacji można otrzymać polimery różniące się masą i strukturą cząsteczkową, polidyspersyjnością, a tym samym właściwościami technologicznymi i użytkowymi.
Wskutek udziału w reakcji zakończenia łańcucha dwóch rodników szybkość polimeryzacji określa się wzorem, zawierającym stężenie inicjatora z potęgą 0,5:
![]()
gdzie: Kp - stała szybkości polimeryzacji, s-1.
[In] i [M] - odpowiednio stężenia inicjatora i monomeru.
Średni stopień polimeryzacji (długość łańcucha) oblicza się za pomocą wzoru [8]:
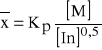
który zmnieja się od podwyższenia temperatury polimeryzacji zgodnie z zależnością:
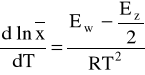
gdzie: Ew i Ez - energia aktywacji wzrostu i zakończenia łańcucha, kJ/mol;
R - stała Boltzmana, kJ;
T - temperatura polimeryzacji, K.
Reakcja przeniesienia łańcucha towarzyszy procesowi hamowania polimeryzacji za pomocą inhibitorów:
∼R•m + AX → ∼Rm−X + A•
Proces ten polega na utworzeniu rodników (A•) niezdolnych do inicjowania. Utworzone rodniki A• uczestniczą tylko w rekombinacji.
a) A• + A• → A−A
b) A• + ∼R•m → A−Rm
Związek AX w równianiu 3.25 występuje jako inhibitor.
Jako inhibitory (AX) najczęście stosuje się fenole, jednozasadowe aminy aromatyczne, sole miedzi, molibdenu, cyny, nitrozwiązki i inne. Inhibitory są dodawane do monomerów przy ich przechowywaniu lub dodaje się je do masy reakcyjnej dla zahamowania polimeryzacji na pewnym stopniu konwersji monomeru w polimer.
3.3. Polimeryzacja jonowa
Polimeryzacja jonowa wyróżnia się tym, że aktywne centrum ma budowę jonu. W przypadku, kiedy ośrodek jest kationem (karbokation), mamy polimeryzację kationową, a przy tworzeniu się karboanionu przebiega polimeryzacja anionowa.
3.3.1. Polimeryzacja kationowa
Zainicjowanie procesu zachodzi poprzes odziaływania kationów powstających przy dysocjacji katalizatora z cząsteczkami monomeru z utworzeniem aktywnych karbokationów. Zwykle stosowanymi katalizatorami w procesach polimeryzacji kationowej są kwasy Lewisa (AlCl3, BF3, SnCl4, TiCl4) jak i kwasy protonowe (siarkowy, fosforowy).
Mechanizm zapoczątkowania polega na dysocjacji katalizatora:
A:B → A+B-;
W przypadku wykorzystania jako katalizatora trójfluorku boru (BF3) inicjowanie zachodzi w wyniku reakcji katalizatora z kokatalizatorami - donorami protonów (woda, alkohol) i dysocjacji utworzonej cząsteczki:
A). Inicjowanie BF3 + HOH → H+[BF3OH]-
BF3 - katalizator; HOH - kokatalizator.
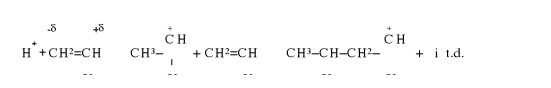
B). Wzrost łańcucha powstaje w skutek przyłączenia utworzonego protonu do cząsteczki monomeru powodującego tworzenie się bardzo aktywnego karbokationu, do którego przyłącza się następna cząsteczka monomeru:
Polimeryzacji kationowej ulegaję się monomery zawierające elektrodonorowe podstawniki boczne (X).
Energja aktywacji reakcji Ea = 50÷60 kJ/mol [8].
Wzrost łańcucha przebiega według mechanizmu przyłączania cząsteczek monomeru podobnie jak w polimeryzacji rodnikowej, dla tego jest to polimeryzacja adycyjna.
Charakterystyczną cechą polimeryzacji kationowej jest niska energia aktywacji i duża szybkość w niskich temperaturach - Tpolim=-50÷-110оC [9]. Polimeryzacja kationowa odróżnia się ujemnym wskaźnikiem temperaturowym wskutek czego szybkość procesu wzrasta przy obniżeniu temperatury. W warunkach takich unika się niepożądanych reakcji ubocznych (np. przeniesienie łancucha na polimer), co umożliwia uzyskanie polimerów regularnych o dużym ciężarze cząsteczkowym. Na przebieg polimeryzacji wpływa również rodzaj rozpuszczalnika, powodującego dysocjację katalizatora w warunkach stabilności kationu.
C). Zakonczenie wzrostu łancucha w polimeryzacji kationowej prowadzi do regeneracji katalizatora w wyniku przeniesienia kationu wodoru na istniejący w roztworze anion:
Przy polimeryzacji kationowej tworzą się polimery liniowe o regularnej budowie łańcucha.
3.3.2. Polimeryzacja anionowa
Reakcja przebiega pod wpływem czynników nukleofilowych, głównie anionów. Katalizatorami polimeryzacji anionowej są metale alkaliczne, ich wodorki, wodorotlenki, amidki i alkoholany: butylolit (Li-C4H9); etylan litu (Li-OC2H5); wodorek potasu (K-H); wodorotlenek sodu (NaOH); amidek sodu (NaNH2); potas (K).
Typowym przypadkiem polimeryzacji anionowej jest polimeryzacja katalizowana przez amidek w ciekłym amoniaku jako rozpuszczalniku [8]:
a) inicjowanie polega na dysocjacji katalizatora na jony:
NaNH2 → Na+ + NH2-
b) wzrost łańcucha zaczyna się wskutek przyłączenia utworzonego anionu do cząsteczki monomeru z utworzeniem karboanionu:
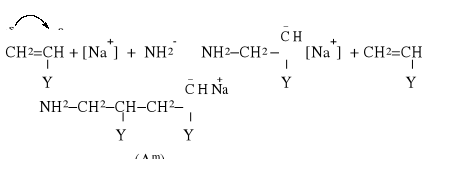
Wzrost łańcucha jest identyczny jak w przypadku polimeryzacji rodnikowej. Do polimeryzacji anionowej podatne są monomery zawierające podstawnik boczny elektronoakceptorowy (Y). Wzrost łańcucha przebiega w temperaturze -40÷50oC z utworzeniem makrocząsteczek o strukturze liniowej i pewnej regularności [10].
W polimeryzacji anionowej możliwe jest otrzymanie tak zwanych polimerów “żyjących” o strukturze (Am), charakteryzujące się brakiem etapu samorzutnego zakończenia łańcucha. Wzrost makrocząsteczki morze przebiegać nadal po dodaniu nowej porcji monomeru. W takim przypadku polimeryzacja musi przebiegać w warunkach całkowicie bezwodnych, bez dostępu tlenu i innych środków wodorodonorowych. W taki sposób syntezuje się blok-kopolimery.
zakonczenie łańcucha przebiega w obecności donora protonów:
3.3.3. Polimeryzacja koordynacyjna jonowa
Odróżnia się od innych rodzajów polimeryzacji wykorzystaniem zespolowych katalizatorów. Najbardziej znane i powszechnie stosowane katalizatory tego typu są to katalizatory Zieglera-Natty, wytwarzane na podstawie trujchlorku tytanu i glinu trujalkilowego. Cząsteczka monomeru zostaje skoordynowana przez atom metalu katalizatora. Dzięki temu umożliwia się powstawanie polimerów o regularnej budowie izotaktycznej lub syndiotaktycznej.
Polimeryzacja koordynacyjna przebiega przez dwa etapy. Na pierwszym etapie (I) odbywa się orientacja cząsteczki monomeru do metalu katalizatora z następującym jej wdrodzeniem w strukturę kompleksu - etap (II):
Wzrost łańcucha można porównać do wzrostu włosa - cząsteczka monomeru przyłącza się ze strony kompleksu katalizatorowego.
3.3.4. Polimeryzacja z przeniesieniem atomu
Do polimeryzacii addycyjnej należy również polimeryzacia w której następuje przeniesieniu atomu (migracyjna), która przebiega za pomocą wędrówce (migracji) atomów o wysokiej ruchliwości, znajdujących się zwykłe w grupach funkcyjnych. Do takiego procesu może być zaliczona reakcja syntezy poliuretanów na podstawie dwuizocyjanianów i dioli:
Proces ten zalicza się do polimeryzacji addycyjnej w wyniku tożsamościowości typu reakcji - przyłączenia bez utworzenia związków ubocznych.
3.4. Kopolimeryzacja
Jest polimeryzacją, w której reagują ze sobą dwa różne monomery. Wytworzone kopolimery w zależności od składu monomerów i wzajemnego ich stosunku ilościowego mają różny skład, strukturę i właściwości.
Kopolimery ogólnie można podzielić na statystyczne, naprzemienne, blokowe i szczepione.
- kopolimery statystyczne są produktami o nieuporządkowanym (przypadkowym) rozmieszczeniu w makrocząsteczce merów pochodzących z użytych monomerów A i B:
−A−B−A−B−B−B−A−A−B−A−
- kopolimery naprzemienne są produktami o budowie regularnej: makrocząsteczki są zbudowane z merów A i B sąsiadujących ze sobą przemiennie:
−A−B−A−B−A−B−A−B−
- kopolimery blokowe mają strukturę łańcuchową, a ich makrocząsteczka składa się z oddzielnych odcinków (bloków) zbudowanych wyłącznie z merów A lub wyłącznie z merów B:
−A−A−A−A−A−A−A−B−B−B−B−B−B−B−
Kopolimery blokowe otrzymuje się przez wytworzenie makrorodników pod działaniem promeni ultrafioletowe w mieszaninie telomerów z monomerem lub polimerem innej budowy, albo przez wykorzystanie otrzymanych metodą polimeryzacji anionowej polimerów “żyjących” i monomerów o innej budowie. Telomery - są to polimery lub oligomery, który powstają podczas polimeryzacji rodników w środowisku czterochlorku węgla:
Polimery blokowe są materiałami o skomplikowanych właściwościach fizykochemicznych, które mogą mieć dwie różne wartości temperatury zeszklenia lub topnienia.
Kopolimery w zależności od składu monomerów i wzajemnego ich stosunku ilościowego mają różny skład, strukturę i właściwości fizyczne lub użytkowe. Mery A i B pochodzące z użytych monomerów w makrocząsteczce kopolimeru mogą być ułożone w sposób regularny, nieregularny, blokowy lub szczepiony. Biorąc to pod uwagę ustalono ich nazewnictwo w sposób następujący:
• Kopolimery z nieokreślonym układem merów. Nieokreślony układ merów przedstawia się jako (A-co-B), a uzyskany kopolimer ma nazwę poli(A-co-B), np. kopolimer nieokreślony styrenu i metakrylanu metylu nazywa się:
poli[styren-co-(metakrylan metylu)]
• Kopolimery statystyczne, w których mery A i B są rozmieszczone przypadkowo, lecz wg praw statystycznych np.:
Stanowią one większość wytwarzanych polimerów, jest nim np. kopolimer butadienu i styrenu. Statystyczny układ merów przedstawia się jako (A-stat-B), przy czym -stat wskazuje, że statystyczny rozkład merów uważa się za znany. Ich nazwa jest następująca:
poli(A-stat-B), np. Poli(styren-stat-butadien)
• Kopolimery przemienne mają budowę regularną, a mery A i B są ułożone przemiennie:
Otrzymuje się je przez polimeryzację kondensacyjną, np. poliamidów i poliestrów. Przemienny układ monomerów przedstawia się jako (A-alt-B), a uzyskany kopolimer przemienny nazywa się:
poli(A-alt-B), np. poli[styren-alt-(bezwodnik maleinowy)]
• Inne rodzaje kopolimerów okresowych, w których mery występują w sposób uporządkowany, np.:
Okresowy układ merów przedstawia się np. jako (A-per-B-per-B), a kopolimer okresowy:
poli(A-per-B-per-B)
• Kopolimery blokowe mają budowę łańcuchową, a makrocząsteczka zawiera odcinki (bloki) zbudowane wyłącznie z merów A lub B, np.
Układ merów w blokach przedstawia się Ak-block-Bm, a uzyskany kopolimer: poliA-block-poliB, np.:
polistyren-block-polibutadien
Kopolimery blokowe mogą mieć dwie wartości temperatury zeszklenia, odpowiadające wartościom temperatury zeszklenia odpowiednich homopolimerów.
• Kopolimery szczepione mają budowę rozgałęzioną, łańcuch główny składa się z merów A, a łańcuchy boczne z merów B, np.:
Kopolimer ten określa się: poliA-graft-poliB, przy czym mery A tworzą łańcuch główny, a mery B - łańcuchy boczne, np. polistyren szczepiony na polibutadienie:
polibutadien-graft-polistyren.
Kopolimery szczepione otrzymuje się w warunkach polimeryzacji szczepionej, w których wytwarza się centra aktywne na makrocząsteczce mogące następnie inicjować polimeryzację innego monomeru (B). Centra aktywne w polimerze A mogą powstawać podczas napromienowania, utleniania, pod wpływem ultradźwięków oraz przez przeniesienie łańcucha podczas polimeryzacji rodnikowej. W wyniku polimeryzacji szczepionej można połączyć ze sobą polimery wzajemnie się nie mieszające, np. [8]:
polietylen-graft-polikwas akrylowy
3.5. Polimeryzacja kondensacyjna
Polimeryzacja kondensacyjna nazywana jest też krótko polikondensacją. Jest ona reakcją podstawienia w grupach funkcyjnych i przebiega stopniowo. W każdym stadium reakcji tworzą się przejściowe, trwałe związki i wydziela się produkt uboczny, np.:
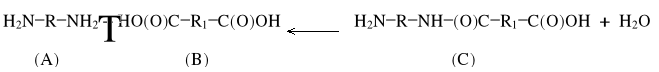
Następnie monomery A i B mogą reagować między sobą lub z C, tworząc D i t.d.
Dla syntezy polimerów metodą polikondensacji wykorzystywane są monomery zawierające dwie lub więcej grupy zdolne do reakcji, nazywane grupami funkcyjnymi. Reakcja odbywa się stopniowo przez podstawienia w grupach funkcyjnych, tworząc związki pośrednie.
Grupami funkcyjnymi są:

Związkami wyjściowymi (monomerami) są: kwasy dikarboksylowe lub ich chlorobezwodniki, diaminy, diole i inne.
Otrzymywane polimery nazywają się polikondensacyjnymi. Mają one mniejszy ciężar cząsteczkowy niż polimery uzyskane w polimeryzacji addycyjnej i odznaczają się dużymi oddziaływaniami międzycząsteczkowymi, co na ogół zwiększa ich krystaliczność i sztywność a także powoduje wysoką wytrzymałość mechaniczną. Do polimerów polikondensacyjnych zaliczamy:
- poliamidy (monomerami są kwasy i diaminy);
- poliestry (monomerami są kwasy i dioli);
- poliwęglany (na podstawie dioli i kwasu chlorowęglowego);
- polisiloksany (poliorganosiloksany i silany na podstawie dihydroorganosiloksanów);
- poliimidy (diaminy i dwubezwodniki kwasów tetrakarboksylowych);
- żywice fenoloformaldehydowe, mocznikowe, melaminowoformaldehydowe, epoksydowe i inne.
Podczas polikondensacji monomer zanika już w początkowym stadium reakcji, a ciężar cząsteczkowy uzyskanego polimeru wolno się zwiększa i wzrasta po konwersji grup funkcyjnych ponad 0,95 (95%).
Polikondensacja w zależności od rodzaju grup funkcyjnych monomerów i warunków reakcji może być równowagowa lub nierównowagowa.
W przypadku polikondensacji równowagowej średni stopień polimeryzacji jest zależny od ilości produktu ubocznego, znajdującego się w strefie reakcji. Te zależność określa się wzorem [11]:
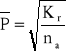
gdzie: Kr - stała równowagi reakcji;
na - ułamek molowy związku małocząsteczkowego, ubocznego, np. H2O.
Średni stopień polimeryzacji jest wprost proporcionalny do pierwiastka kwadratowego ze stałej równowagi i odwrotnie proporcjonalny do ułamka molowego wydzielającego się związku małocząsteczkowego. Stała równowagi określa się za pomocą zależności:
![]()
gdzie: [-NHCO-] i [H2O] - stężenie polimeru utworzonego i związku ubocznego;
[-COOH] i [-NH2] - stężenie grup funkcyjnych w cząsteczkach monomerów wyjściowych.
Dla polikondensacii równowagowej Kr znajduje się w zakresie od 10 do 1000.
Ze wzoru 3.38 wynika że wrost stopnia polimeryzacji uwarunkowany jest znmiejszeniem ułamku związku ubocznego (na) w strefie reakcji, co wymusza przeprowadzenie polikondensacji w warunkach szybkiego usuwania (odparowania) tego związku.
Ważną wielkością w procesie polikondensacji jest stopień przereagowania grup funkcyjnych, który określa się wzorem:
![]()
gdzie: ![]()
- średni stopień polimeryzacji;
fsr - średnia funkcyjność monomerów:
![]()
gdzie: fA i fB - zawartości grup funkcyjnych w cząsteczkach A i B;
NA i NB - liczba cząsteczek monomerów A i B (ułamki molowe).
Polikondensacja prowadzi do powstania związków wielkocząsteczkowych w przypadku, gdy stopień przereagowania grup sięga ponad 0,99. Na przykład ![]()
=0,998 stopień polimeryzacji sięga wartości![]()
= 500, gdy fsr = 2. Ze wzrostem fsr średni stopień przereagowania końcowy (![]()
) maleje, a (![]()
) rośnie wskutek utworzenia się polimeru o strukturze usieciowanej, która zawiera nieprzereagowane grupy funkcyjne, np. przy f = 2,4 stopień polmeryzacji ![]()
sięga nieskończoności gdy ![]()
=0,8.
Niektóre cechy polimeryzacji addycyjnej i kondensacyjnej zestawiono w tabeli 3.2.
Tabela 3.2
Porównanie reakcii
Cecha |
Polimeryzacja addycyjna |
Polimeryzacja kondensacyjna |
Typ reakcji |
przyłączenia, łańcuchowa |
podstawienia, stopniowa |
Zmiana ciężaru cząsteczkowego (M) w ciągu reakcji (rys.3.2) |
niezmienna |
rośnie |
Charakter przebiegu |
nierównowagowa |
równowagowa |
Zmiana stężenia monomeru w ciągu reakcji |
maleje stopniowo |
zanika na początkowym etapie |
Porównanie kinetyk wymienionych reakcji
Polimeryzacja rodnikowa [8]:
Vp=K[M][In]1/2
gdzie Vp - szybkośći reakcji, zwiększa się z podwyższeniem T, na skutek tego że:
K=Ae-E/RT
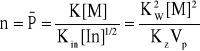
2. Polimeryzacja jonowa:
Vp=K[kat][M]n
gdzie n=1÷3.
[kat] - stężenie katalizatora.
Vp rósnie z obniżeniem T.
3. Polikondensacja równowagowa [11]:
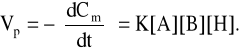
gdzie: Cm - stężenie grup funkcyjnych;
[A] i [B] - stężenia monomerów;
[H] - stężenie katalizatora kwasowego.
O różnym charakterze polimeryzacji addycyjnej i polikondensacji świadczy zróżnicowany charakter zmiany ciężaru cząsteczkowego (M) podczas reakcji (rys.3.2.).
3.6. Modyfikacja chemiczna polimerów
Modyfikacja chemiczna polega na zmianie budowy chemicznej polimeru przez reakcję podstawienia w makrocząsteczkach wyjściowych polimerów.
Metodę tę opracowano na początku dwudziestego wieku dla uzyskania polimerów o niezbędnych właściwościach na podstawie polimerów naturalnych i najpierw celulozy. Celuloza jest rozpowszechniony polimer naturalny cechuje się właściwościami wytrzymałościowymi, jednak nie poddaje się rozpuszczeniu i topnieniu przy ogrzewaniu na skutek dużej sztywności ją makrocząsteczek, spowodowanej budową chemichną:
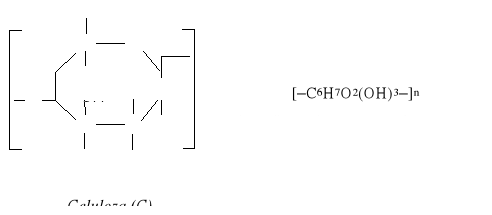
Celuloza nie poddaje się przetwórstwu wiadomymi metodami poprzez topnienie lub rozpuszczanie.
Produkty modyfikacji celulozy to: estry (azotany, octany, pochodne metoksylowe i benzoksylowe) lub etery (metylo-, etyloceluloza) - są produktami przetwarzalnymi i znajdują zastosowanie np: celuloid, jedwab i folia i inne.
Działając kwasem azotowym na celulozę otrzymujemy azotan:
[−C6H7O2(OH)3−]n + mHNO2 ![]()
[−C6H7O2(OH)(ONO2)m−]n azotan (C)
rozpuszczalny, sprężysty(folia)
Octan celulozy wytwarza się w wyniku działania na celulozę bezwodnikiem kwasu octowego:
[−C6H7O2(OH)3−]n + m(CH3C−O−)2 → [−C6H7O2(OH)(O−CCH3)m−]n octan (C)
|| ||
O O
topliwy, rozpuszczalny, elastyczny (włókna), folia
Do modyfikacji używa się też polimerów syntetycznych. Na przykład polimer taki jak poli(alkohol winylu) (PWA) otrzymywany jest poprzez hydrolizę polimeru syntetycznego poli(octanu winylu) (PWO) w środowisku metanolu:
Poli(alkohol winylu) - polimer wysokokrystaliczny, który rozpuszcza się w wodzie ale jest bardzo benzo- i olejoodporny charakteryzuje się wysoką wytrzymałością mechaniczną, natomiast poli(octan winylu) jest polimerem amorficznym, plastycznym o niskiej wytrzymałości mechanicznej i dobrej rozpuszczalności w alkoholach. Poli(alkohol winylu) jest stosowany do celów medycznych i spożywczych, np. w postaci rozpuszczalnych nici chirurgicznych.
Metodą modyfikacji otrzymuje się wiele znanych polimerów o wysokich właściwościach fizyko-mechanicznych i łatwej przetwarzalności. Do polimerów takiego rodzaju można zaliczyć kauczuk i polietylen chlorowane lub chlorosulfonowane, żywice jonowymienne i inne. Poniżej przedstawiono schemat otrzymywania polietylenu chlorowanego:
⋅⋅⋅ −CH2−CH2−CH2−CH2− ⋅⋅⋅ + Cl2 → −CH2−CH−CH2−CH−
| |
Cl Cl
krystaliczny (Sk>60%)ρw - niska; amorficzny, rozpuszczalny,
polimer nierozpuszczalny ρw - podwyższona
Otrzymany polietylen chlorowany wyróżnia się podwyższoną gestością, strukturą amorficzną i dobrą rozpuszczalnością. Natomiast polietylen wyjściowy jest polimerem nierozpuszczalnym, częściowo krystaliczny (Sk>60 %) o niskiej gęstości.
41
(3.1)
![]()
gdzie y = O lub N.
−C=C− −C≡C− lub −C=O a także
(3.2)
(3.3)
(3.4)
(3.6)
(3.7)
(3.15)
~CH2−CH−CH2−CH~ + R• → ~CH2−C•− CH2 −CH~ + RmH
|
X X X X
R•1m
X
|
(CH−CH2−)m
|
~CH2−C•−CH2−CH~ + m CH2=CH → ~CH2−C−CH2−CH~
| | | | |
X X X X X
~CH2−![]()
+ [BF3OH]- → ~CH=CH + H[BF3OH].
X X
~CH2−![]()
Na+ + ROH → ~CH2−CH−R + NaOH
| |
X X
(3.38)
(3.37)
(3.35)
(3.19)
Rys.3.1. Zależność stopnia polimeryzacji ![]()
od stopnia
przereagowania grup funkcyjnych [11]
(3.39)
(3.40)
(3.41)
(3.42)
(3.45)
(3.44)
(3.43)
(3.46)
(3.36)
Rys.3.2. Zmiana stopnia polimeryzacji w czasie:
1 - polimeryzacja addycyjna [8]; 2 - polikondensacja [11]
(−CH2−CH−)n (−CH2−CH−)n + nCH3OCCH3
| | ||
O−C−CH3 OH O
||
O
(PWO) (PWA)
+nCH3OH
(3.5)
(3.8)
(3.9)
(3.10)
(3.11)
(3.12)
(3.13)
(3.14)
(3.16)
(3.17)
(3.18)
(3.20)
(3.21)
(3.22)
(3.23)
(3.25)
(3.26)
(3.24)
(3.27)
(3.28)
(3.29)
(3.30)
(3.31)
(3.32)
(3.33)
(3.34)
Wyszukiwarka
Podobne podstrony:
FChP-tab11 3, Fizykochemia - książka
FChP-R13, Fizykochemia - książka
FChP-R4, Fizykochemia - książka
FChP-R5, Fizykochemia - książka
FChP-R12, Fizykochemia - książka
FChP-R6, Fizykochemia - książka
Spis trecsi, Fizykochemia - książka
Książka, FIZJOTERAPIA, Fizykoterapia, Książka
FChP-Liter, Tworzywa sztuczne, Fizykochemia
FChP-R2, Tworzywa sztuczne, Fizykochemia
FChP-R1, Tworzywa sztuczne, Fizykochemia
Historia książki 4
fizykoterapia 4
1 WSTEP kineza i fizykot (2)
wyklad 13nowy Wyznaczanie wielkości fizykochemicznych z pomiarów SEM
Właściwości fizykochemiczne białek
2 Fizyko KRIOTERAPIA 2008
Historia książki
więcej podobnych podstron