1
Opracował dr inż. Tadeusz Janiak
Ćwiczenie 5
Fluorymetryczne oznaczanie tryptofanu w białkach
(instrukcja poprawiona)
I Cel ćwiczenia
Celem ćwiczenia jest zapoznanie się ze zjawiskiem fluorescencji. Student zapozna się
również z metodą fluorescencyjnego oznaczania zawartości tryptofanu w białkach. W czasie
ćwiczenia wykonuje się:
1. Widmo absorpcyjne tryptofanu
2. Widmo wzbudzenia tryptofanu
3. Widmo fluorescencyjne tryptofanu
4. Pomiar zależności intensywności fluorescencji od stężenia tryptofanu. Służy ono do
wyznaczania krzywej wzorcowej
5. Pomiar zależności intensywności fluorescencji białka (np. albuminy) od ilości dodanego
tryptofanu (wzorca wewnętrznego). Służy do oznaczenia zawartości tryptofanu w białku.
II Wstęp teoretyczny
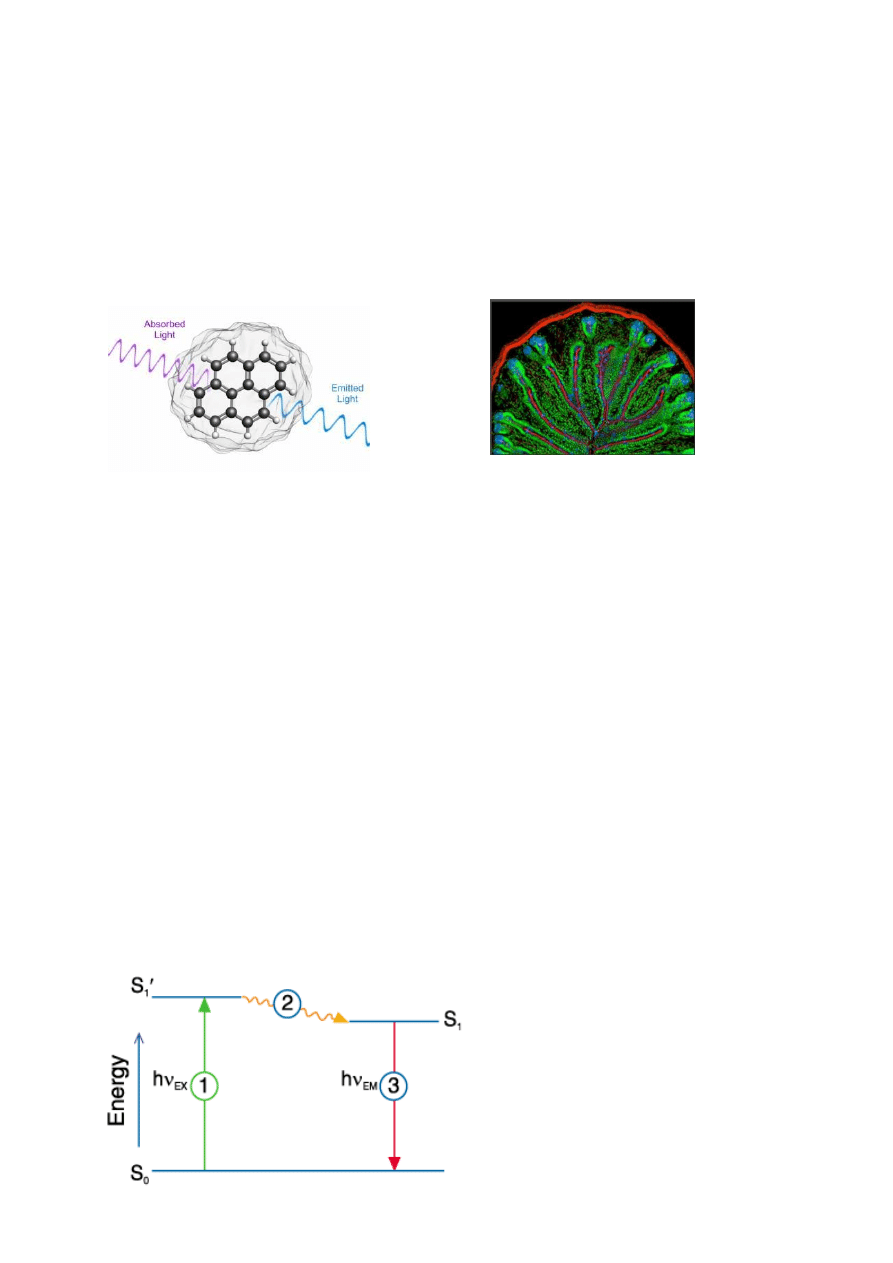
Zjawisko fluorescencji polega na emisji promieniowania elektromagnetycznego
cząsteczek wzbudzonych elektronowo. Można je wyjaśnić w oparciu o diagram
przedstawiony na Rys. 1. Na diagramie przedstawiono, co dzieje się jeżeli cząsteczka związku
o
właściwościach
fluorescencyjnych
zostaje
naświetlona
promieniowaniem
elektromagnetycznym. Przy odpowiedniej długości fali cząsteczka ze stanu podstawowego S
0
zostaje przeniesiona do stanu wzbudzonego elektronowo i wibracyjnie S
1
’
(przejście 1).
2
Rys. 1 Diagram ilustrujący powstawanie fluorescencji (diagram Jabłońskiego)
Stan taki jest nietrwały i następuje utrata energii poprzez przejście na niższy poziom
wibracyjny S
1
(przejście 2). Pozostałej części energii cząsteczka pozbywa się emitując kwant
promieniowania i przechodzi ze wzbudzonego poziomu S
1
na poziom podstawowy S
0.
Pomiary fluorescencji wykonuje się za pomocą fluorymetrów lub spektrofluorymetrów.
Pierwsze z nich służą do pomiarów punktowych, drugie dają możliwość ciągłego
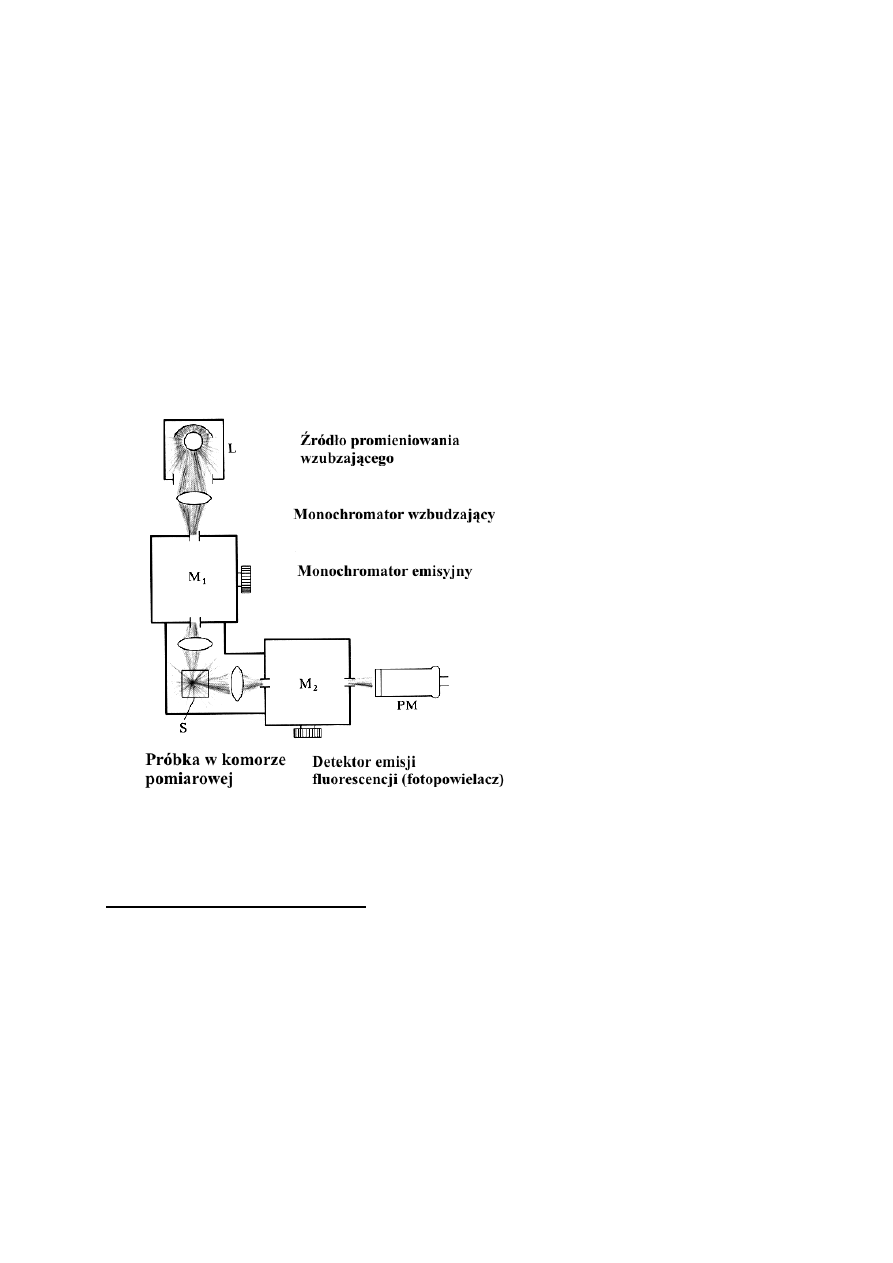
„przemiatania” promieniowania wzbudzającego i promieniowania emitowanego. Schemat
spektrofluorymetru przedstawiony jest na Rys. 2. Promieniowanie elektromagnetyczne z
odpowiedniego źródła światła (lampa) dostaje się do monochromatora M
1
(najczęściej siatka
dyfrakcyjna). Z monochromatora wiązka światła o zadanej długości przechodzi przez kuwetę
zawierającą substancję o właściwościach fluorescencyjnych. Monochromator emisyjny M
2
analizuje wiązkę światła, emitowanego przez wzbudzone cząsteczki, prostopadle do wiązki
światła wzbudzającego. Fluorescencja dla wydzielonej długości fali mierzona jest za pomocą
detektora natężenia promieniowania (najczęściej jest to fotopowielacz).
Rys. 2 Schemat poglądowy spektrofluorymetru.
Rodzaje widm fluorescencyjnych:
Widmo wzbudzenia
Monochromator emisyjny M
2
ustawiony jest na określoną długość fali w zakresie
fluorescencji,
natomiast
monochromator
wzbudzający
przemiata długością fali
promieniowania wzbudzającego
(λ
em
= constant, λ
wzb
zmienia się)
Widmo emisji
Monochromator wzbudzający M
1
ustawiony na określoną długość fali absorbowaną przez
próbkę, natomiast monochromator emisyjny przemiata i analizuje emitowane światło
(λ
wzb
= constant, λ
em
zmienia się)

3
Zastosowanie spektroskopii fluorescencyjnej
Bardzo niskie granice oznaczalności (do 10
-10
M) sprawiają, że metody fluorescencyjne
znajdują zastosowanie w dziedzinach takich jak:
1) Medycyna i analiza kliniczna –oznaczanie witamin, enzymów, hormonów, środków
dopingujących.
2) Farmacja –badania metabolizmu (barbiturany, amfetamina, LSD)
3) Biochemia – detekcja i oznaczanie śladów enzymów, koenzymów, lipidów, kwasów
nukleinowych, protein, chlorofilu
4) żywność – detekcja śladowych komponentów w produktach spożywczych
(aminokwasy, witaminy, proteiny, toksyny)
5) Środowisko –powietrze, woda i gleby (policykliczne węglowodory aromatyczne PAH,
aflatoksyny, PCB, fenole, pestycydy)
6) Analiza organiczna i nieorganiczna – oznaczanie wszelkich substancji fluoryzujących
Inne – detekcja w HPLC, sensory fluorescencyjne, badania fotochemiczne i fotofizyczne
stanów wzbudzonych, spektroskopia czasów życia - techniki impulsowe
Fluorymetryczne oznaczanie zawartości tryptofanu w białkach
Fluorescencyjne właściwości reszt tryptofanu są uzależnione od otoczenia chromoforu, w tym
przede wszystkim od lokalizacji w łańcuchu polipeptydowym oraz ładunku sąsiednich reszt
aminokwasowych. Analiza widm fluorescencji tryptofanu uzyskanych dla białek natywnych o
różnorodnej strukturze wykazała istnienie istotnych różnic dotyczących położenia maksimum
wzbudzenia, kwantowej wydajności fluorescencji oraz szerokości widma. Zjawisko to,
stanowiące poważne utrudnienie w praktycznym zastosowaniu metod fluorymetrycznych do
ilościowego oznaczania zawartości tryptofanu w białkach, można wyeliminować, poddając
badane białka uprzedniej denaturacji w 6 mol/l roztworze chlorowodorku guanidyny z
dodatkiem 2-merkaptoetanolu. Ponadto zastosowanie wewnętrznego standardu pozwala na
wykorzystanie omawianej metody do ilościowego oznaczania tryptofanu w białkach
zawierających ugrupowania pochłaniające w zakresie 290-370 nm, takie jak nukleotyd
flawinowy, hem lub fosforan pirydoksalu.
III Wykonanie ćwiczenia
A Wykonanie widma absorpcyjnego L-tryptofanu
1. Wykonać widmo absorpcyjne tryptofanu o stężeniu 0,5mM za pomocą przyrządu UV-VIS.
Perkin Elmer Lambda 40. Pomiar wykonać w kuwetach kwarcowych w zakresie długości fali
220-330 nm w sposób przedstawiony poniżej.

4
a)
Uruchomić spektrofotometr, komputer oraz monitor przyciskami zasilania.
b)
Jeżeli komputer wyświetli okienko do wprowadzenia hasła sieciowego, to kliknąć
myszą na przycisk Anuluj.
c)
Uruchomić program Lambda 40 ikoną na pulpicie lub poprzez Start-Programy-
Lambda 40-Lambda 40.
d)
W
menu
Utilities-Configuration
w
ścieżce
Data
wpisać
C:\UVWINLAB\DATA\Lab i potwierdzić klikając myszą na przycisk OK.
e)
W oknie Methods uruchomić metodę Lab.
f)
Sprawdzić ustawienie następujących parametrów: w zakładce Scan w polu Start
wavelength - 330 nm, w polu End wavelength - 220 nm. Pole Autosave - On, Autoprint - Off,
pole Display - Overlay. W, zakładce Inst. pole Ordinate mode - A, pola: LamUV i LampVis -
ON. W polu Scen speed -120 nm/min., a Smooth - O. W zakładce Sample w polu Result
Filename podać nazwę pliku, a w polu Number of Samples wpisać liczbę roztworów. W polu
Sample Identity wpisać nazwę kolejnych roztworów oraz ewentualnie ich opis w polu Sample
Info.
g)
Włożyć puste kuwety kwarcowe do obu uchwytów w komorze próbek. Matowe
ścianki kuwet muszą być skierowane w stronę bocznych ścianek uchwytów. Zamknąć
pokrywę oraz kliknąć myszą na Autozero. Pojawi się okno Sample Change z prośbą o
umieszczenie odnośnika Blank. Kliknąć przycisk OK. Ponownie wykonać pomiar linii
zerowej.
h)
Pobrać do jednej z kuwet kwarcowych 3 ml roztworu tryptofanu o stężeniu 0,5 mM w
buforze fosforanowym pH=7 a do drugiej 3 ml roztworu buforowego fosforanowego pH=7.
Włożyć kuwetę z 3 ml 0,5 mM roztworu tryptofanu do uchwytu bliżej położonego a kuwetę
z odnośnikiem, roztwór buforu fosforanowego pH=7 do drugiego chwytu położonego dalej.
Kliknąć myszą na przycisk Start. Pojawi się okienko Sample Change z prośbą o umieszczenie
kolejnej próbki. Kliknąć myszą na przycisk OK. Zostanie zarejestrowane widmo absorpcyjne.
i)
Z menu View wybrać Vertical Cursor Cont. Pojawi się pionowa linia, która służy do
odczytywania długości fali oraz odpowiadającej jej absorbancji (na dole ekranu). Linię tę
przesuwa się wzdłuż widm za pomocą myszy. Znaleźć maksimum na krzywej absorpcji.
k)
Po zakończeniu pomiarów wyjść z programu przez File-Exit. Jeżeli wyświetli się
okno(a) z prośba o zapisanie metody lub danych to kliknąć na przyciski Cancel lub Exit
i)
Wyłączyć komputer i spektrofotometr.
B Pomiary fluoroscencyjne
Uwaga! Nie można dotykać palcami ścianek kuwety fluorescencyjnej gdyż
tłuszcz z opuszek spowoduje zmianę charakterystyki widm.
Pomiary widma fluorescencyjnego dla krzywej wzorcowej tryptofanu i dla
albuminy z wzorcem wewnętrznym (tryptofanem) wykonać dla takich samych
ustawień przyrządu (szczególnie ważne są szczeliny).
1. Pobrać do kuwety do fluorescencji 3 ml roztworu tryptofanu o stężeniu 0,5 mM w
buforze fosforanowym pH=7 i umieścić w komorze spektrofluorymetru.
2. Wykonać widmo wzbudzenia pobranego roztworu stosując: wzbudzenie (excitation) w
zakresie 230-340 nm oraz emisję (emission) λ=354 nm.

5
3. Wykonać widmo fluorescencji tego samego roztworu stosując: wzbudzenie (excitation)
λ=295 nm oraz emisję (emission) w zakresie 320-420 nm.
4. Opróżnić kuwetę. Przemyć ją kilkakrotnie wodą dejonizowaną. Delikatnie usunąć całą
wodę. Przepłukać kuwetę alkoholem etylowym. Osuszyć ją ręcznikiem papierowym.
5. Pobrać 3 ml buforu fosforanowego do kuwety fluoroscencyjnej. Dodać, za pomocą
mikropipety lub mikrostrzykawki, 15 μl roztworu tryptofanu o stężeniu 0,5 mM w buforze
fosforanowym pH=7. Wymieszać zawartość kuwety.
6. Wykonać widmo fluorescencji roztworu tryptofanu stosując: wzbudzenie (excitation)
λ=295 nm oraz emisję (emission) w zakresie 320-420 nm i odczytać, korzystając z kursora,
emisję dla długości fali λ=354 nm.
7. Dodać jeszcze 5 razy po 15 μl roztworu tryptofanu o stężeniu 0,5 mM w buforze
fosforanowym pH=7. Po każdym dodaniu roztworu tryptofanu do kuwety zawartość jej
dobrze wymieszać i wykonać widmo fluorescencji. Odczytać, korzystając z kursora, emisję
dla długości fali λ=354 nm.
8. Wyniki zapisać w tabeli 1
Tabela 1. Krzywa wzorcowa emisji fluorescencji tryptofanu.
Dodana objętość
roztworu tryptofanu
0,5 mM
[μl]
Emisja fluorescencji
dla λ=354
[j.u.]
Stężenie końcowe
tryptofanu bez
korekcji
[mM]
Stężenie końcowe
tryptofanu z korekcją
[mM]
15
30
45
60
75
90
9. Opróżnić kuwetę. Przemyć ją kilkakrotnie wodą dejonizowaną. Delikatnie usunąć całą
wodę. Przemyć kuwetę alkoholem i osuszyć ją ręcznikiem papierowym.

6
10. Odpipetować 3 ml roztworu białka o stężeniu 0,05 g/l (roztwór wodny w 6 M
chlorowodorku guanidyny z dodatkiem β-merkaptoetanolu) do kuwety fluorescencyjnej.
11. Wykonać widmo fluorescencji roztworu białka stosując: wzbudzenie (excitation) λ=294
nm oraz emisji (emission) w zakresie 320-420 nm.
12. Dodać 10 μl roztworu tryptofanu o stężeniu 0,5 mM w buforze fosforanowym pH=7 do
kuwety fluoroscencyjnej zawierającej badane białko. Wymieszać zawartość kuwety.
13. Wykonać widmo fluorescencji otrzymanego roztworu stosując wzbudzenie (excitation)
λ=294 nm dla emisji (emission) w zakresie 320-420 nm i odczytać, korzystając z kursora,
emisję dla długości fali λ=354 nm.
14. Dodać jeszcze 5 razy po 10 μl roztworu tryptofanu o stężeniu 0,5 mM w buforze
fosforanowym pH=7. Po każdym dodaniu roztworu tryptofanu do kuwety zawartość jej
dobrze wymieszać i wykonać widmo fluorescencji, odczytać, korzystając z kursora, emisję
dla długości fali λ=354 nm.
15. Wyniki zapisać w tabeli 2.
16. Powtórzyć czynności z pkt.11-15 dla roztworu drugiego białka.
Tabela 2. Emisja fluorescencji roztworu białka o stężeniu 0,05 g/l (roztwór wodny w 6 M
chlorowodorku guanidyny) z wzorcem wewnętrznym (tryptofanem)
Białko I …………………………………………..
Dodana objętość
roztworu tryptofanu
0,5 mM [μl]
Emisja fluorescencji
dla λ=354
[j.u.]
Stężenie końcowe
tryptofanu
bez korekcji [mM]
Stężenie końcowe
tryptofanu
z korekcją [mM]
10
20
30
40
50
60

7
Białko II …………………………………………..
Dodana objętość
roztworu tryptofanu
0,5 mM [μl]
Emisja fluorescencji
dla λ=354
[j.u.]
Stężenie końcowe
tryptofanu
bez korekcji [mM]
Stężenie końcowe
tryptofanu
z korekcją [mM]
10
20
30
40
50
60
Wykonanie pomiarów fluoroscencyjnych
1. Włączyć komputer, włączyć monitor, włączyć fluorymetr.
2. Uruchomić program Cary Eclipse.
3. Wybrać opcje scan – otwiera się okno programu.
4. Wybrać opcję set up. Pojawia się okno z zakładkami Excitation oraz Emission.
Wykonywanie widma wzbudzenia:
a) włożyć kuwetę z badanym roztworem do uchwytu
b) przejść do zakładki Excitation,
c) ustawić nastawy według tabeli
Pole zakładki
Długość fali
Szczeliny
Emission
354 nm
Emission slit 5
Start 230 nm
Excitation slit 2.5
Stop 340 nm
c) wcisnąć O.K.
d) przycisnąć przycisk START na górnym pasku programu,
e) pojawia się nowe okno wpisać nazwę próbki, wcisnąć O.K.
f) po wykonaniu widma zapisać go
8
Wykonywanie widma fluorescencji (emisji fluorescencji):
a) przejść do zakładki Emission,
b) ustawić nastawy według tabeli
Pole zakładki
Długość fali
Szczeliny
(wykonanie widma
tryptofanu)
Szczeliny
(krzywa wzorcowa i
pomiary w obecności
białka)
Excitation
294 nm
Excitation slit 2.5
Excitation slit 5
Start 320 nm
Emission slit 5
Emission slit 10
Stop 420 nm
c) wcisnąć O.K.
d) Przycisnąć przycisk START na górnym pasku programu
e) Pojawia się nowe okno wpisać nazwę próbki, wcisnąć O.K.
f) Odczytać za pomocą kursora wartość emisji dla λ=354 nm.
4. Opracowanie wyników
1. Na papierze milimetrowym zaznaczyć punkty do krzywej wzorcowej fluorymetrycznego
oznaczania zawartości tryptofanu w próbie, zależność natężenia fluorescencji od
skorygowanego stężenia tryptofanu (wyniki z Tabeli 1). Wyznaczyć, metodą najmniejszych
kwadratów prostą opisującą tą zależność. Sporządzić wykres prostej na papierze
milimetrowym. Podać współczynnik kierunkowy prostej (a
w
).
2. Wykreślić zależność natężenia fluorescencji I
fl
od stężenia dodanego do roztworu białka
tryptofanu C
tr
- dla wyników z Tabeli 2. Obliczyć liniową regresję metodą najmniejszych
kwadratów. Podać współczynnik kierunkowy prostej (a
oz
). Wykres zawierający punkty
pomiarowe oraz prostą (l
oz
) umieścić na papierze milimetrowym.
3. Ekstrapolować prostą (l
oz
) do stężenia tryptofanu (c
tr
) równego zeru. Odczytać z wykresu
wartość natężenia fluorescencji pochodzącej od reszt tryptofanu zawartych w badanym białku
4. Korzystając z krzywej wzorcowej, wyznaczyć stężenie tryptofanu w badanym białku.
5. Skorygować odczytaną wartość, mnożąc ją przez empiryczny współczynnik
proporcjonalności równy a
w
/a
oz
.
6. Wyznaczyć liczbę reszt tryptofanu R
tr
przypadających na jedną cząsteczkę białka,
korzystając z zależności:
R
tr
= c
tr
/c
b
gdzie: C
b
- stężenie molowe białka w próbie.
Masę molową białka poda prowadzący zajęcia.
Literatura
1. http://analiza.ovh.org/wyk/3.pdf
2. http://www.fuw.edu.pl/IIPRACOWNIA/home/Opisy-cwiczen/SF5.pdf
3.
Fluorescence of Proteins in 6-M Guanidine Hydrochloride A Method for the Quantitative
Determination of Tryptophan
Patrick PAJOT European Journal of Biochemistry Volume 63 Issue 1, Pages263-269
Wyszukiwarka
Podobne podstrony:
05a Spektrochemia , Fluorescenc Nieznany
2 Spektrofotometryczne oznzczan Nieznany (2)
FIG 05A id 169833 Nieznany
2 Spektrofotometryczne oznzczan Nieznany (2)
Spektrometr fluorescencji atomowej
Spektroskopia fluorescencyjna Ula Radziwanowska
spektroskopia fluorescencyjna
biologia spektroskopia2 id 8805 Nieznany
Fluorescencja id 178235 Nieznany
Spektroskopia NMR
SPEKTROSKOPIA ROTACYJNA
Gor±czka o nieznanej etiologii
Fluorescencja chlorofilu
Spektrometria mas NMAZ
02 VIC 10 Days Cumulative A D O Nieznany (2)
Abolicja podatkowa id 50334 Nieznany (2)
45 sekundowa prezentacja w 4 ro Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
więcej podobnych podstron