
WYDZIAŁ NAUK O ŻYWNOŚCI I RYBACTWA
CENTRUM BIOIMMOBILIZACJI I INNOWACYJNYCH
MATERIAŁÓW OPAKOWANIOWYCH
ENZYMOLOGIA
Kierunek:
Technologia Żywności
i Żywienie Człowieka
semestr I
Wykład 4
Kinetyka reakcji enzymatycznych
oraz inhibitory

Zakres materiału ENZYMOLOGIA
1. Biochemia, Autor: Jeremy Berg, Lubert Stryer, John L. Tymoczko,
PWN Warszawa (2005)
Rozdziały:
8. Enzymy:: podstawowe pojęcia i kinetyka
9. Strategie katalityczne
10. Strategie regulacyjne:: enzymy i hemoglobina
2. Ćwiczenia z enzymologii i technik biochemicznych. Bartoszewska, Niziołek,
Paszowski, Wydawnictwo SGGW
3. Handbook of Food Enzymology – ed. John R. Whitaker et al., CRC Press; (2002)
4. Enzymes in Food Technology -‐ ROBERT J. WHITEHURST, BARRY A.
LAW, Editors Sheffield Academic Press CRC Press (2002)
3. Food chemistry -‐ Hans-‐Dieter Belitz, Werner Grosch, Peter
Schieberle, Springer (2004)
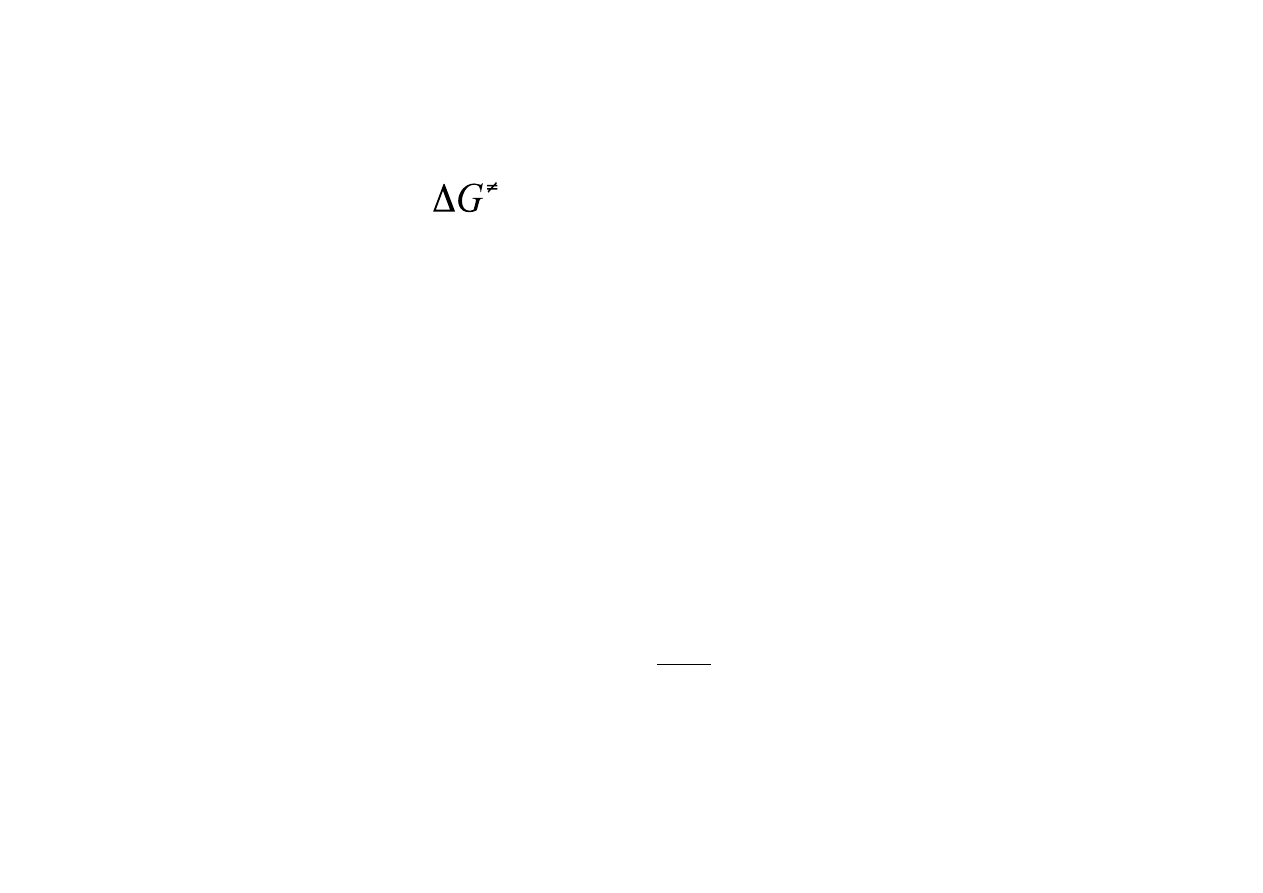
€
k = ve
−ΔG
RT
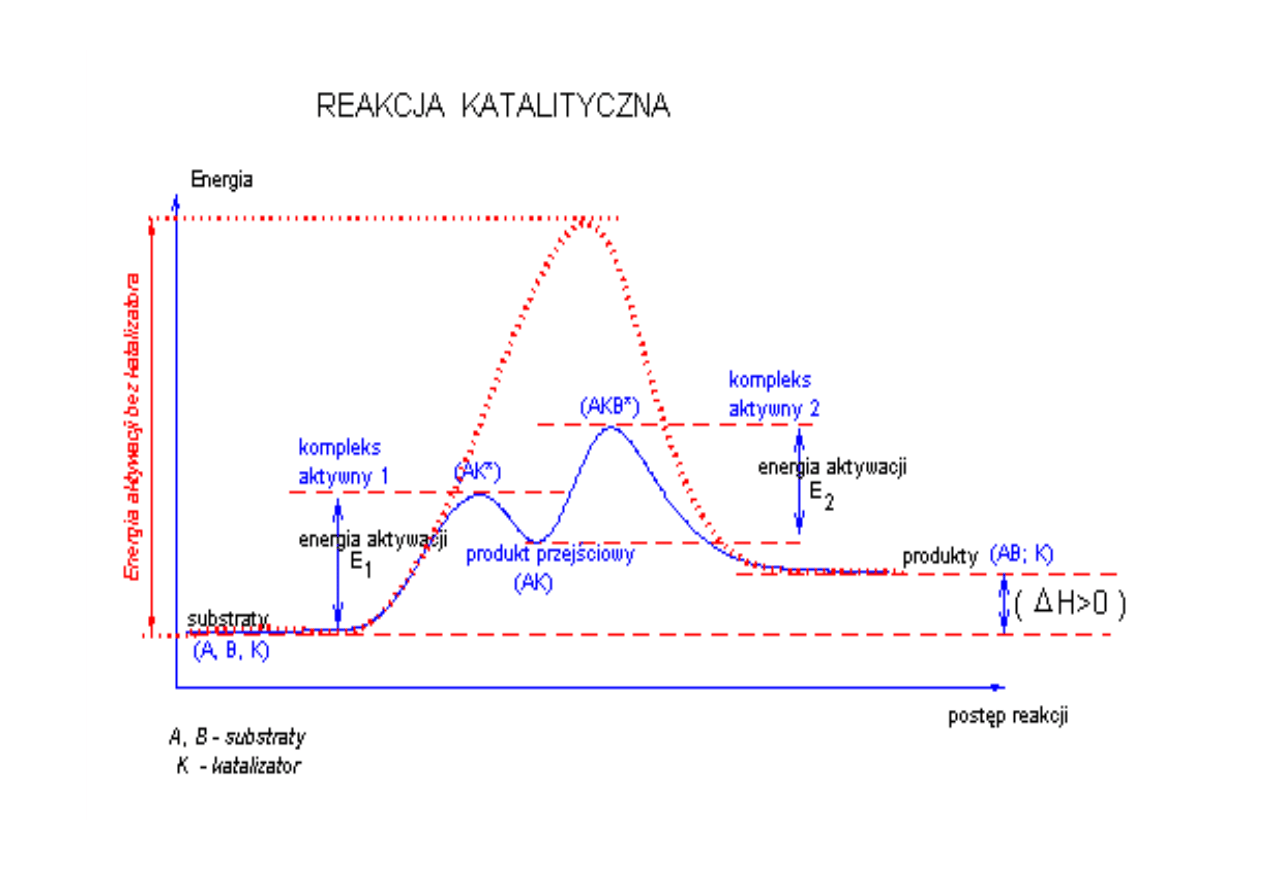
Energia aktywacji
Energia aktywacji -‐ to różnica energii swobodnej między
stanem przejściowym a substratem. Minimalna energia jaką
musi posiadać substrat, aby mógł przereagować.
Stan przejściowy – stan o maksymalnej energii na wykresie
postępu reakcji. Dla reakcji enzymatycznych stanem
przejściowym jest kompleks enzym-‐substrat ES.
Stałą szybkości opisujemy wówczas równaniem:
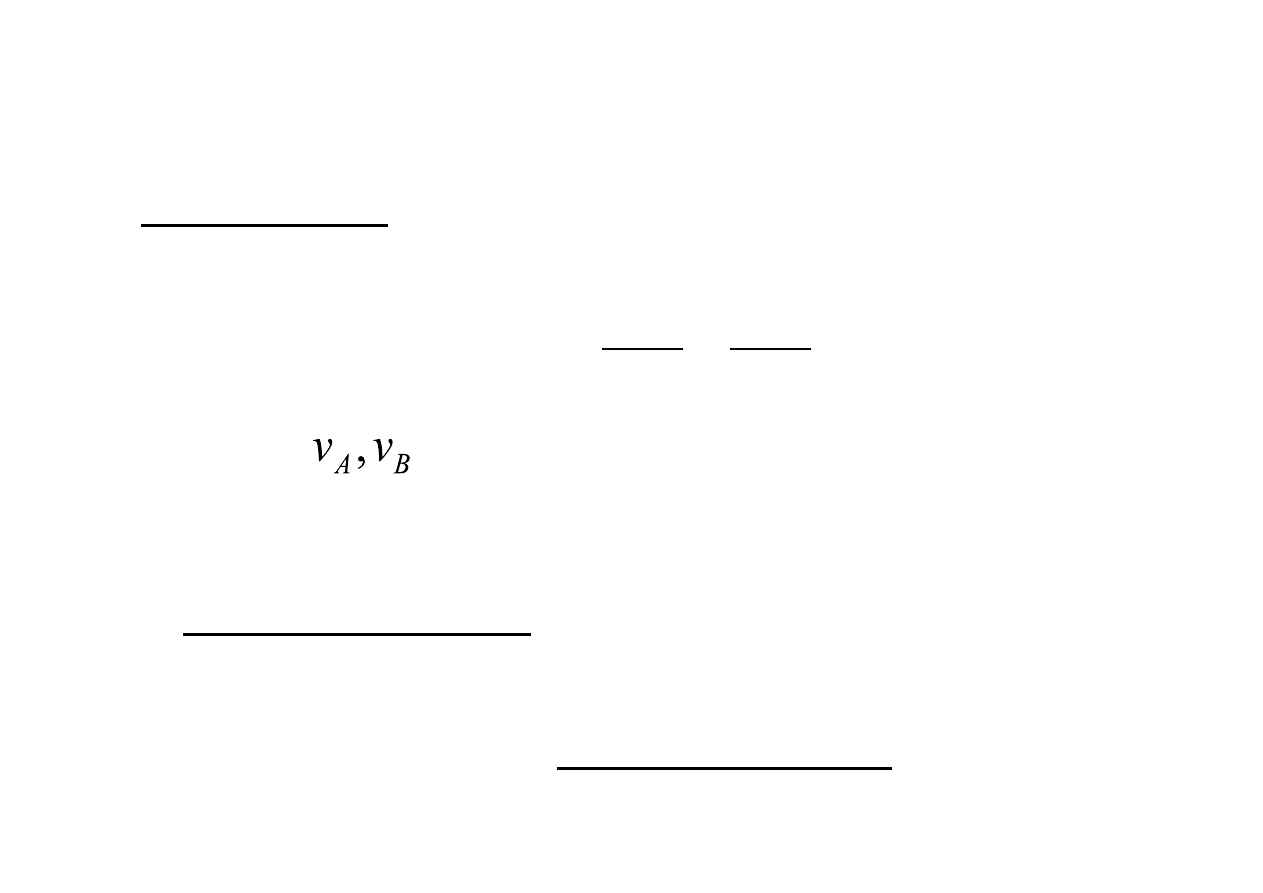
Szybkość reakcji jest miara ilości zużywanych substratów lub
powstających produktów w jednostce czasu. Opisujemy ją
wzorem:
•
Gdzie: -‐ współczynniki stechiometryczne reagentów
A -‐ substraty
B -‐ produkty
Doświadczalnie wyznaczone równanie tego typu nosi nazwę
równania kinetycznego, przyjmuje ono postać:
Współczynnik k nosi nazwę stałej szybkości reakcji.
€
v = −
dc
A
v
A
dt
=
dc
B
v
B
dt
€
v = k A
[ ]
a
B
[ ]
b
...
Kinetyka reakcji
ν
A
A
ν
B
B

Przez scałkowanie powyższego równania otrzymamy:
Szybkość reakcji chemicznej a stałą szybkości reakcji
Co z tego
wynika?

Rzędowość reakcji chemicznych
Rząd
reakcji
względem
danego
składnika,
to
wykładnik
potęgi,
do
której
podniesione
jest
stężenie
reagenta
w
równaniu
kinetycznym.
Całkowity
rząd
reakcji
to
suma
poszczególnych
rzędów
a
+
b
+...
w
równaniu
kinetycznym.
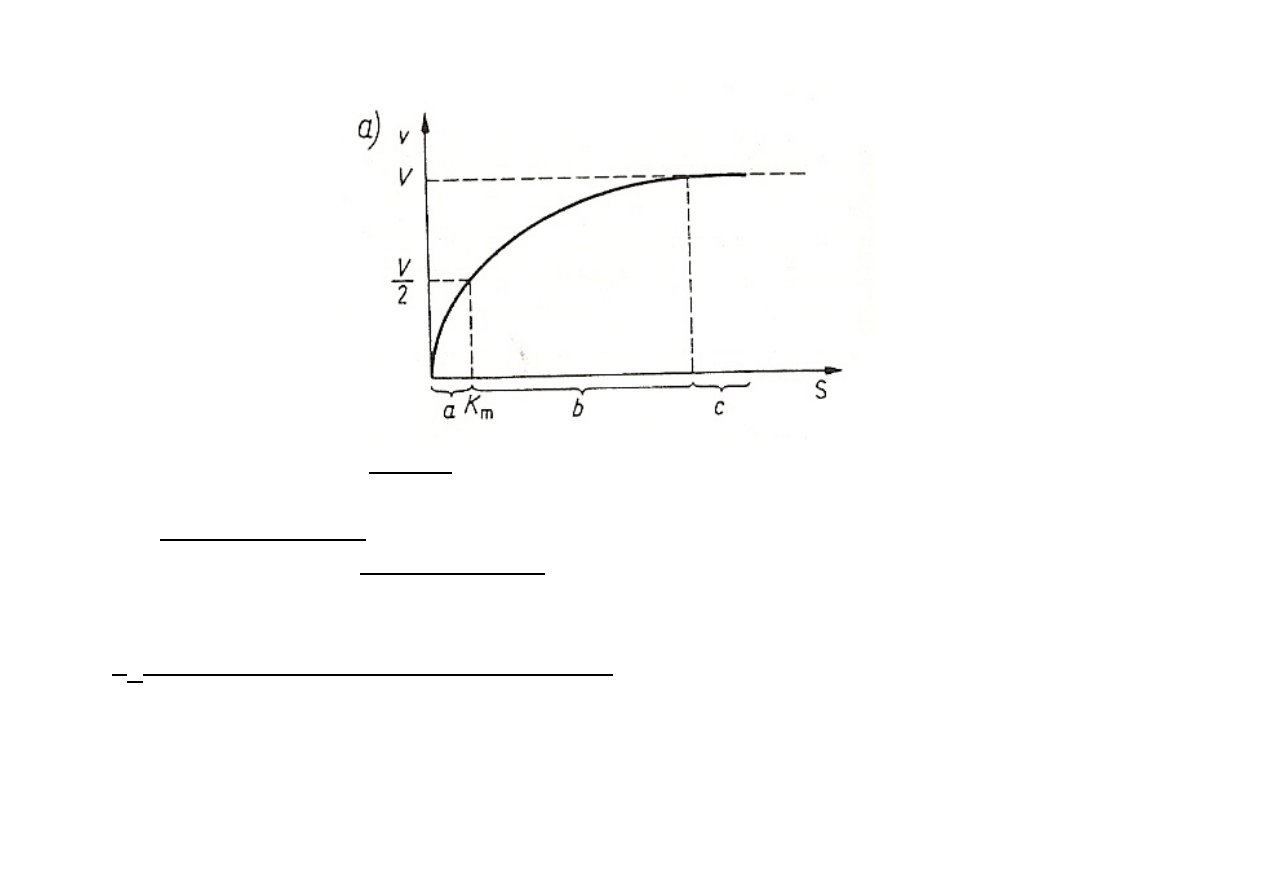
a – kinetyka reakcji I rzędu (linia prosta – zależność wprost proporcjonalna pomiędzy
badanymi czynnikami)
b
–
kinetyka
mieszana
zależność
nieproporcjonalna
c
–
kinetyka
reakcji
zerowego
rzędu
(szybkość
reakcji
osiąga
V
max
– reakcja przebiega
zgodnie z kinetyką zerowego rzędu)
K
m
-‐ charakteryzuje daną aktywność enzymu
-‐ jest to takie stężenie substratu [mol/l], przy którym reakcja biegnie z szybkością równą
połowie szybkości maksymalnej
-‐ jest to takie stężenie substratu, przy którym połowa miejsc aktywnych jest obsadzona
substratem
-‐ dla większości enzymów wartości leżą w przedziale od 10
-‐1
do 10
-‐7
M
Reakcja enzymatyczna – szybkość zależy od stężenia substratu

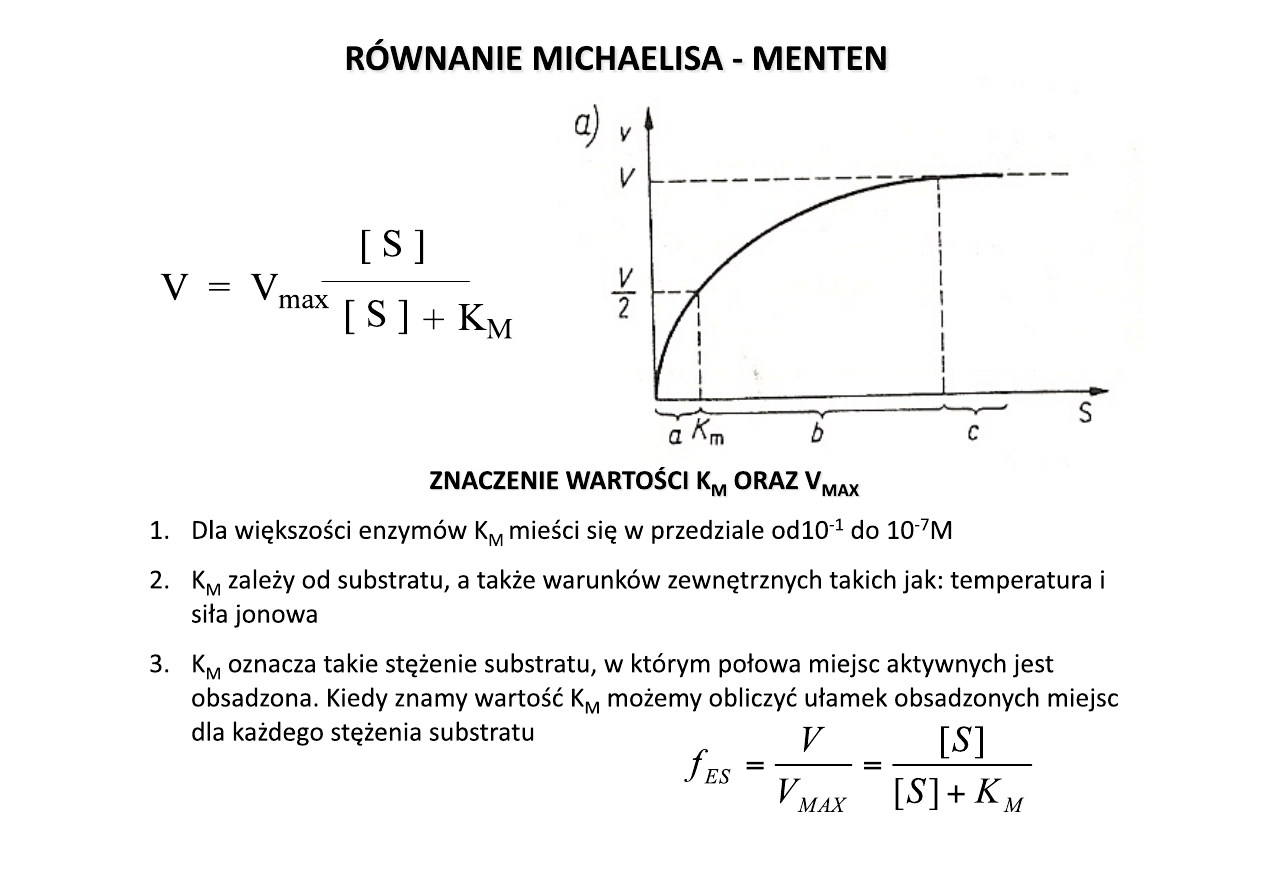
Model Michaelisa-‐Menten opisuje właściwości
kinetyczne wielu enzymów
1. Dla wielu enzymów szybkość katalizy V zmienia się ze stężeniem substratu [S].
V jest definiowane jako liczba moli produktu utworzonego w czasie 1 sekundy.
2. Przy stałym stężeniu enzymu, V prawie liniowo zależy od [S], kiedy [S] jest
małe. Przy dużych [S], V jest prawie niezależne od [S],
3. W 1913 roku Leonor Michaelis i Maud Menten zaproponowali prosty model,
odpowiadający takiej charakterystyce kinetycznej. Podstawową cechą tego
modelu jest założenie, że kompleks ES jest koniecznym etapem pośrednim
procesu katalitycznego. Najprostszy model, który odpowiada właściwościom
kinetycznym wielu enzymów, wygląda następująco
Mechanizm
katalizowanej
reakcji
z
udziałem
jednego
substratu.
E
–
enzym,
S
–
substrat,
P
–
produkt, k
1
i k
3
– stałe szybkości reakcji
przebiegających w prawą stronę, k
2
– stała
szybkości reakcji zachodzącej w lewą stronę.
Enzym
E
łączy
się
z
substratem
S
tworząc
kompleks
ES;
reakcję
charakteryzuje
stała
szybkości k
1
. Istnieją dwie możliwe drogi
rozpadu kompleksu ES. Może on
dysocjować do E i S ze stałą szybkości k
2
lub
może
się
przekształcać,
tworząc
produkt
P
ze
stałą
szybkości
k
2
. Zakłada
się,
że
produkt
reakcji
nie
może
ulec
powrotnemu
przekształceniu
w
wyjściowy
substrat,
warunek,
który
jest
spełniany
w
początkowym
etapie
reakcji,
zanim
stężenie
produktu
stanie
się
znaczące.
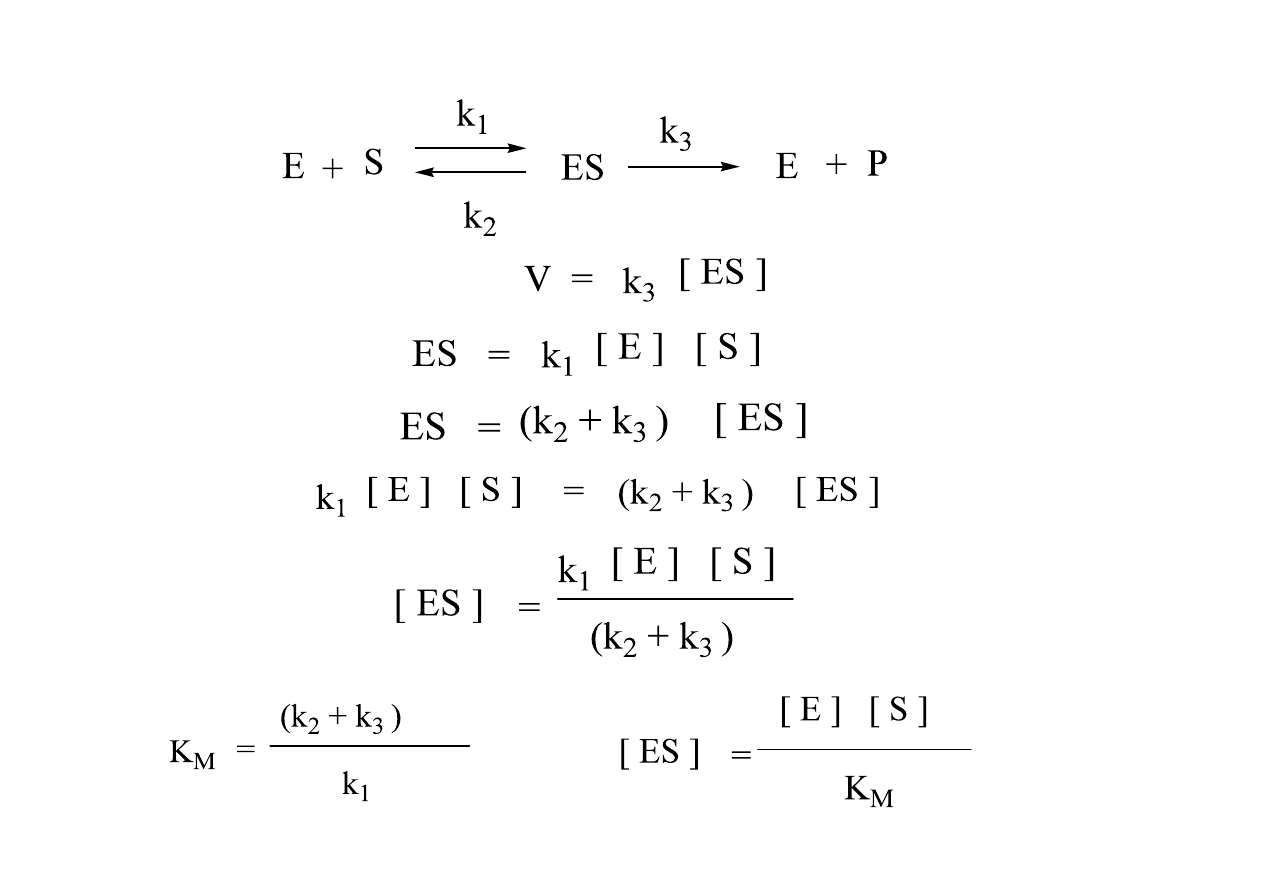
Rekacja enzymatyczna (równanie kinetyczne)
Stała Michaelisa:
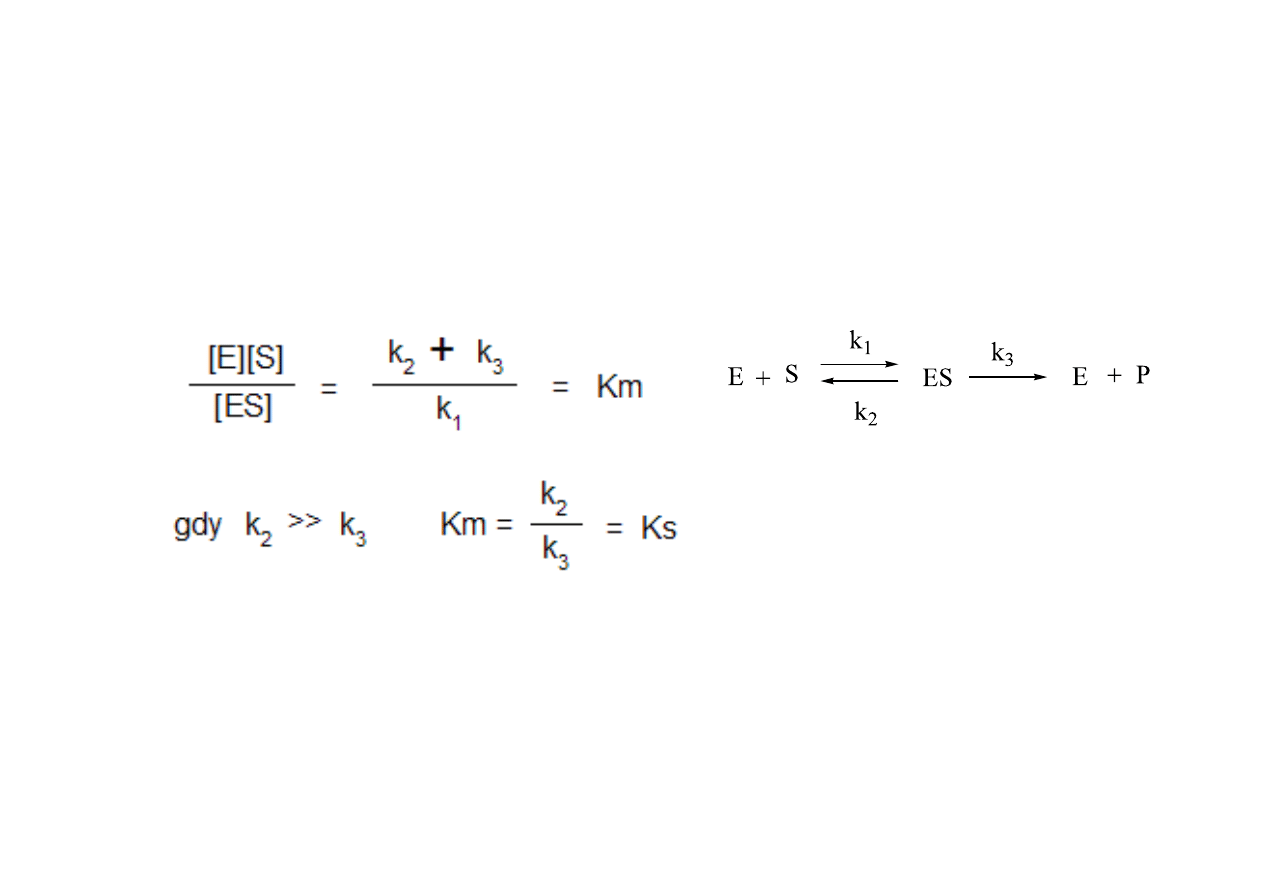
Ks
-‐
stała
dysocjacji
kompleksu
ES
do
E
i
S
-‐
właściwa
miara
siły
tworzenia
kompleksu
ES,
czyli
powinowactwa
enzymu
do
substratu
Ks -‐ miara powinowactwa E do S, ale tylko w pewnych warunkach (tylko wtedy, kiedy k
2
> k
3
) = k
2
/k
1
= Ks
•
K
m
(stała Michaelisa) opisuje tworzenie i rozpad kompleksu ES
•
jest miarą powinowactwa enzymu do substratu
•
niska wartość K
m
– silne powinowactwo enzymu do substratu
•
wysoka wartość K
m
– słabe powinowactwo enzymu do substratu
Stała Michaelisa
Gdy to:
więc:
Stała
Michaelisa-‐Menten
jest
to
stężenie
substratu
(mol/dm
3
), przy którym szybkość reakcji
enzymatycznej jest równa połowie jej szybkości maksymalnej.
Stała Michaelisa -‐ znaczenie
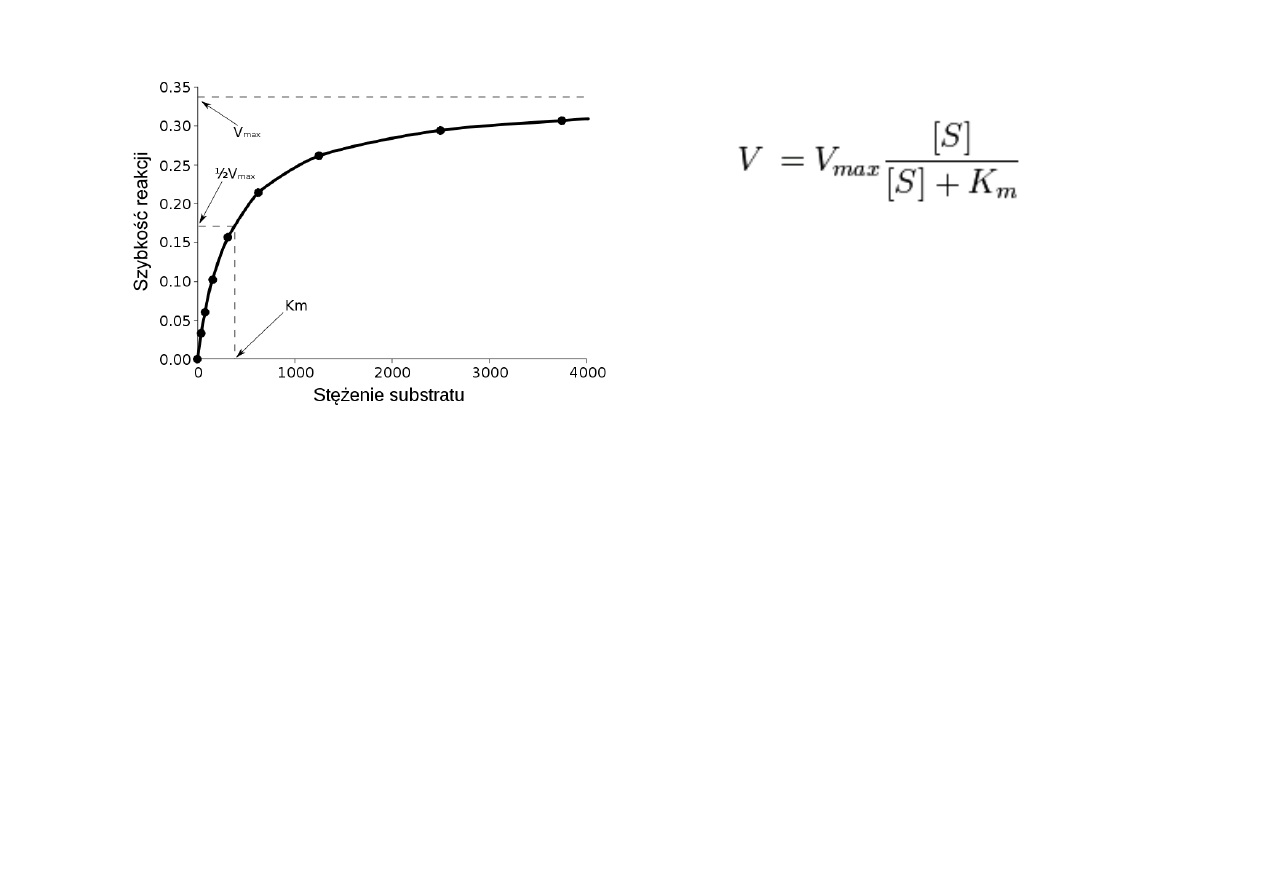
Równaniem
Michaelisa-‐Menten:
gdzie:
V
–
szybkość
początkowa,
S
–
stężenie
substratu,
K
m
– stała Michaelisa-‐Menten.
Równanie Michaelisa-‐Mentena
1. Przy niewielkich stężeniach substratu, kiedy [S] jest znacznie mniejsze niż K
M
, V= [S] V
max
/
K
M ;
oznacza to, że szybkość reakcji jest wprost proporcjonalna do stężenia substratu.
2. Przy dużych stężeniach substratu, kiedy [S] jest dużo większe od K
M
, V = V
max
, co oznacza,
że szybkość jest maksymalna i niezależna od stężenia substratu.
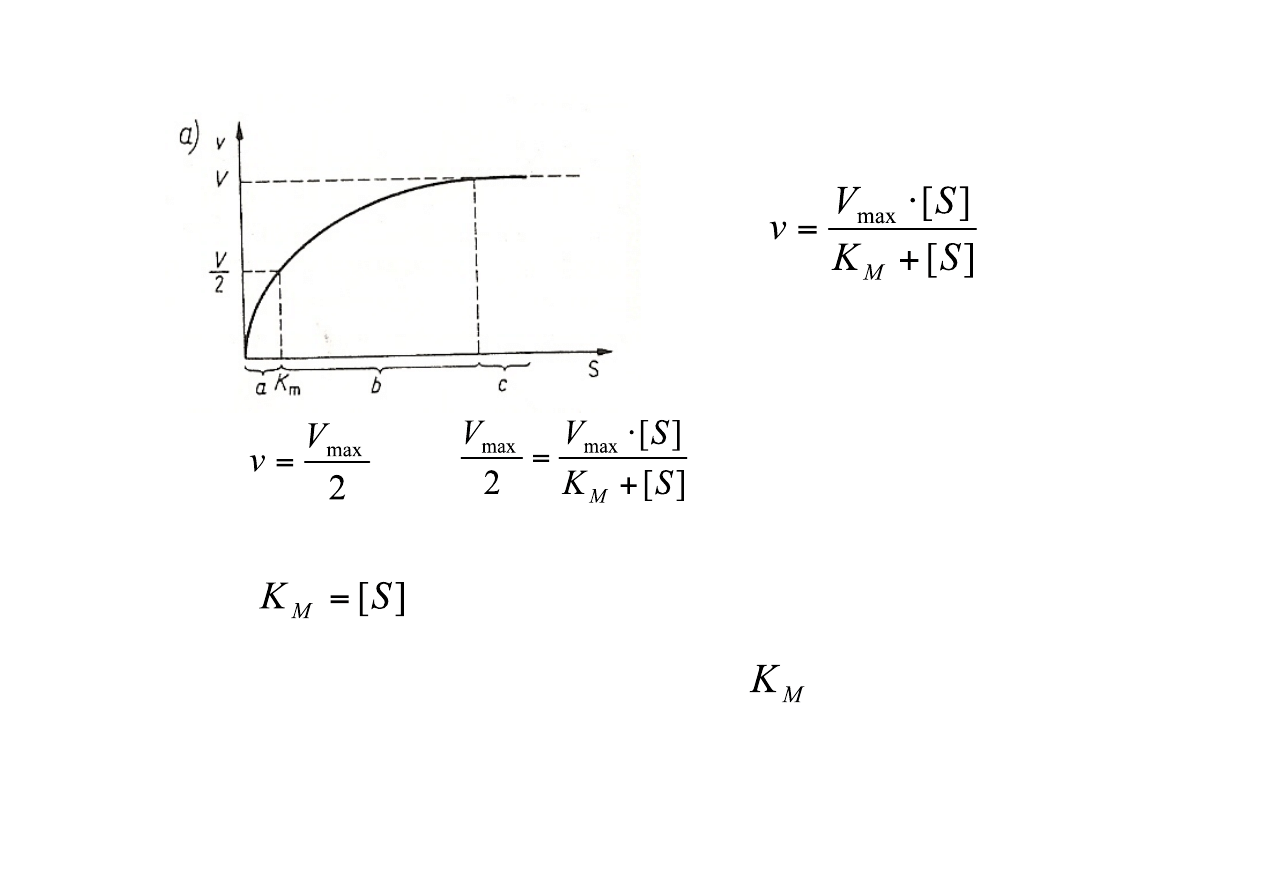
3. Znaczenie K
M
wynika z równania przedstawionego wyżej. Kiedy [S] = K
M
, wtedy V = V
max
/2.
Stąd K
m
jest równe takiemu stężeniu substratu, dla którego szybkość reakcji osiąga połowę
swojej wartości maksymalnej.
4. Inaczej mówiąc K
M
jest to takie stężenie substratu, w którym połowa miejsc aktywnych jest
obsadzona.
5. Liczba obrotów enzymu oznacza liczbę cząsteczek substratu przekształconych w produkt
reakcji na jednostkę czasu, w warunkach pełnego wysycenia enzymu substratem.
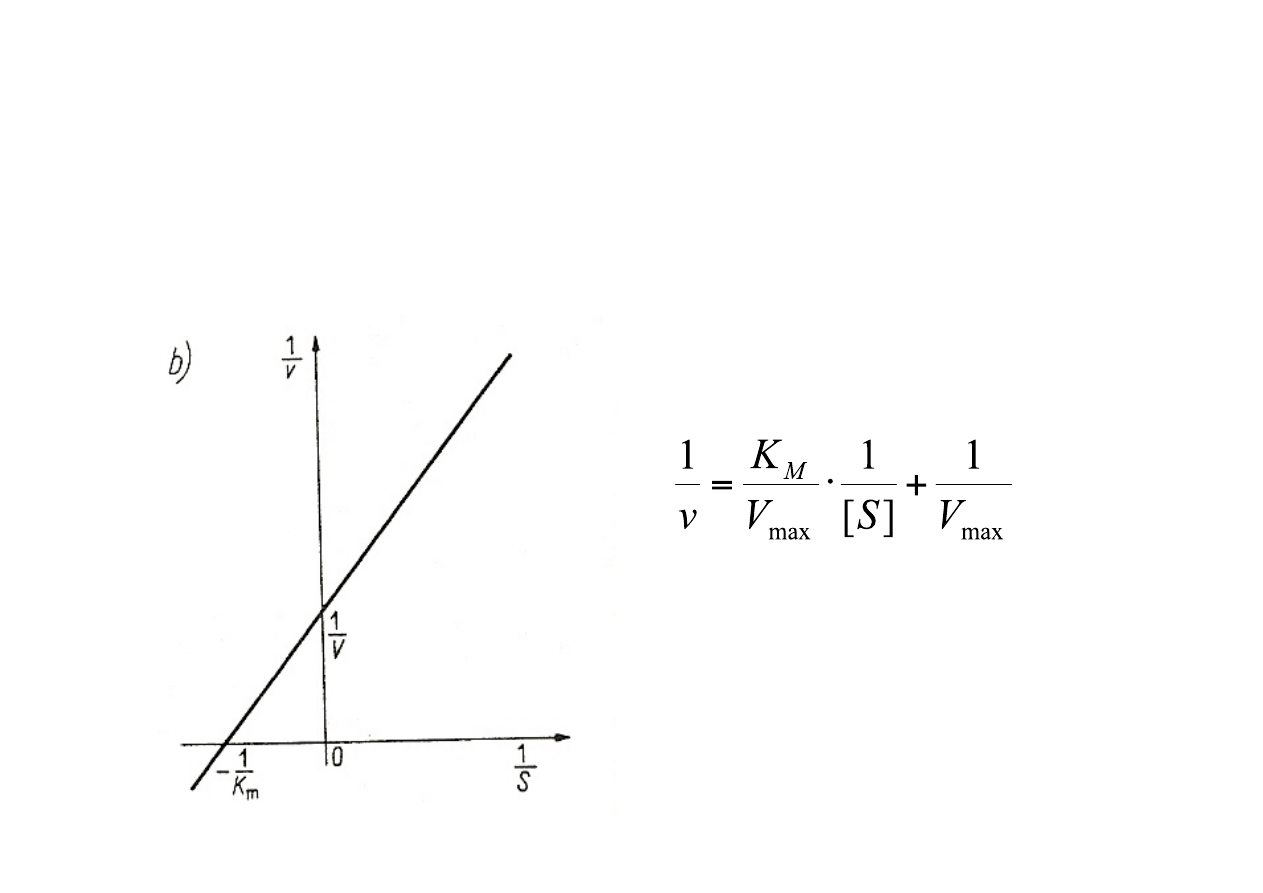
Wpływ stężenia substratu na szybkość reakcji przy stałym stężeniu enzymu -‐
krzywa wg Lineweavera-‐Burka.
Teoria Lineweavera – Burka
max

Pomiar aktywności enzymu – polega na określeniu albo zaniku substratu,
albo powstania produktu w zależności od czasu. Aby wyznaczyć aktywność
enzymu należy więc zmierzyć szybkość reakcji.
Jednostka aktywności enzymatycznej – katal; 1 katal (kat) wywołuje w
określonych warunkach reakcji przemianę 1 mola substancji w ciągu jednej
sekundy.
Liczba obrotów – określa liczbę cząstek substratu, które w jednostce czasu
(minuta lub sekunda) ulegają przekształceniu przez jedną cząstkę enzymu.
€
v = −
dc
A
v
A
dt
=
dc
B
v
B
dt
Jednostki enzymatyczne
Jednostka enzymu (jednostka enzymatyczna), U (ang. unit, w pol. także J) –
jednostka aktywności enzymów wprowadzona w 1964 przez Komitet Nazewnictwa
Międzynarodowej Unii Biochemii i Biologii Molekularnej. Zdefiniowana przez
Komisję
Enzymową
w
1961.
Jeden
U
to
ilość
enzymu
katalizująca
przemianę
1
μmola
substratu
w
czasie
1
minuty. Zwykle przyjmuje się określone warunki: temperaturę 30 °C, dane pH i
maksymalne
wysycenie
enzymu
substratem.
Ponieważ
minuta
nie
jest
jednostką
SI,
do
określania
aktywności
enzymów
zaleca
się
stosowanie katala (od 1978).
1 U = 1 μmol/min = 1/60 μkatala = 16,67 nanokatala
UWAGA: Jednostka enzymu U, nie ma nic wspólnego z IU (Internawonal Unit;
Jednostka Międzynarodowa), miarą aktywności substancji biologicznie czynnych.
Inhibicja
Inhibicja polega na blokowaniu przez inhibitor współdziałania składników
uczestniczących w reakcji enzymatycznej:
-‐ enzym,
-‐ substrat(y),
-‐ koenzym.
Szybkość reakcji enzymatycznej zależy od czynników hamujących, które dzieli się
na działające niespecyficznie i specyficznie.
PODZIAŁ INHIBICJI
1. Nieodwracalna
2. Odwracalna
-‐ kompetycyjna
-‐ niekompetycyjna
-‐ akompetycyjna
-‐ mieszana
INHIBICJA AKTYWNOŚCI ENZYMÓW umożliwia:
-‐ identyfikację reszt istotnych dla aktywności enzymów
-‐ poznanie mechanizmów działania enzymów
-‐ działanie leków i czynników toksycznych
-‐ mechanizm kontroli aktywności enzymów
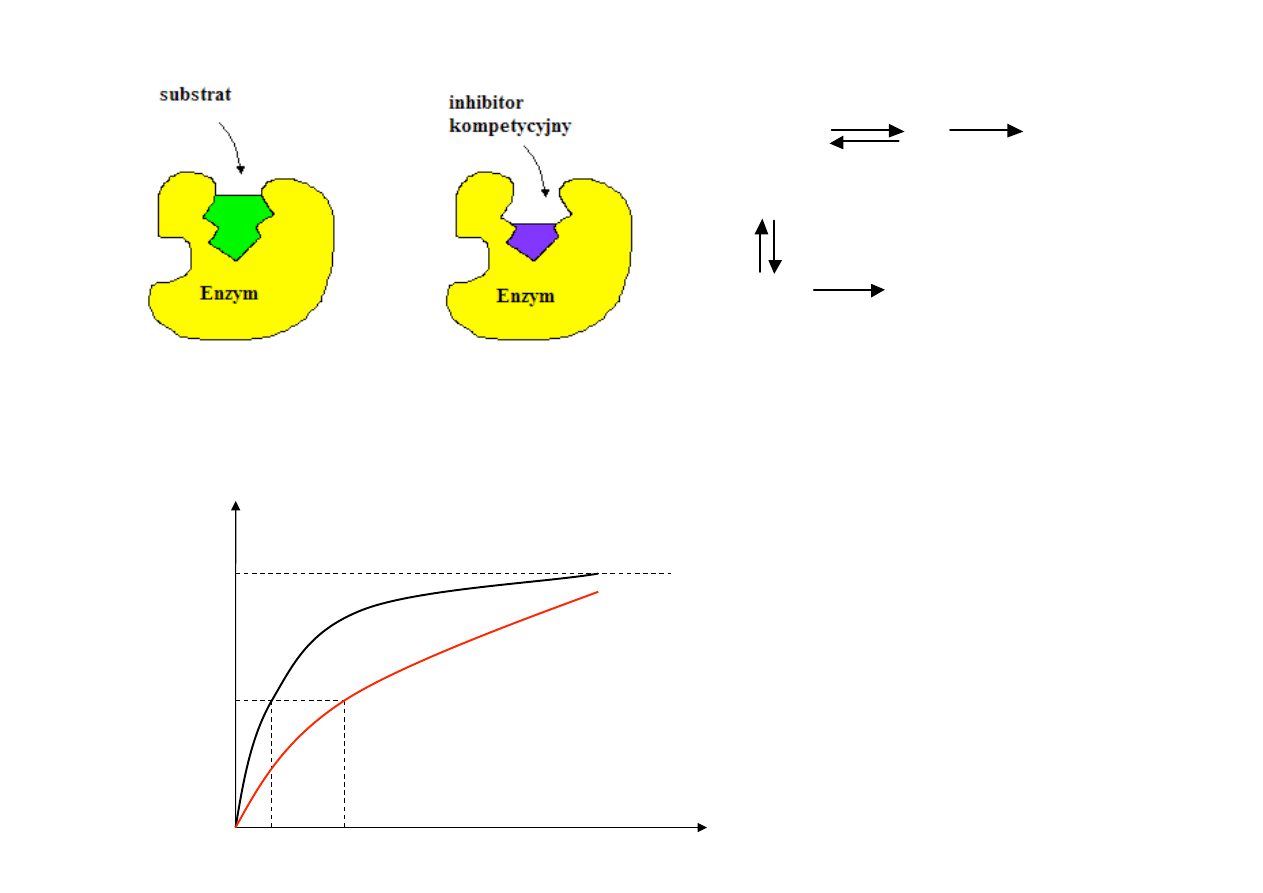
1. INHIBICJA KOMPETYCYJNA (WSPÓŁZAWODNICZĄCA)
!
!
"!#!$!
"$!
"!#!%!
#!
&!
"$!
'()*!(+)*,-.!
/
&
!
[S
o
]
V
o
K
m
’
K
m
V
max
K
m
(K
m
spp
)
nowa tzw. pozorna
wartość K
m
wyznaczona w
obecności
inhibitora
jest
wyższa
w
porównaniu
z
rzeczywistą
K
m
Wykres Michaelisa-‐Menten
• inhibitor kompetycyjny zmniejsza szybkość katalizy enzymatycznej przez zmniejszenie liczby
cząsteczek enzymu wiążących substrat,
•
inhibicję
kompetycyjną
można
znieść
przez
zwiększenie
stężenia
substratu
•
V
max
nie ulega zmianie, zwiększa się K
m
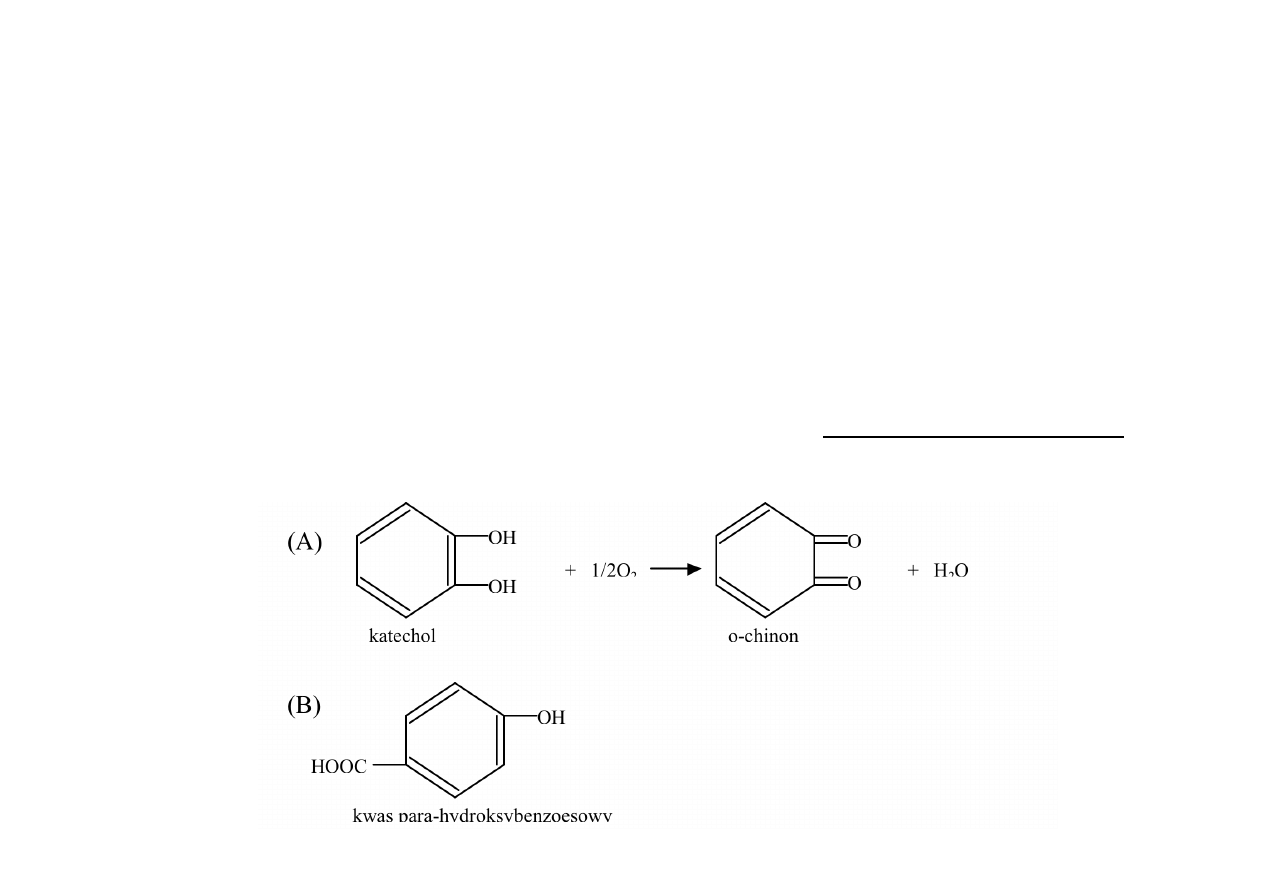
PRZYKŁADY:
a)
działanie
malonianu
CH
2
(COOH)
2
na dehydrogenazę bursztynianową -‐ (CH
2
)
2
(COOH)
2
Przykład ten jest wyjątkowym przypadkiem. Inne przypadki inhibitorów
konkurencyjnych
wykazują
zazwyczaj
mieszane
działanie,
inhibitora
konkurencyjnego
i
akompetycyjnego.
b) hamowanie syntezy białka przez analogi aminokwasów białkowych (aminokwasy
niebiałkowe), np. niektóre leki, niektóre pestycydy, antybiotyki
c) hamowanie oksydazy polifenolowej przez kwas para-‐hydroksybenzoesowy
(brązowienie warzyw i owoców)
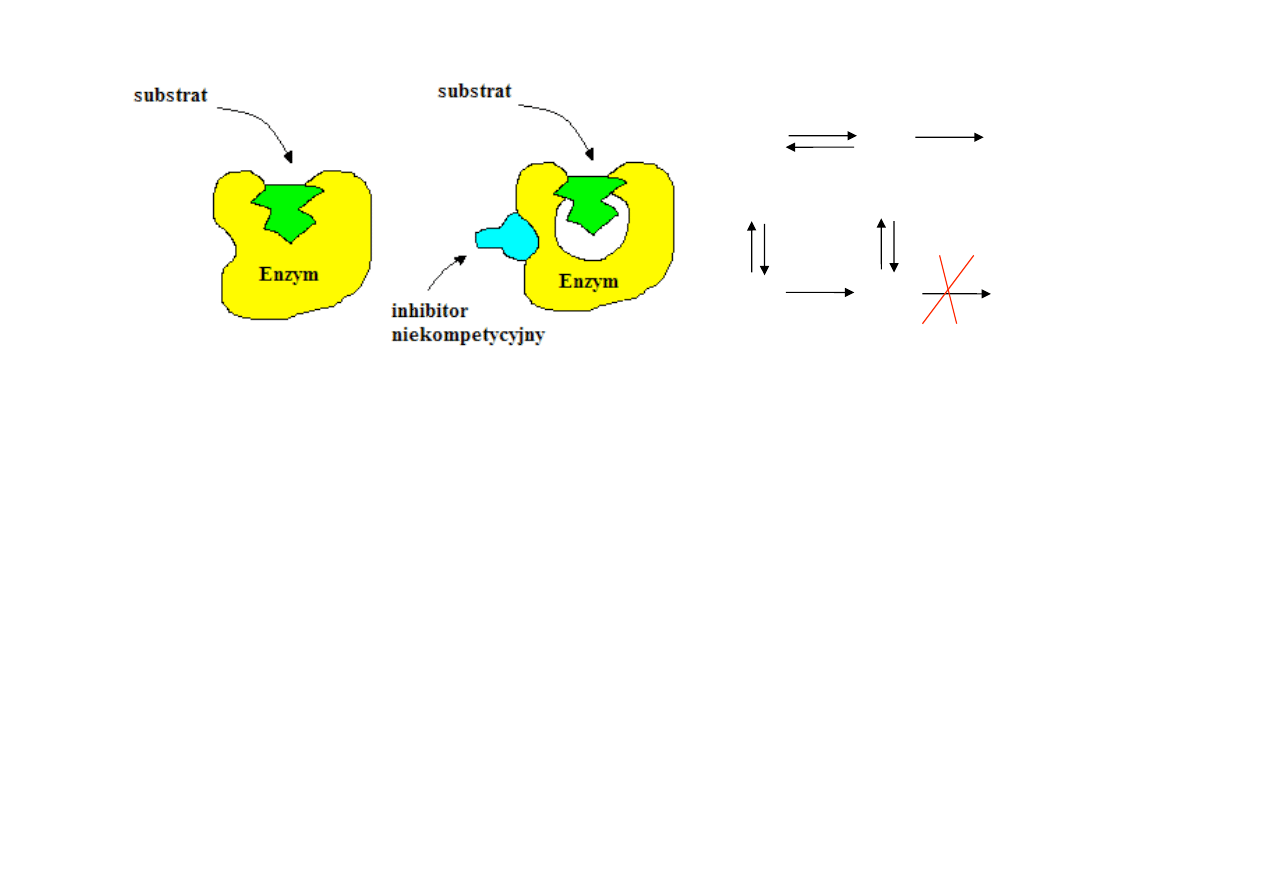
2. INHIBICJA NIEKOMPETYCYJNA (NIEWSPÓŁZAWODNICZĄCA)
!
E
ES
E + P
+ I
EI
K
I
+ S
+ I
+ S
K
I
E + P
ESI
• wiąże się w miejscu katalitycznym (nie w miejscu wiązania substratu) lub poza nim,
wywołując zmiany konformacyjne niekorzystne dla miejsca katalitycznego
• inhibitor nie jest strukturalnie podobny do substratu, nie może wiązać się w miejscu
wiązania substratu, tylko w miejscu katalitycznym lub poza nim => obniża szybkość
maksymalną katalizy
• zmniejsza szybkość katalizy enzymatycznej poprzez zmniejszenie liczby obrotów enzymu
(obniża stężenie funkcjonalnego enzymu, pozostała część funkcjonalnego enzymu
zachowuje się jak bardziej rozcieńczony roztwór enzymu)
• inhibitor niekompetycyjny nie wpływa na przyłączanie substratu do enzymu – nie wpływa
na powinowactwo E do S
• nie zmienia K
m
, obniża V
max
Nie można znieść inhibicji niekompetycyjnej zwiększając stężenie substratu, lecz poprzez
związanie inhibitora przez inny związek
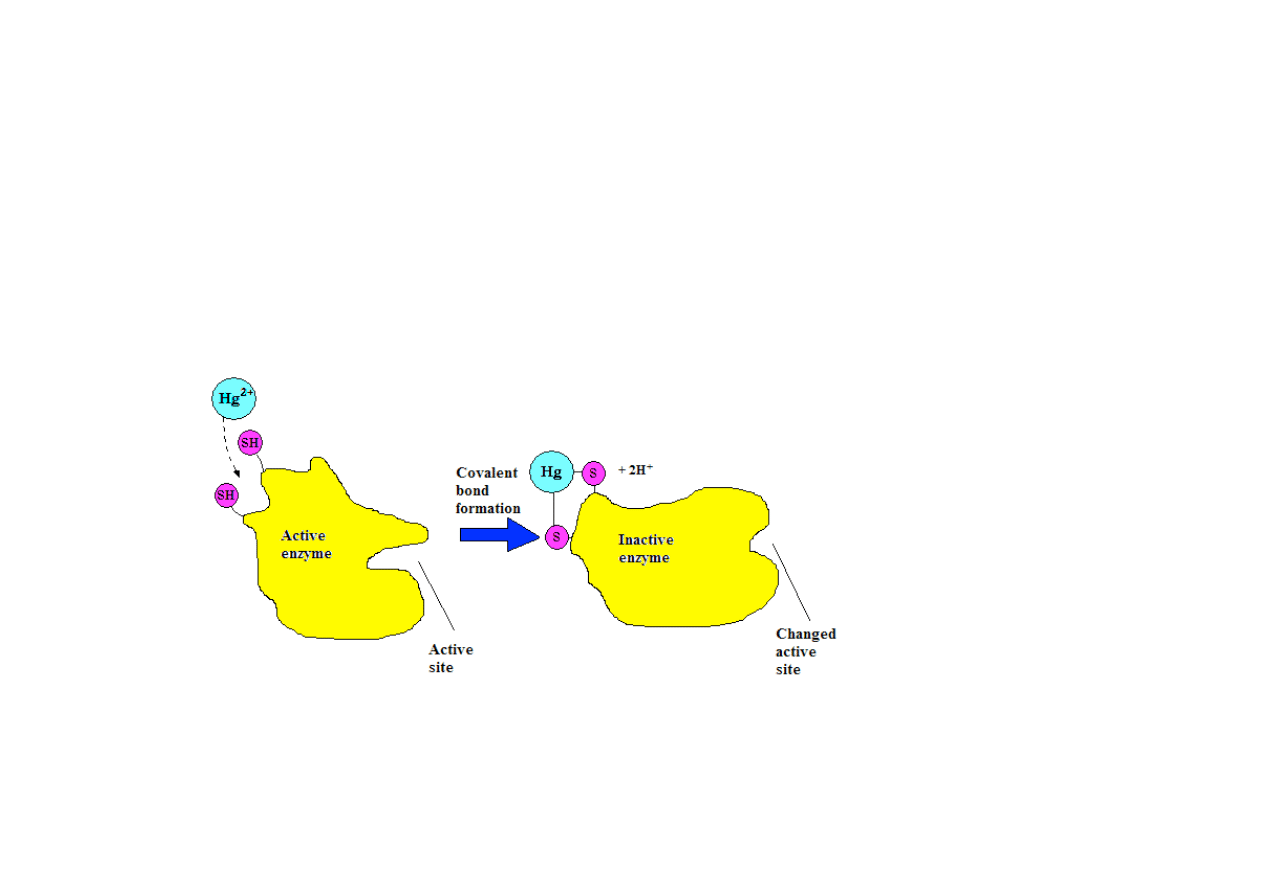
PRZYKŁADY:
a)
działanie
jonów
metali
ciężkich,
np.
Cu
2+
, Hg
2+
, Ag
2+
, które wiążą się z gr. –SH cysteiny w
miejscu katalitycznym lub poza nim (tworzą się merkaptydy)
b) działanie cyjanków, azydków i fluorków, które wiążą jony metali np. Fe
2+
lub Fe
3+
(tworzą one kompleksy z jonami Fe podobne do żelazicyjanków i żelazocyjanków)
c) działanie czynników chelatujących dwuwartościowe kawony, np. EDTA
(etylenodiaminotetraoctan) wiążący jony Mg
2+
.
!
W
wielu
przypadkach
dla
inhibitorów
nieodwracalnych
otrzymuje
się
parametry
kinetyczne
(K
m
, V
max
) charakterystyczne dla inhibitorów odwracalnych niekompetycyjnych,
stąd wiele zamieszania np. co do działania cyjanków.
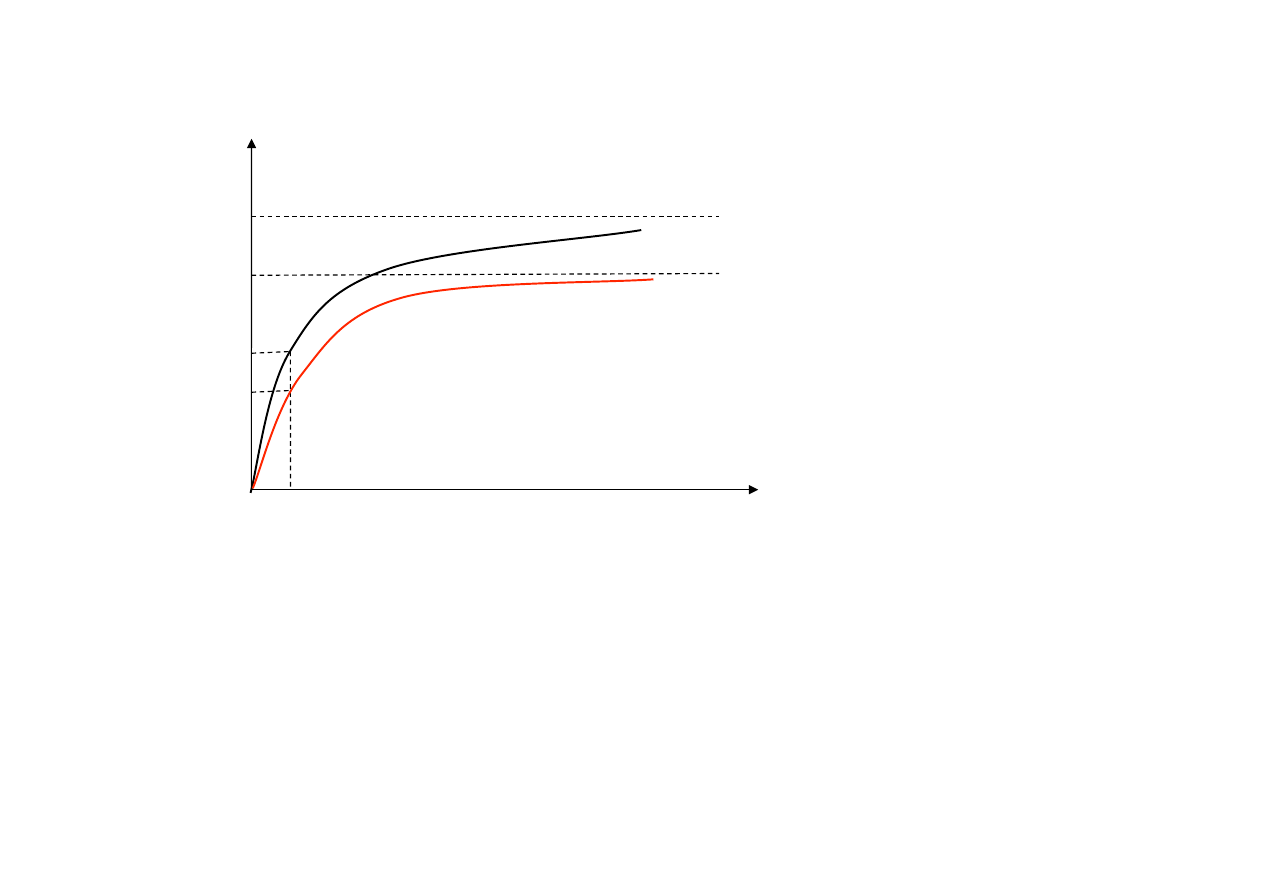
[S
o
]
V
o
K
m
=
K
m
’
V
max
’
V
max
’ (V
max
spp
)
nowa tzw.
pozorna
wartość
V
max
wyznaczona
w
obecności
inhibitora
jest
niższa
w
porównaniu
z
rzeczywistą
V
max
V
max
V
max
’/2
V
max
/2
2. INHIBICJA NIEKOMPETYCYJNA (NIEWSPÓŁZAWODNICZĄCA)
V
max
nie może być osiągnięta nawet przy dużym stężeniu substratu
V
max
’ maleje proporcjonalnie do wzrostu stężenia inhibitora (im wyższe stężenie inhibitora tym
niższa V
max
’)
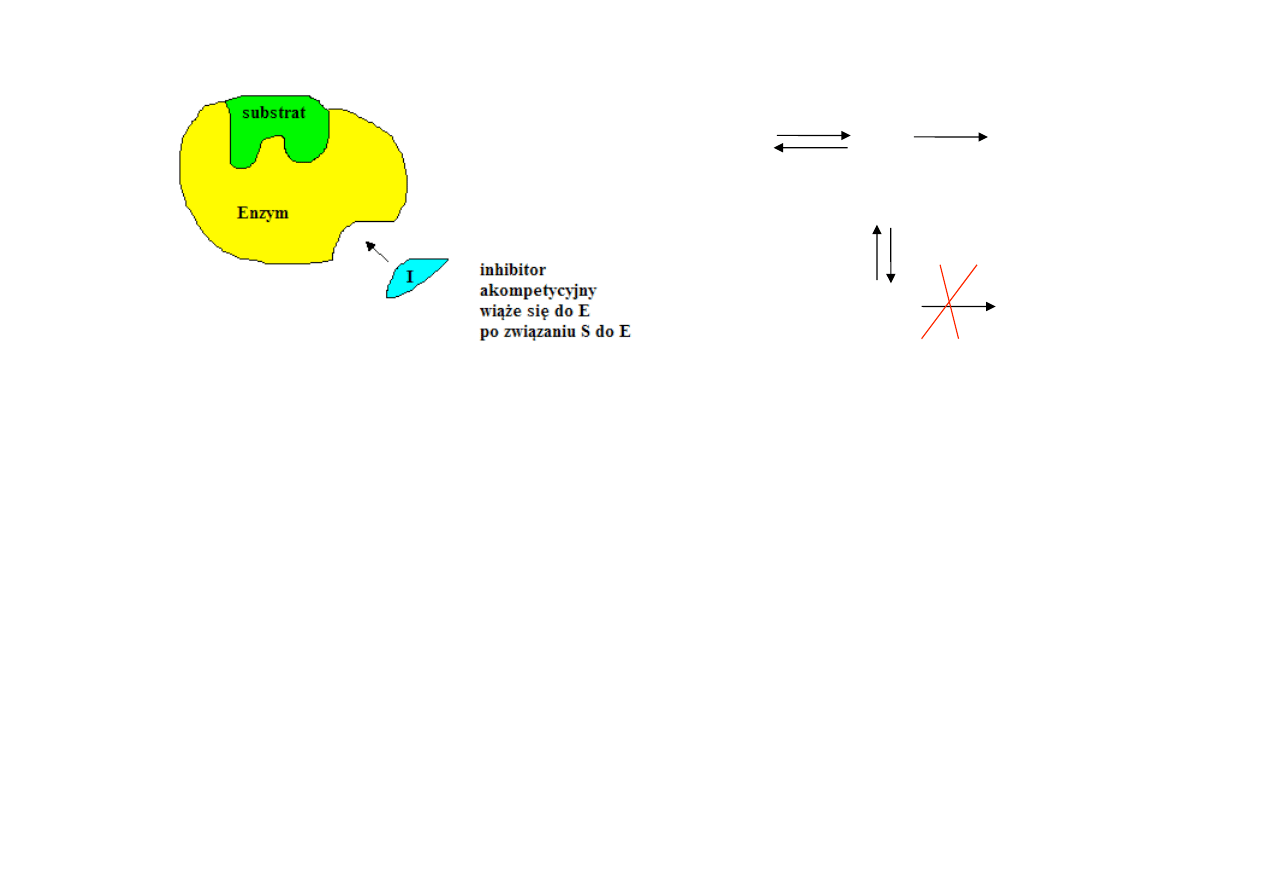
3. INHIBICJA AKOMPETYCYJNA
!
E
ES
E + P
+ S
+ I
K
I
E + P
ESI
-‐ wiąże się w innym miejscu niż miejsce wiązania substratu, wiąże się do kompleksu ES
tworząc nieaktywny katalitycznie kompleks ESI
-‐
związanie
substratu
przez
enzym
odsłania
miejsce
wiązania
inhibitora
-‐
obniża
V
max
i K
m
-‐ nie można go znieść przez zwiększenie stężenia substratu
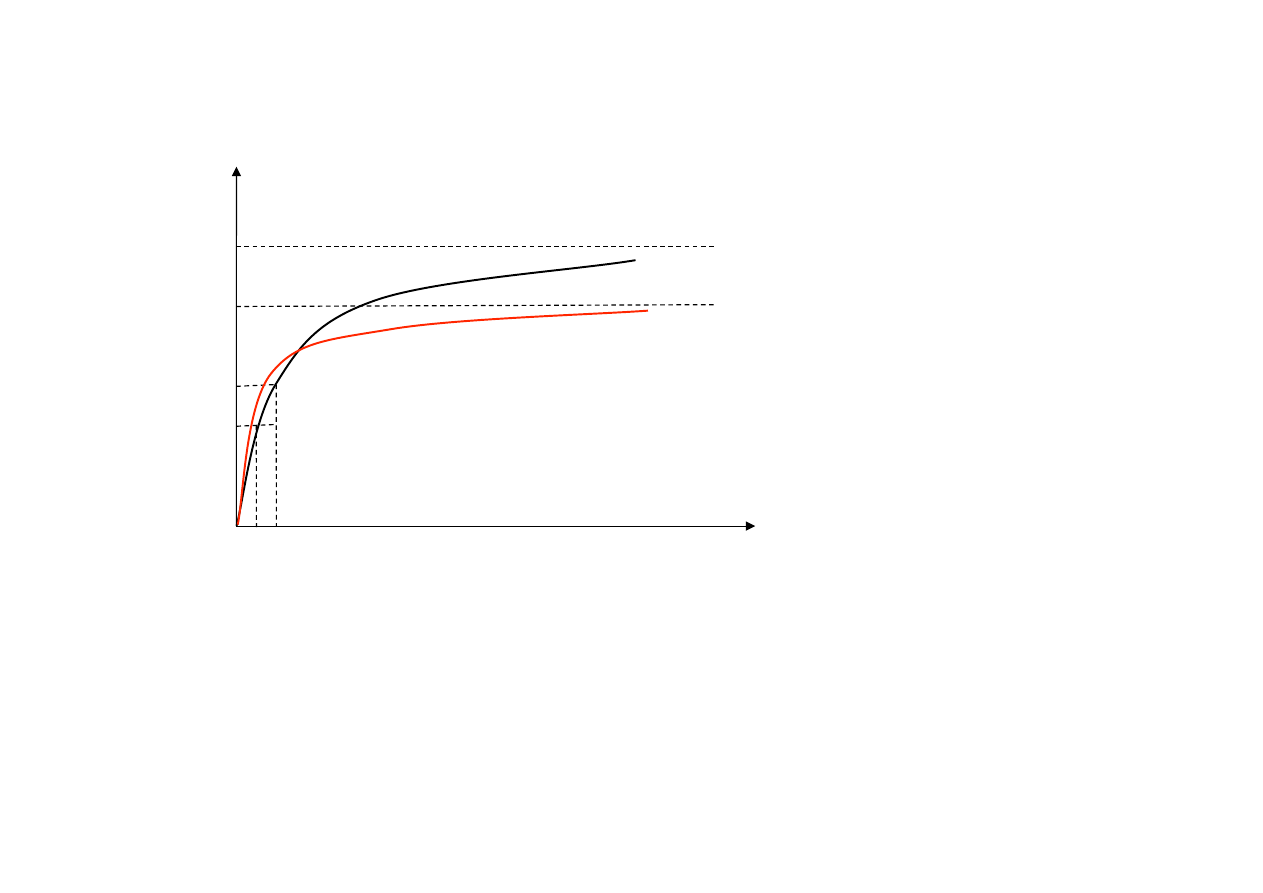
[S
o
]
V
o
K
m
V
max
’
V
max
’ (V
max
spp
)
nowa tzw.
pozorna
wartość
V
max
wyznaczona
w
obecności
inhibitora
jest
niższa
w
porównaniu z rzeczywistą V
max
V
max
V
max
’/2
V
max
/2
K
m
’
3. INHIBICJA AKOMPETYCYJNA
Ten
typ
inhibicji
jest
dość
rzadki
i
może
dotyczyć
niektórych
enzymów
mulwmerycznych,
katalizujących
reakcje
z
udziałem
wielu
substratów.

4. INHIBICJA MIESZANA
E
ES
E + P
+ I
EI
K
i
+ S
+ I
K
I
E + P
ESI
E + P
K
i
≠ K
I
K
i
– stała dysocjacji EI
K
I
– stała dysocjacji ESI
•
inhibitor
może
wiązać
się
do
E
lub
kompleksu
ES
•
nie
ważne
założenie
o
niezależności
miejsc
wiązania
S
i
I,
tak
jak
w
inhibicji
niekompetycyjnej
• inhibitor może się wiązać w centrum aktywnym (miejscu wiązania substratu lub miejscu
katalitycznym) lub poza nim
• inhibitor wpływa jednocześnie na wiązanie substratu, jak również liczbę obrotów
enzymu
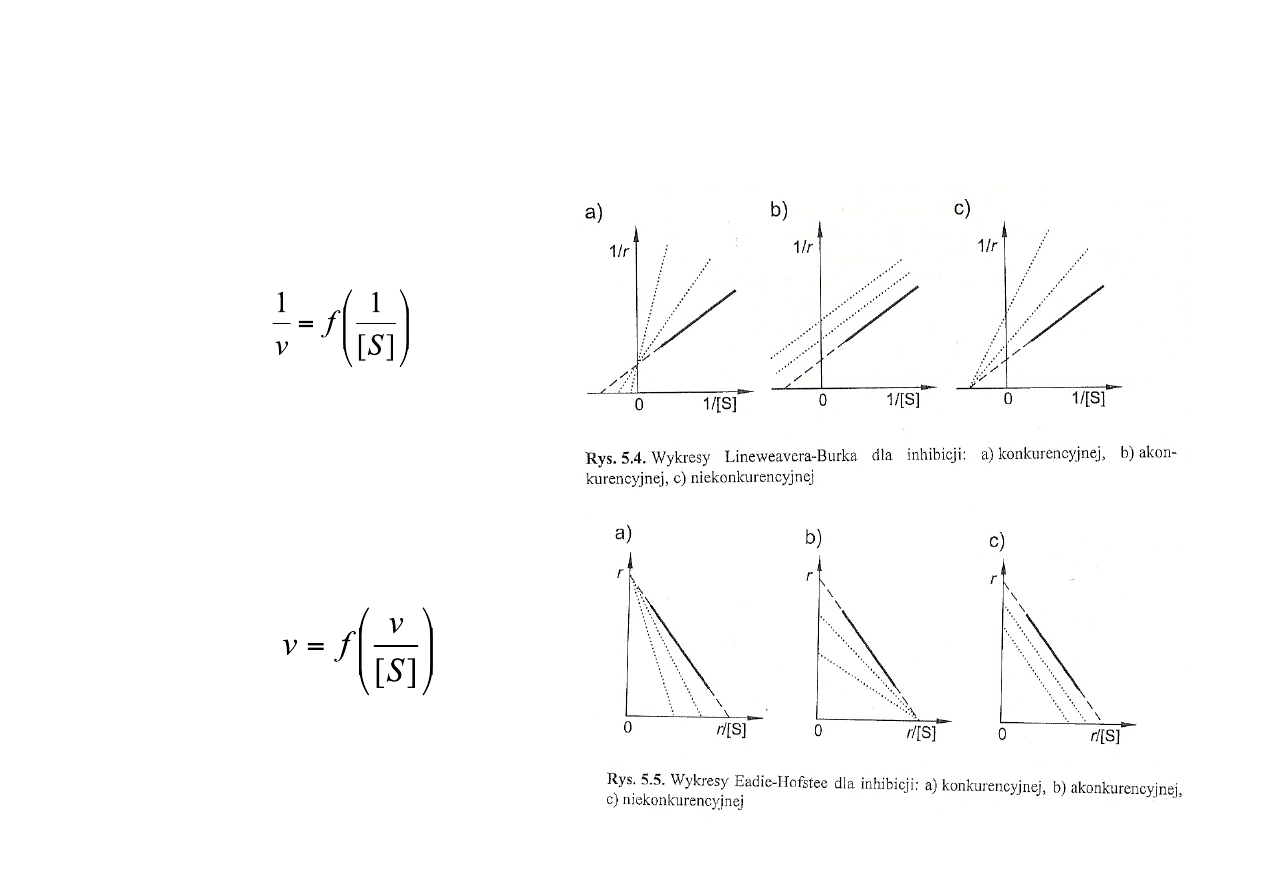
Rozróżnianie rodzajów inhibicji
Wykresy Lineweavera-‐Burka
Wykresy Eadie-‐Hofsteego
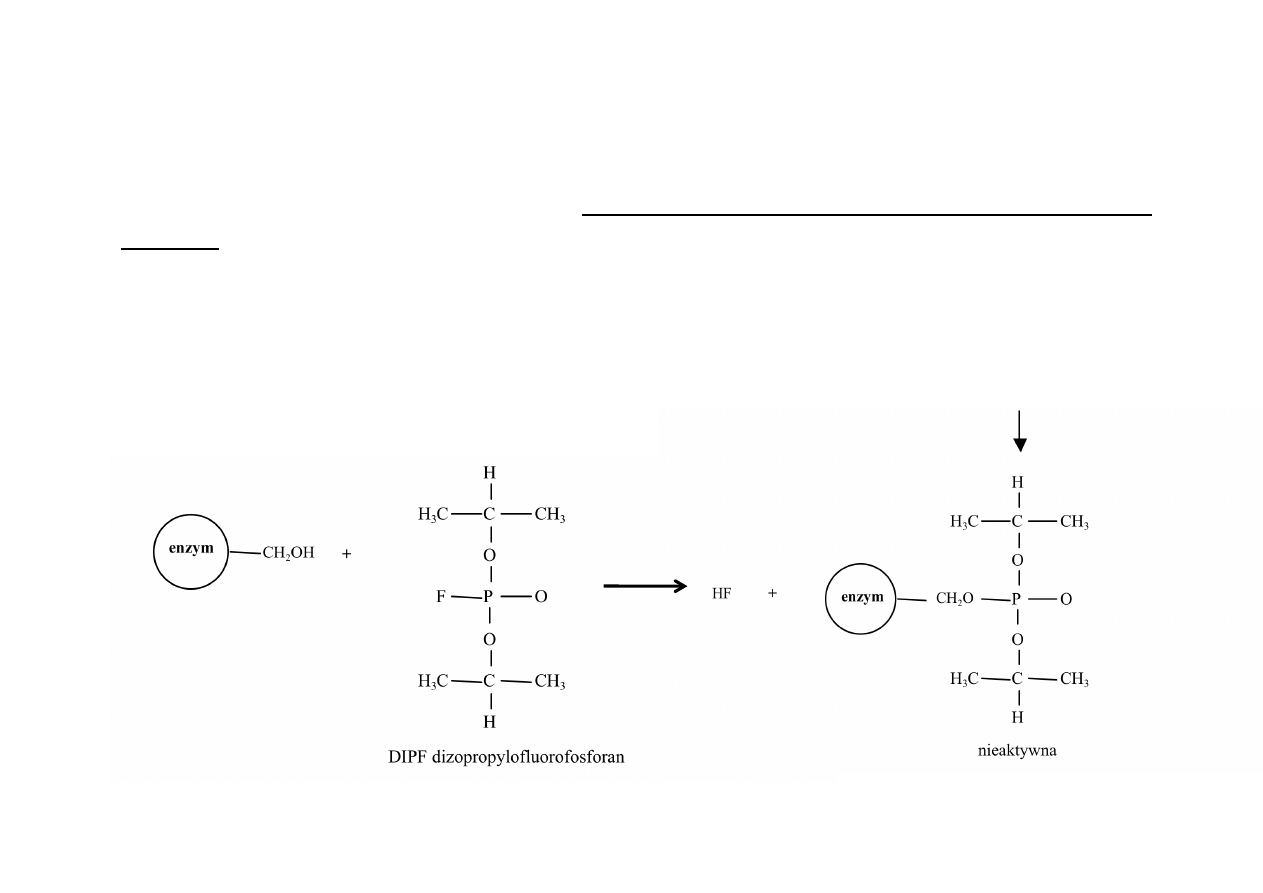
INHIBICJA NIEODWRACALNA
inhibitor
nieodwracalny
zwykle
tworzy
kowalencyjnie
wiązanie
z
grupą
funkcyjną
enzymu
lub
wiąże
się
tak
silnie,
że
jego
dysocjacja
jest
bardzo
powolna
w
przypadku
inhibicji
nieodwracalnej
nie
może
być
stosowana
zależność
Michaelisa-‐
Menten.
PRZYKŁADY:
działanie
gazów
paraliżujących
układ
nerwowy,
które
polega
na
inaktywacji
acetylocholinoesterazy
Wyszukiwarka
Podobne podstrony:
Enzymologia wykłady ściąga
Wyklad 1 z enzymologii, Studia, Przetwórstwo mięsa - Semestr 1, Enzymologia, Wykłady
Enzymologia wykład, V sem
Enzymologia wyklad 30 11
WYKŁAD 7, Studia Biologia, Enzymologia, wykłady
EnzymologiaTZ wyklad 3
EnzymologiaTZ wyklad 7
enzymologia w8, studia, bio, 3rok, 6sem, enzymologia, enzymologia wykłady
WYKŁAD 3, Studia Biologia, Enzymologia, wykłady
wyklad w11, studia, bio, 3rok, 6sem, enzymologia, enzymologia wykłady
WYKŁAD 6, Studia Biologia, Enzymologia, wykłady
enzymologia w10, studia, bio, 3rok, 6sem, enzymologia, enzymologia wykłady
wykłady z enzymologii, Studia Biologia, Enzymologia, wykłady
enzymologia w7, studia, bio, 3rok, 6sem, enzymologia, enzymologia wykłady
Enzymologia wykłady ściąga
EnzymologiaTZ wyklad 7
więcej podobnych podstron