BIOLOGIA MOLEKULARNA
Wykład 1, 6.10.2006
„Genomy” Brown
„Genom człowieka” PWN
„Molecular biology of the cell”
Biologia molekularna - nauka o zapisie genetycznym i mechanizmach jego odczytu. To młoda nauka, zaczęła się kształtować mniej więcej w połowie XX wieku. Datą przełomową jest rok 1953.
Genom - kompletny zestaw genów danego organizmu lub komórki (tak naprawdę to cała sekwencja nukleotydowa DNA zawartego w danym organizmie czy komórce)
Genomika - nauka o kompletnych genomach, pojawiła się w latach 90 XX wieku, w 1995 roku poznano pierwszy kompletny genom
*odnośnie terminów kończących się na -om (stare pojęcia) - oznaczają kompletny zestaw wszystkich elementów danej klasy w obiekcie, który badamy
Pewne prawidłowości w rozwoju nauki są stare. Nigdy żadne prognozy naukowe się nie sprawdzają.
Trochę historii:
1859, Darwin - człowiek, który stworzył wielkie prawo biologiczne; jego dzieło „O powstawaniu gatunków” to rok 1859. W jego czasach uważało się, że cechy potomstwa są mieszaniną cech rodziców - ewolucja przez dobór naturalny nie mogła mieć miejsca. Pogląd ten stanowił fundamentalny problem jego teorii ewolucji.
1866, Mendel - badając groszek zauważył pewne prawidłowości, które uzupełniały koncepcję Darwina. Pokazał, że determinanty cech są skwantowane. Narodziła się genetyka, oraz nabrała prawdziwego sensu teoria ewolucji.
1868, Friedrich Miescher - badał ropę z bandaży szpitalnych; wyizolował nową substancję, którą nazwał nukleiną. Była to substancja bogata w kwas fosforowy i zawierała związki azotowe (był to brudny kompleks nukleoproteinowy, chociaż my utożsamiamy to z DNA). 1868 rok przyjmuje się jako datę odkrycia DNA. Przez wiele lat związek odkryty przez Mitchella uchodził za substancję mało interesującą.
1911, Thomas Morgan - badając Drosophila i obserwując korelację między cechami fenotypowymi a pewnymi obrazami chromosomów, postulował koncepcję, że determinanty cech organizmu zawarte są w chromosomach. Jest to chromosomowa teoria dziedziczności, za którą dostał Nobla. Już wtedy wiadomo było, że chromosomy zawierają białka i kwasy nukleinowe, przy czym uważano białka za część funkcjonalną, odpowiedzialną za funkcję chromosomów. Kwasy nukleinowe wydawały się pełnić funkcje strukturalne (łączniki, rusztowanie).
1944, Oswald Avery - zrobił doświadczenie pokazujące, że oczyszczony DNA z jednej bakterii, włożony do innej, niesie za sobą cechy szczepu, z którego został wyizolowany. Udowodnił, że DNA jest nośnikiem dziedziczności (cech dziedzicznych). Od tamtego momentu zaczęto się niesłychanie DNA interesować. Wiadomo było, że są w nim zapisane cechy, ale nadal nie znano jego budowy.
1951 - niezwykle intensywne badania budowy, cech i właściwości DNA prowadzono w wielu ośrodkach, między innymi w Cambridge, gdzie zaczął się tym zajmować fizyk Francis Crick. W tym samym czasie w innym ośrodku, w King's College, DNA badała Rosalind Franklin, wybitna krystalografka. Uzyskała ona słynny obraz dyfrakcyjny bardzo stężonego roztworu DNA, który stał się podstawą do rozwikłania struktury DNA.
1948, Linus Pauling - chemik, dwukrotny laureat nagrody Nobla, wymyślił α-heliks, ale absurdalny model tej cząsteczki.
1950, Erwin Chargraff - o chemii DNA wiedział wszystko: przebadał DNA setek organizmów i zauważył, że liczba zasad purynowych równa jest liczbie zasad pirymidynowych, liczba adenin równa jest liczbie tymin, a liczba cytozyn - guaninom. Ale nic mu to nie powiedziało.
1953, Watson i Crick - nie zrobili żadnych doświadczeń ani eksperymentów, zebrali fakty i zinterpretowali dane Rosalindy Franklin i opracowali strukturę podwójnej helisy DNA. Model ten sugerował, jak ta cząsteczka może się namnażać. Wiadomo już było, że w tym długim heliksie zawarta jest informacja genetyczna, determinująca cechy organizmów. Pozostawał problem, jak te długie cząsteczki odczytać (wiedziano, że nawet krótsze cząsteczki, np. DNA bakteriofagów, mają kilka tysięcy nukleotydów).
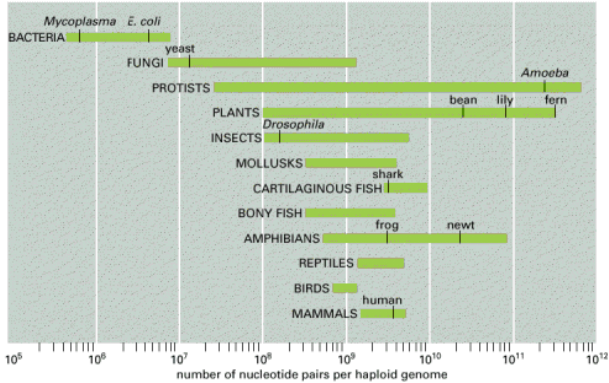
Wiadomo było, że zawartość DNA w komórkach jest dużo większa. Tę zawartość DNA w jądrze komórkowym określa się często jako c. Mamy tu wykres, w skali logarytmicznej w parach nukleotydów, określający wielkości genomów organizmów z poszczególnych grup taksonomicznych. Wraz ze wzrostem zaawansowania ewolucyjnego czy złożoności organizmów wielkość c rośnie: bakterie przeciętnie mają mniejsze genomy niż Eukariota, ssaki mają większe genomy niż grzyby.
Występuje też zjawisko paradoksu wielkości c, które mówi, że niezależnie od opisanej wcześniej prawidłowości, to we wszystkich taksonach grupujących podobne do siebie organizmy w sensie ewolucyjnym, zróżnicowanie zawartości DNA u poszczególnych gatunków jest gigantyczne (potrafi być o 3-5 rzędów wielkości); np. ameba i najprostszy pierwotniak mogą mieć 100 tysięcy razy więcej DNA jeden od drugiego, chociaż ich złożoność ewolucyjna nie różni się. Dlaczego istnieje taka różnica? Nie wiadomo.
Drugim aspektem paradoksu wielkości c jest fakt, że mamy wiele organizmów nie przewyższających ewolucyjnie człowieka, a mających np. 10 razy więcej DNA (np. traszka, paproć).
1977 - dwie grupy uczonych opracowały dość różne pod względem koncepcji metody stosunkowo prostego badania sekwencji nukleotydowych DNA. Dawniej potrafiono to robić na RNA - był to proces trudny i żmudny, odczytywano po jednym nukleotydzie przy pomocy chromatografii.
Maxam i Gilbert, USA - stosowali czysto chemiczne metody
Sanger, GB - metoda enzymatyczna; wcześniej opracował też metodę sekwencjonowaina białek
Opracowali oni metody sekwencjonowania DNA. W 1977 Sanger swoją metodą podał sekwencję nukleotydową pierwszego kompletnego genomu - genomu bakteriofaga φx174, mającego 5383 nukleotydy. Zarówno metoda Gilberta jak i Sangera, polegała na tym, że po reakcjach chemicznych/enzymatycznych DNA poddawało się elektroforezie w żelu poliakrylamidowym. U Maxama i Gilberta reakcję przeprowadzało się w kapilarach. Po rozdziale elektroforetycznym przykładana była klisza (DNA wyznakowany był radioaktywnie) i ręcznie sczytywało się sekwencję. Z jednej takiej elektoforezy 2 razy puszczonej można było odczytać 300 - 400 nukleotydów. Była to metoda czasochłonna i nie nadawała się do stosowania na dużą skalę, ale dość zaawansowana. Uruchomiono maszyny, które robiły to szybciej - odczytywały sekwencję z fluorescencyjnie wyznakowanego DNA za pomocą skanerów laserowych. Za pomocą zautomatyzowanego procesu można odczytać 500-800, czasem 1000 nukleotydów z jednej rundy sekwencjonowania.
Niespełna rok temu pojawiła się nowa metoda, która pozwala w sposób zautomatyzowany odczytywać miliony nukleotydów na raz. Jest to metoda oparta na PCRze - równolegle, jednocześnie przeprowadza się 1,5 miliona reakcji, każda w objętości rzędu pikolitrów (10^-12 litra). Przy pomocy tej metody na raz można odczytać około 100 nukleotydów - ale jeżeli to są setki różnych nukleotydów w milionie reakcji, to otrzymujemy niesłychaną masę danych. Problemem jest uporządkowanie tego.
W tej dziedzinie postęp jest ogromny - w ciągu dnia można odczytać miliony par zasad.
Jaką przyjąć strategię przy odczytywaniu sekwencji nukleotydowej kawałka DNA mającego setki tysięcy nukleotydów, gdy na raz potrafimy odczytać, w najlepszym przypadku, 1000 nukleotydów? Podejścia są dwa:
Robimy coraz dokładniejszą mapę fizyczną kawałka DNA, który chcemy sekwencjonować - aż sklonujemy małe kawałki rzędu 500-1000 nukleotydów, każdy z nich możemy zsekwencjonować. Ponieważ mamy dokładną mapę, wiemy jak ułożyć te kawałki - który jest pierwszy, który drugi itd. Skonstruowanie mapy fizycznej i uporządkowanie sklonowanych odcinków DNA jest bardzo trudne, żmudne i długotrwałe. Ale odczyt sekwencji jest rzeczą trywialną - sekwencja sama nam się układa. Pierwsze próby sekwencjonowania dłuższych odcinków DNA były oparte na takim uporządkowanym klonowaniu i sekwencjonowaniu kawałków - tzw. clone by clone. Ta metoda w praktyce nie bywa stosowana, bo etap przygotowania klonów jest tak długotrwały i skomplikowany, że się to nie opłaca.
Shotgun sequencing (sekwencjonowanie losowe/statystyczne)- polega na tym, że sekwencjonuje się coś ;), czyli jakiś DNA, który nas interesuje, wszystko jedno jaki. DNA fragmentujemy mechanicznie i klonujemy wszystkie kawałeczki. Następnie wszystkie te kawałeczki sekwencjonujemy i otrzymujemy mnóstwo różnych sekwencji, które w pewien sposób reprezentują DNA. Znajdujemy taką sekwencję, na której końcu jest ciąg nukleotydów występujący na początku innej sekwencji. Prawdopodobnie więc odcinki te na siebie zachodzą. Jeżeli odpowiednio dużo razy przeczytamy sekwencje nukleotydowe, to statystycznie rzecz biorąc prawie na pewno prawie każdy fragment DNA przynajmniej raz przeczytamy. Pokrycie sekwencjonowania wynoszące między 5 a 10 razy zapewnia nam, że 99,x% sekwencji w ten sposób poznamy. Czyli gdy mamy cząsteczkę DNA mającą milion zasad, to siekamy ją na fragmenty około 500 nukleotydów i sekwencjonujemy. Gdy przesekwencjonujemy 5-7 milionów par zasad, to z ogromnym prawdopodobieństwem 995 tysięcy nukleotydów z miliona odczytamy. Te ok. 0,5% nukleotydów mogło (ale nie musiało) zostać nie trafione - wtedy domykamy nieciągłości/przerwy w sposób świadomy je klonując, namnażając PCRem. Metoda w pełni losowego sekwencjonowania też jest trochę nieopłacalna, bo składanie sekwencji (informatyczne) jest bardzo trudne.
Metoda shotgun sequencing stosowana jest powszechnie, wymyślił ją Sanger i przy jej użyciu zsekwencjonował faga λ (prawie 50 tysięcy par nukleotydów)
W praktyce używa się metody pośredniej - najczęściej cały genomowy DNA klonujemy w bardzo dużych wektorach (nawet miliony par zasad w jednym klonie) i staramy się sporządzić mapę fizyczną tych klonów. Następnie najczęściej pojedyncze klony mające setki tysięcy czy miliony par zasad sekwencjonujemy w sposób czysto losowy metodą shotgun. Następnie składamy sekwencje nukleotydowe całych klonów, zaś mapa fizyczna tych klonów na DNA genomowym pozwala nam na złożenie ich w sposób uporządkowany.
Najprostsze genomy prokariotyczne są rzędu milionów par nukleotydów. U Eukariontów to: u grzybów co najmniej 10 milionów par nukleotydów; u kręgowców rzędu miliardów par nukleotydów. Czyli między bakterią a ssakiem różnica w zawartości DNA w genomie wynosi około 1000 razy.
Pokazane były dwa wykresy ilustrujące dwie tendencje w latach, gdy mowa była o sekwencjonowaniu na dużą skalę. Pierwszą była tendencja do redukcji kosztu sekwencjonowania (na początku lat 90 koszt wynosił około 10$ za zasadę, teraz kilka centów) - obserwujemy wykładniczy spadek kosztów. Druga tendencja wynika z tego, że pojawiła się masa danych, co spowodowało problem jak i gdzie zapisać sekwencje. Powstały liczne bazy danych, w których deponuje się wszystkie sekwencje nukleotydowe jakie zostały poznane. Większość z tych baz utrzymywana jest z pieniędzy publicznych i jest dostępna za darmo. W Gene Bank (najbardziej podstawowa baza) długość sekwencji nukleotydowych tam zdeponowanych rośnie wykładniczo (to jest ta druga tendencja - wykładniczy przyrost ilości informacji). W tej chwili w bazach danych są zapisane setki miliardów nukleotydów.
W 1995 roku pojawiła się pierwsza kompletna sekwencja nukleotydowa genomu komórki bakteryjnej (Haemophilus influenzae). Niespełna dwa lata później otrzymano sekwencję pierwszego Eukariota (Saccharomyces cerevisiae). W 1999 podano sekwencję 22. chromosomu człowieka. W 2000 roku zsekwencjonowano genom Arabidopsis thaliana. W 2001 roku ogłoszono zsekwencjonowanie genomu człowieka - była to niekompletna wersja, wersja ostateczna ukazała się w 2004 roku.
Dla dużych genomów, takich jak genomy roślin czy kręgowców, stosunkowo łatwo jest podać przybliżoną wersję sekwencji nukleotydowej (taką na 95% dokładną). Bardzo trudne i skomplikowane jest domknięcie tej sekwencji, uporządkowanie jej do dokładności 99,99%. Ogromna większość genomów Eukariotycznych, o których mówi się, że są znane, jest tak naprawdę poznana na poziomie tej przybliżonej wersji, z kilkoma procentami sekwencji nieznanych i błędnych.
Stan zaawansowania genomiki można śledzić na bieżąco na stronie Genomes OnLine (http://www.genomesonline.org/) - można tu sprawdzić genomy już zsekwencjonowane, będące w trakcie sekwencjonowania itp.
Generalnie bada się genomy dwóch typów organizmów:
organizmów modelowych, czyli tych, które z pewnych powodów są intensywnie badane (E.coli, Drosophila, drożdże, Caenorhabditis elegans, mysz, Arabidopsis thaliana). Organizmy modelowe zazwyczaj mają jakąś cechę, czyniącą je bardzo atrakcyjnym obiektem badawczym. Najczęściej ta sama cecha powoduje, że są one wybitnie nietypowe jak na przedstawiciela grupy, którą mają modelować. Wnioski wyciągane na podstawie analizy organizmów modelowych trzeba przyjmować z ogromną ostrożnością - prawie wszystkie organizmy modelowe są bardzo nietypowe i bardzo niereprezentatywne. Np. Arabidopsis thaliana wybrano do badań, gdyż ma malutki jak na roślinę genom - jest unikatową rośliną wśród roślin, a bada się ją jako przedstawiciela tej grupy.
organizmów istotnych z przyczyn głównie praktycznych - medycznych, gospodarczych; czasem są to organizmy mające bardzo interesującą pozycję ewolucyjną. Są to więc genomy patogenów, roślin uprawnych, zwierząt powodujących choroby itp., lub genomy badane poznawczo np. szympansa, żachwy czy gąbki.
Wielkości tych genomów są następujące:
S. cerevisiae 316 kbp
Drosophila 120 Mbp lub 180 Mbp*
E. coli 4639 kbp
H. sapiens 2,8 Gbp lub 3,1 Gbp
C.elegans 97 Mbp (w 1998r jako pierwszy genom organizmu tkankowego)
*U Eukariota DNA jest w postaci chromatyny „rozluźnionej” (euchromatyny) i „zbitej” (heterochromatyny). Wiemy, że prawie wszystkie interesujące sekwencje i geny siedzą w euchromatynie, w heterochromatynie zaś są jakieś sekwencje powtarzalne, dość nieistotne z punktu widzenia wiedzy o genomie. Poza tym DNA zawarte w heterochromatynie bardzo trudno jest sekwencjonować, przede wszystkim z powodu monotonnych sekwencji przez które maszyny głupieją. Dlatego gdy mówimy o wielkości genomu Eukariota, to prawie zawsze nie mówimy o wielkości całego DNA zawartego w komórce, tylko o części euchromatynowej. W przypadku Drosophila to ogromna różnica.
W przypadku organizmów diploidalnych mówiąc genom, mamy na myśli połowę DNA z komórki (haploidalny garnitur chromosomów); druga połowa jest mniej więcej identyczna.
Table 1-1. Some Genomes That Have Been Completely Sequenced SPECIES SPECIAL FEATURES HABITAT GENOME SIZE (1000s OF NUCLEOTIDE PAIRS PER HAPLOID GENOME) NUMBER OF GENES (PROTEINS)
EUBACTERIA
Escherichia coli laboratory favorite human gut 4639 4289
EUCARYOTES
Saccharomyces cerevisiae (budding yeast) minimal model eucaryote grape skins, beer 12,069 ~6300
Arabidopsis thaliana (wall cress) model organism for flowering plants soil and air ~142,000 ~26,000
Caenorhabditis elegans (nematode worm) simple animal with perfectly predictable development soil ~97,000 ~19,000
Drosophila melanogaster (fruit fly) key to the genetics of animal development rotting fruit ~137,000 ~14,000
Homo sapiens (human) most intensively studied mammal houses ~3,200,000 ~30,000
|
Następnie pokazane było zdjęcie chromosomów D.melanogaster, z na ciemno zaznaczonymi fragmentami heterochromatynowymi, stanowiącymi ogromną część genomu, a których sekwencji nie znamy.
Genomy organelli
Figure 14-55. Various sizes of mitochondrial genomes. The complete DNA sequences for more than 200 mitochondrial genomes have been determined. The lengths of a few of these mitochondrial DNAs are shown to scale as circles for circular genomes and lines for linear genomes. The largest circle represents the genome of Rickettsia prowazekii, a small pathogenic bacterium whose genome most closely resembles that of mitochondria. The size of mitochondrial genomes does not correlate well with the number of proteins encoded in them: while human mitochondrial DNA encodes 13 proteins, the 22-fold larger mitochondrial DNA of Arabidopsis encodes only 32 proteins - that is, about 2.5-fold as many as human mitochondrial DNA. The extra DNA that is found in Arabidopsis, Marchantia, and other plant mitochondria may be "junk DNA". The mitochondrial DNA of the protozoan Reclinomonas americana has 97 genes, more than the mitochondrion of any other organism analyzed so far. (Adapted from M.W. Gray et al., Science 283:1476 - 1481, 1999.)
Semiautonomiczne organelle, takie jak mitochondria i chloroplasty, mają własny genom. Genom mitochondrialny jest prawie zawsze kolisty i zazwyczaj dość niewielki, ale różnice w wielkościach poszczególnych genomów mitochondrialnych są znaczące - od kilkunastu tysięcy par nukleotydów u człowieka (mitochondrialny genom ssaczy jest jednym z najmniejszych), przez Reclinomonas americana (największy genom mitochondrialny zwierzęcy), do milionów par zasad u roślin. Redukcja wielkości genomu zaszła niesłychanie daleko, genomy mitochondrialne są małe i proste. Kodują one od 13-15 białek (człowiek) do 90 kilku u Reclinomonas americana, czyli mało. Gdy porównamy sobie zestaw genów mitochondrialnych z różnych organizmów, to tylko 5 z nich jest wspólne dla wszystkich mitochondriów:
Figure 14-57. Comparison of mitochondrial genomes. Less complex mitochondrial genomes encode subsets of the proteins and ribosomal RNAs that are encoded by larger mitochondrial genomes. The five genes present in all known mitochondrial genomes encode ribosomal RNAs (rns and rnl), cytochrome b (cob), and two cytochrome oxidase subunits (cox1 and cox3). (Adapted from M.W. Gray et al., Science 283:1476-1481, 1999.)
Ogólnie mówiąc w mitochondriach różnych organizmów jest od kilkunastu do kilkudziesięciu genów kodujących białka, przy czym u wszystkich organizmów tylko 5 genów zawsze występuje (pozostałe się różnią). Dowodzi to tego, że w ewolucji była bardzo duża swoboda utraty genów mitochondrialnych na rzecz genomu jądrowego.
*na wykładzie gen=gen kodujący produkt białkowy. Geny kodujące różnego rodzaju RNA mają inne właściwości, omawiane są osobno, jest ich niewiele.
W plastydach jest znacznie większa różnorodność. W chloroplastach różnych roślin genomy mogą być zarówno koliste, jak i liniowe; zazwyczaj jest wiele cząsteczek w jednym organellum. Mogą być one różnej wielkości: od setek tysięcy do milionów par nukleotydów, mogą także zawierać dość różne zestawy genów - najczęściej między kilkadziesiąt a małe kilkaset (w okolicach stu), czyli znacząco więcej niż genom mitochondrialny.
Genomy organelli badane były od stosunkowo wielu lat, wiemy o nich dość dużo.
Craig Venter - robi rzeczy, których się nie da zrobić . Ma wiele prywatnych firm. W połowie lat 90 złożył do National Institutes of Health (NIH) projekt zsekwencjonowania genomu bakterii metodą shotgun (w pełni). Gdy eksperci zobaczyli projekt stwierdzili, że jest on niemożliwy do realizacji (około 2 milionów par nukleotydów) i odmówili jego zesponsorowania. Venter znalazł prywatnych sponsorów i wykonał pracę w 3 miesiące.
Jedną z firm, które założył jest TIGR (The Institute for Genomic Research) - na jej stronach można znaleźć mnóstwo interesujących informacji o genomice.
Właśnie w TIGRze Venter ze współpracownikami zrobili pierwszy kompletny genom komórkowy (Haemophilus influenzae) metodą shotgun w ciągu trzech miesięcy (8 osób na 14 maszynach). Haemophilus influenzae to bakteria gramujemna powodująca nieżyty górnych dróg oddechowych (też bakteryjne zapalenie opon mózgowych). Jej genom ma około 1,800,000 bp a jego zsekwencjonowanie kosztowało niecały milion dolarów. W tych niespełna 2 milionach par nukleotydów znaleziono 1700 genów kodujących białka, kilka genów kodujących rRNA, 58 genów kodujących tRNA.
Statistics for Haemophilus influenzae KW20 Rd |
|
Źródło: TIGR
ORFy (Open Reading Frames) w sekwencji H.influenzae zajmują 85% sekwencji nukleotydowej. Gdy dodamy do tego rDNA i geny kodujące tRNA, to około 90% sekwencji genomu tej bakterii stanowi zapis informacji genetycznej. W takim razie na wszelkie funkcje regulatorowe itp. przypada nam około 10% genomu - genom ten jest bardzo oszczędny, wypakowany informacją, nie ma w nim miejsca na niewiadomo do czego służące sekwencje (co w przypadku bakterii generalnie się sprawdza).
Łatwo zidentyfikować sekwencję kodującą polipeptyd (od AUG do STOP). Zdefiniowanie genu natomiast nie jest możliwe. Stwierdzenie gdzie się gen zaczyna, a gdzie kończy, jest bardzo trudne, dlatego na wykładach przez gen rozumiemy ORF. To, co jest przed otwartą ramką odczytu i za nią do genu na pewno należy, ale nie wiadomo, gdzie się kończy… Czyli gen=gen kodujący białko, gen kodujący białko=ORF.
Kolejnym genomem, opublikowanym także przez zespół Ventera, był genom Mycoplasma genitalium - bakterii występującej m.in. w płucach, pasożyta mogącego występować także poza komórką. To prosty organizm, o najmniejszym znanym genomie bakteryjnym mającym 580 tysięcy par nukleotydów. Znaleziono w nim około 470 genów (=ORFów). ORFy te się praktycznie ze sobą stykają (typowe dla bakterii), w genomie tym nie ma pustych sekwencji (niekodujących). Występuje tu bardzo duża zgodność kierunku transkrypcji genów z kierunkiem wędrówki widełek replikacyjnych (w kolistym genomie replikacja zachodzi z jednego punktu, widełki wędrują od ori w przeciwnych kierunkach).
Genom Mycoplasma genitalium stał się w ostatnich latach obiektem bardzo interesujących doświadczeń mających na celu stworzenie minimalnej komórki - ludzie próbują wyrzucać z genomu tej bakterii kolejne geny sprawdzając, które tak naprawdę są absolutnie niezbędne do funkcjonowania komórki. Eksperymenty te budzą wiele wątpliwości etycznych.
Statistics for Mycoplasma genitalium G-37 |
|
Genom bakteryjny właściwie bez przerwy jest w trakcie replikacji. Nie jest dobre dla maszynerii enzymatycznej replikacji i transkrypcji, gdy polimeraza DNA zderza się z polimerazą RNA. Natomiast gdy widełki replikacyjne i maszyneria transkrypcyjna idą w tym samym kierunku, to jakoś sobie dają radę. Dlatego w wielu (ale nie wszystkich!) bakteriach jest dość dobra zgodność kierunku transkrypcji z kierunkiem replikacji DNA.
Schemat genomu Archeona - Archeoglobus fulgidus - inny sposób przedstawiania genomu: ponad 2 miliony par nukleotydów, geny zaznaczone na niciach Watsona i Cricka, na kolejnych pierścieniach zaznaczone są interesujące sekwencje. Genom Archebakterii jest generalnie podobny do genomu Eubakteryjnego, ma tylko trochę krótsze geny.
Genom najlepiej poznanego obiektu doświadczalnego (fizjologia, funkcjonowanie) - Escherichii coli - opublikowany został w 1997 roku. Sekwencjonowały go dwie niezależne grupy badawcze - amerykańska i japońska. Pokazana została mapka tego genomu, mającego 4,600,000 par nukleotydów.
Widzimy, co typowe dla bakterii, że między ORFami prawie nie ma przerw i 88% sekwencji zajętej jest przez ORFy, stanowiące niecałe 4300 genów (sekwencje powtarzające się, geny regulacyjne itp. to około 10% genomu). W E.coli prawie nie ma zgodności kierunku transkrypcji i kierunku replikacji, nie wiadomo dlaczego. Geny kodujące rRNA są transkrybowane w tym samym kierunku, w którym poruszają się widełki replikacyjne, zaś geny kodujące białka transkrybowane są w zasadzie losowo.
73% operonów bakteryjnych w E.coli zawiera 1 gen - połowa genów tej bakterii nie tworzy (nie wchodzi w skład) operonów, są to geny „pojedynczo transkrybowane”. Czyli ogromna większość operonów zawiera w sobie tylko 1 ramkę odczytu, są też dwu/trzy genowe, czyli są to układy proste (chociaż operon to w zasadzie specyficzny wynalazek bakteryjny). Operony kilkunasto genowe zdarzają się rzadko. Ogromna większość operonów E.coli w sekwencjach na lewo od ORFów zawiera miejsce wiązania dla jednego tylko czynnika transkrypcyjnego - możliwość regulacyjna jest tu bardzo ograniczona. Wynika z tego, że większość genów regulowana jest przez jeden tylko czynnik transkrypcyjny (regulacja na zasadzie gen włączony/wyłączony, nie ma miejsca na jakieś kombinacje, także z tego względu, że tylko 10% sekwencji nie koduje białek).
Statistics for Escherichia coli K12-MG1655 |
|
Porównano genów z E.coli z genami z innych znanych genomów (genom E.coli był zsekwencjonowany jako szósty). W ten sposób próbowano poznać funkcje białek kodowanych przez geny E.coli - porównywano i szukano podobnych genów w innych organizmach. Jeżeli sekwencja jest mocno konserwowana ewolucyjnie, to prawdopodobnie cecha jest bardzo istotna, gen koduje jakieś podstawowe funkcje. Poza tym gdy nie wiemy do czego służy białko w jednym organizmie, ale znamy funkcję podobnego (=homologicznego) białka w innym organizmie, to być może ma ono tę samą funkcję.
Takich różnych zestawień w czasach „genomicznych” robi się masę i wyciąga się z nich różne wnioski. Gdy porównano te 6 znanych genomów, wśród których były bakterie, archebakterie, drożdż, to okazało się, że w tych genomach (każdy co najmniej paręset/parę tysięcy genów) porządnie konserwowanych genów, czyli takich, które dało się zidentyfikować we wszystkich organizmach, było 16. Niby organizmy te są do siebie podobne, jednak białka realizujące niektóre ich „wspólne” funkcje okazały się do siebie kompletnie niepodobne. Czyli życie jest dużo bardziej zróżnicowane na poziomie molekularnym, niż nam się to wydawało…
W następnej kolejności próbuje się przypisać jakieś funkcje zidentyfikowanym genom. Czasem jest to oczywiste i potrafimy konkretnemu białku przypisać funkcję. Czasem nie wiemy co to jest konkretnie za białko, ale widać, że jest to np. białko błonowe, ma w środku kanał, więc jest odpowiedzialne za transport itp. Postąpiono tak z tymi ponad 4000 genów E.coli (najintensywniej, najdłużej i najlepiej zbadany organizm). Dla 40% białek tej bakterii nie istnieją nawet domysły ich funkcji, a niektóre z tych białek są niezbędne do życia. Nie są one podobne do żadnych innych białek w żadnym innym organizmie.
Jest jedna kategoria szczególnie mocno reprezentowana - są to białka związane z transportem. Białek zaangażowanych w transport w obrębie komórki czy przez błonę jest mnóstwo (co występuje w większości komórek).
Pokazane były przewidywane funkcje genów E.coli (1997r).
Gdy zestawimy kilkadziesiąt znanych genomów prokariotycznych, jakie da się zauważyć prawidłowości? Prawie dokładnie 90% genomów bakteryjnych (i Eubacteria i Archebacteria) zajętych jest przez ORFy (sekwencje kodujące sekwencje polipeptydowe), wszystkim innym funkcjom pozostaje około 10% genomu. Prawie wszystkie genomy bakteryjne są bardzo upakowane, bardzo zwarte, bardzo oszczędne i ogromna większość służy zapisaniu sekwencji aminokwasowej białka. W 2000 roku mamy wciąż średnio 43% białek o nieznanej funkcji (przy większej ilości danych), czyli o 40% białek bakteryjnych nie wiemy, do czego służą. Co więcej, gdy sekwencjonujemy nowy genom i przyglądamy się nowo odkrytym ORFom, próbując znaleźć reprezentantów już wcześniej poznanych, okazuje się, że mniej więcej 25% ORFów to ORFy występujące tylko w tym gatunku (dla niego unikatowe, chociaż w ewolucji powstały z jakiś innych genów, to są tak zmienione, że nie da się zauważyć homologów w żadnym innym gatunku). Różnorodność genetyczna jest ogromna.
Kolejna prawidłowość, będąca niemal idealnie przestrzegana w świecie bakteryjnym, jest taka, że im większy genom (od setek tysięcy par nukleotydów do prawie 10 milionów par nukleotydów), tym więcej genów. Znaczy to, że przeciętny gen w różnych bakteriach jest mniej więcej zawsze tej samej wielkości. Wyjątkiem jest tu Mycobacterium leprae - bakteria mająca spory genom (około 3,5 miliona par zasad), a mało genów. Połowę jej genomu stanowią ORFy. Jest to bakteria będąca w trakcie uwsteczniania się - traci ona geny na ogromną skalę (to pasożyt głównie siedzący w komórkach).
Przeciętne białko bakteryjne jest rzędu trzystu kilkudziesięciu aminokwasów (średnio 336aa, od 291 do 380, większość od 310 do 340) i nie zauważa się od tego jakiś większych odstępstw. Strategia ewolucyjna w przypadku organizacji genomu u Eubakterii i Archebakterii jest właściwie identyczna.
Warto, abyśmy wiedzieli ;): jeśli mamy bakterię, to ma ona na pewno kilka milionów par zasad (0,58-8,7 Mbp), w nich kilka tysięcy genów, z których każdy to około 1000 nukleotydów, czyli ponad 300 aminokwasów. Mniej więcej 90% genomu to sekwencje kodujące polipeptydy.
Bakterie są niebywale różnorodne, różnice dotyczą wszystkiego, od wielkości, przez tryb życia, kształt, po sposób funkcjonowania. W laboratorium potrafimy hodować
0.01-0.1% bakterii, o których wiemy, że istnieją. Pozostałych 99% nie sposób hodować, więc i nie sposób badać, więc nic o nich nie wiemy. Można je identyfikować molekularnie (PCR), przypisywać do gatunku, ale nie da się zbadać ich morfologii, fizjologii itp. i prawdopodobnie są to bakterie najciekawsze ze względu na ich wyszukane potrzeby, których nie znamy.
Także z punktu widzenia genomiki bakterie są bardzo różne:
nie zawsze mają jeden chromosom kolisty - np. Borrelia Burgdorferi. Jest to bakteria często spotykana, powoduje boreliozę, jest przenoszona przez kleszcze. Jej genom jest liniowy, ma około 1 miliona par zasad. Bakteria ta ma także kilkanaście plazmidów różnej wielkości (genom B.burgdorferi = liniowy chromosom + kilkanaście plazmidów).
Vibrio cholerae ma dwa chromosomy koliste: jeden ma 3 miliony par zasad, drugi zaś milion par zasad
Streptomyces coelicolor ma największy zsekwencjonowany do tej pory genom prokariotyczny, mający prawie 9 milionów par zasad. Jest to genom liniowy. Ma prawie 8 tysięcy genów
Rickettsia prowazekii - to bakteria wywołująca tyfus plamisty. Jest dość nietypowa, gdyż koduje tylko 75% jej genomu, przy czym niekodujące 25% genomu zawiera bardzo mało pseudogenów (pseudogeny - sekwencje, po których widać, że były genami, ale już nie funkcjonują). Czyli pseudogenów jest mało, a mimo to aż 25% genomu nie koduje białek. Możliwe że są to pseudogeny tak zdezorganizowane, że ich nie rozpoznajemy.
Mycobacterium leprae - mniej więcej tylko połowa genomu tej bakterii koduje białka, prawie połowa genów niedawnego jej przodka uległa zepsuciu - przestały one funkcjonować jako geny, nie kodują białka i nie są odczytywane. Są jej niepotrzebne - to pasożyt wewnątrzkomórkowy. Ale jeszcze niedawno były funkcjonalne, gdyż rozpoznajemy je jako pseudogeny.
M.tuberculosis i M.leprae (gruźlica i trąd) - porównanie: są blisko spokrewnione, mają podobnej wielkości genomy, ale liczba genów jest diametralnie różna. M.tuberculosis ma 4 tysiące genów i prawie żadnych pseudogenów; M.leprae ma niecałe 2 tysiące genów i ponad tysiąc pseudogenów. Widać, że M.leprae to bakteria ulegająca uproszczeniu ewolucyjnemu - traci zbędne geny.
Thermotoga maritima - to bakteria (Eubakteria) żyjąca w gorących wodach morskich. Ma niecałe 2 miliony par nukleotydów, typowy genom archebakteryjny - gdy porównamy sekwencje nukleotydowe genów tej bakterii z innymi to okazuje się, że sporo jej genów to geny typowe dla Archebakterii. Genów wybitnie archebakteryjnych w genomie T.maritima jest prawie 500 (umiejscowione na czwartym okręgu w postaci tzw. `wysp archebakteryjnych'), zaś genów wybitnie Eubakteryjnych około 1000. To mozaika, geny archebakteryjne prawdopodobnie wniknęły do tej Eubakterii. Właściwie wszystkie genomy wszystkich organizmów to mniej lub bardziej mozaiki składane na skutek horyzontalnego transferu genów - w skali ewolucyjnej do genomu komórki wchodzą geny z innych gatunków, na tyle niedawno, że ciągle zachowują charakter typowy dla genomu, z którego pochodzą. Takich genów, ewidentnie pojawiających się na skutek horyzontalnego transferu genów, we wszystkich genomach trochę można znaleźć (u człowieka też). Tu mamy przykład skrajny, gdzie 1/3 genów to geny z innej grupy taksonomicznej.
*Gdy próbujemy odtwarzać filogenezę organizmów, wcale nie powstają nam „drzewka” tylko „krzaki”, sieć. Organizmy, które znamy w tej chwili, wcale nie są potomkami jednego organizmu z przeszłości - to najczęściej mozaiki wielu gatunków z zamierzchłych czasów.
Neisseria meningitidis - powoduje zapalenie opon mózgowych. Bakteria ta ma świetne mechanizmy uciekania systemowi immunologicznemu żywiciela, dzięki temu, że ma niezwykłą zmienność antygenów powierzchniowych. Zmienność ta zaś wynika z tego, że rekombinuje ona swoim genomem, wymienia różne kawałki. Dzieje się tak, gdyż bakteria ta posiada dużo sekwencji powtarzalnych w genomie (na okręgach 4-9), które ułatwiają rekombinację. Duża ilość sekwencji powtarzalnych jest nietypowa dla bakterii, ale dzięki niej N.meningitidis może szybko ewoluować.
13