
Mechanizmy
powstawania bólu
Jerzy Wordliczek, Jan Dobrogowski
Ból jest doznaniem czuciowym, związanym zarówno z działaniem uszka
dzającego bodźca (lub też bodźca, którego działanie może spowodować
wystąpienie takiego uszkodzenia), jak i spostrzeżeniem powstającym na
podstawie psychicznej interpretacji zachodzących zjawisk, zmodyfikowa
nym przez wcześniejsze doświadczenia i psychosomatyczne uwarunkowa
nia. Nocycepcja jest więc jedynie fizjologicznym procesem odczuwania
bólu, natomiast jego klinicznym wykładnikiem jest cierpienie, będące psy
chicznym komponentem zachowania bólowego. Ból może powstawać
w wyniku podrażnienia receptorów bólowych - nocyceptorów lub obniże
nia progu ich pobudliwości, albo też w następstwie uszkodzenia układu
nerwowego. Może pojawiać się także bez towarzyszącego uszkodzenia
tkanek, jednak jest odnoszony przez chorego do takiego uszkodzenia (ból
psychogenny).
Podstawową funkcją bólu w ustroju jest jego ostrzegająco-ochronne
działanie. Przemijający ból fizjologiczny (wywołany przez bodziec nie-
uszkadzający tkanek) pojawia się wtedy, gdy np. dotkniemy gorącego
przedmiotu - wtedy naszą natychmiastową reakcją jest usunięcie się z ob
szaru zagrożenia w obawie przed uszkodzeniem. Z kolei w przypadku ura
zów obejmujących głębokie struktury somatyczne (skręcenia, złamania),
towarzyszący im ból wymusza ograniczenie aktywności, a związana z nim
nadwrażliwość dodatkowo eliminuje możliwość jakiegokolwiek kontaktu,
przez co zmniejsza się potencjalne ryzyko dalszego uszkodzenia lub nasi
lenia zmian patofizjologicznych. Wzrost wrażliwości ułatwia także zdro-

12 Mechanizmy pov.'s:av. ai a bólu
wienie poprzez minimalizowanie ryzyka wystąpienia dalszych uszkodzeń
na drodze eliminacji wszystkich bodźców, nie tylko tych szkodliwych.
Ból występujący w czasie uszkodzenia lub choroby inicjuje segmentar-
ną i ponadsegmentarną odpowiedź ośrodkowego układu nerwowego
(OUN), która pomaga utrzymać ustrojowi homeostazę w okresie rozwoju
procesu patologicznego. Reakcje dotyczą przede wszystkim zmian w krą
żeniu (przyśpieszenie akcji serca, wzrost rzutu serca, a także przepływu
w mózgu i mięśniach) oraz oddychaniu (przyśpieszenie i pogłębienie od
dechów) - są one określane mianem reakcji atawistycznych, a ich celem
jest przystosowanie organizmu do działań o charakterze „walki lub uciecz
ki". Jednak utrzymywanie się tych zmian przez dłuższy czas. jak i fakt, że
ostry ból jest jednym z istotnych czynników generujących odpowiedź
ustroju na uraz, sprawia, że pomimo korzystnego dla organizmu działania
stymulacji bólowej w początkowym okresie rozwoju procesu patologicz
nego jej utrzymywanie się (brak skutecznego uśmierzenia bólu) jest przy
czyną występowania wielu powikłań.
W przeważającej większości przypadków prawidłowo prowadzone po
stępowanie przeciwbólowe oraz procesy naturalnego zdrowienia sprawia
ją, że ostry ból zwykle zanika po upływie kilku lub kilkunastu dni. Jednak
w przypadku braku lub też nieskutecznej terapii przeciwbólowej utrzymu
jący się ból powoduje narastanie zmian patofizjologicznych w OUN (pla
styczność OUN), zaś ostra postać bólu może przekształcić się w przewle
kły zespół bólowy (np. przetrwały ból pooperacyjny lub pourazowy).
Z uwagi na rodzaj bodźca wywołującego stymulację nocyceptywną
rozróżnia się ból fizjologiczny i ból kliniczny.
Ból fizjologiczny pojawia się jako wynik działania stymulacji nocy-
ceptywnej, która nie jest spowodowana uszkodzeniem tkanek. Jest on
związany z aktywacją nocyceptorów bodźcami o wysokiej intensywności.
Po przekroczeniu progu pobudliwości nocyceptorów informacja nocycep
tywną jest przekazywana włóknami A8 i C do rdzenia kręgowego
i wywołuje m.in. aktywację somatycznych motoneuronów, konwergencję
stymulacji ze skórnych i trzewnych zakończeń pierwotnych oraz przewo
dzenie tej informacji do rogu przedniego i w konsekwencji wzrost napię
cia mięśniowego oraz aktywację współczulnych neuronów przedzwojo-
wych w rogu przednio-bocznym, manifestującą się odpowiedzią uogólnio
ną (wzrost ciśnienia tętniczego krwi i tętna) i segmentarną (zmiany
w narządowym przepływie krwi, potliwość, reakcja pilomotoryczna,
skurcz mięśni gładkich).
Z kolei w przypadku uszkodzenia tkanek wystąpieniu bólu towarzyszy
pojawienie się nadwrażliwości, związane ze zmianą właściwości i wrażli
wości pierwotnych zakończeń nerwowych (sensytyzacja obwodowa) oraz
Patomechanizm bólu ostrego
13
zmianami w OUN (sensytyzacja ośrodkowa). Zjawisko to przejawia się
obniżeniem progu bólowego (alodynia - ból pojawia się po zadziałaniu
bodźca nienocyceptywnego, np. dotyku), wzrostem odpowiedzi na stymu
lację (hiperalgezja), występowaniem bólów spontanicznych oraz bólu rzu
towanego. Ten rodzaj stymulacji bólowej, charakterystyczny m.in. dla bó
lu pooperacyjnego, nazwany jest bólem klinicznym.
PATOMECHANIZM BÓLU OSTREGO
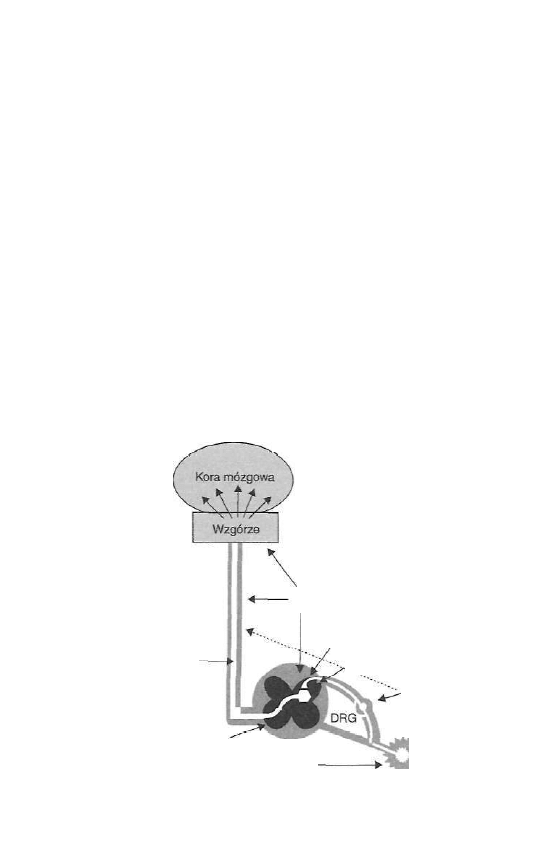
Proces powstawania odczucia bólowego nosi nazwę nocycepcji i obejmu
je cztery etapy: transdukcję, przewodzenie, modulację i percepcje (ryc.
1.1).
W procesie transdukcji zamiana energii działającego bodźca uszkadza
jącego (mechanicznego, termicznego, chemicznego) na impuls elektrycz
ny, przewodzony włóknami nerwowymi, odbywa się w obwodowych za-
PERCEPCJA
MODULACJA
Drogi rdzeniowo-wzgórzowe
Ośrodkowe zakończenia
neuronu nooyceptorowego
Róg tylny rdzenia kręgowego
PRZEWODZENIE
Rdzeń kręgowy'
Obwodowe zakończenia -
neuronu nocyceptorowego
(tzw. pierwotne zakończenia nerwowe)
TRANSDUKCJA
Rycina 1.1. Proces nocycepcji.

14
Mechanizmy powstawania bólu
kończeniach neuronu nocyceptorowego (I neuron „drogi bólowej"), tj.
nocyceptorach, zlokalizowanych w pierwotnych (obwodowych) zakończe
niach nerwowych sieci włókien (włókna A5 i C) wyspecjalizowanej
w przekazywaniu informacji nocyceptywnej.
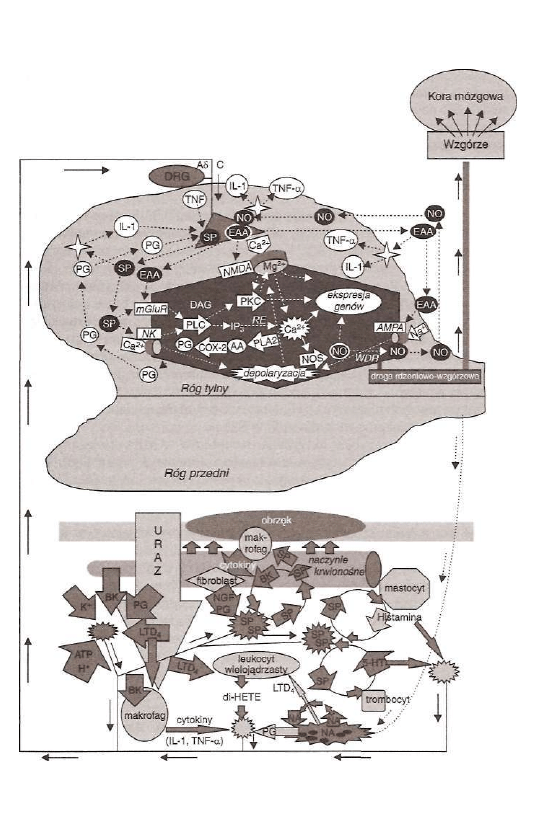
Bezpośredni uraz tkanek powoduje wzrost poziomu potasu oraz uwol
nienie bradykininy (BK) i prostanoidów w uszkodzonych tkankach. Afe-
renty pierwotnych zakończeń włókien A8 i C, poza ortodromową transmi
sją informacji nocyceptywnej do OUN, pobudzają też na drodze antydro-
mowej uwalnianie substancji P (SP) z pierwotnych zakończeń nerwowych.
Prowadzi to do rozszerzenia łożyska naczyniowego i wzrostu przepusz
czalności kapilar, a następnie obrzęku i zaczerwienienia w miejscu urazu.
Stymulowany antydromowo wzrost uwalniania SP z pierwotnych zakoń
czeń nerwowych prowadzi także do uwalniania bradykininy (i wzrostu
przepuszczalności naczyń), jak i serotoniny (5-HT) z płytek krwi, a ponad
to histaminy z mastocytów oraz prostanoidów, cytokin i NGF (ang. nerve
growth factor - czynnik wzrostowy nerwu). Mediatory te z kolei wtórnie
zwiększają przepuszczalność naczyń i uwalnianie SP. Dlatego też już na
poziomie tkankowym powstają dodatnie sprzężenia zwrotne, tzw. błędne
koła bólowe, będące przyczyną nadwrażliwości na ból w miejscu urazu
(pierwotnej hiperalgezji).
W konsekwencji działania opisanych powyżej mechanizmów dochodzi
do powstania w tkankach, w miejscu działania urazu, sensytyzującej
(uwrażliwiającej) „mieszaniny" wywołującej stan zapalenia neurogenne-
go.
W odpowiedzi na stymulację nocyceptywną, na poziomie tkankowym,
biorą udział również zakończenia współczulne. Noradrenalina (NA) uwal
niająca się z zakończeń pozazwojowych włókien współczulnych stymuluje
autoreceptory na tych samych zakończeniach, powodując uwalnianie pro-
stacykliny (PGI,) sensytyzującej nocyceptory oraz leukotrienu D
4
(LTD
4
),
który aktywuje leukocyty wielojądrzaste do produkcji algogenu di-HETE
(kwasu dihydroksyeikozatetraenowego).
Proces sensytyzacji wywoływany przez omówione powyżej mediatory
tkankowe nosi nazwę sensytyzacji obwodowej (patrz ryc. 1.2).
Proces transdukcji jest więc inicjowany i nasilany przez:
• bezpośrednią aktywację nocyceptorów,
• sensytyzację nocyceptorów, połączoną z następowym wzrostem ich
aktywności,
• „wynaczynienie" czynników algezjogennych i sensytyzujących.
Informacja nocyceptywną zakodowana w postaci impulsu elektryczne
go dociera w procesie przewodzenia do zwoju rdzeniowego (DRG, ang.
Patomechanizm bólu ostrego
15
Rycina 1.2. Mechanizmy transdukcji i sensytyzacji.

16
Mechanizmy powstawania bólu
dorsal root ganglion) i powoduje uwalnianie tzw. aminokwasów pobudza
jących - glutaminianów i asparaginianów (EAA, ang. excitatory amino-
acids), SP, neurokininy A (NKA) oraz prawdopodobnie innych peptydów,
które są transportowane poprzez dendryty komórki z DRG do synaps two
rzonych przez ośrodkowe zakończenia pierwotnego aferentnego neuronu
nocyceptorowego w rogu tylnym rdzenia kręgowego (RT). Razem z inny
mi czynnikami pełnią one tam funkcję neuroprzekaźników lub modulato
rów.
W warunkach fizjologicznych docierające do OUN bodźce nocycep-
tywne mają za niską amplitudę, aby wywołać potencjał czynnościowy
w dużej liczbie neuronów w RT. W warunkach patologii (np. uraz) wystę
puje natomiast czasowe i przestrzenne sumowanie postsynaptyczne, pro
wadzące do depolaryzacji większej liczby neuronów w RT (poszerzenie
neuronalnych pól odbiorczych) - w wyniku tego procesu bodźce dotych
czas podprogowe stają się nadprogowymi, co klinicznie manifestuje się
długotrwałą hiperalgezją, utrzymującą się, mimo braku stymulacji nocy
ceptywnej, jeszcze po okresie gojenia tkanek. Poszerzenie neuronalnych
pól odbiorczych w RT jest wynikiem dużego i/lub przedłużającego się afe
rentnego napływu informacji nocyceptywnej, powodującego uwolnienie
aminokwasów pobudzających (glutaminianów i asparaginianów) oraz SP
z ośrodkowych zakończeń włókien C w RT.
Powtarzalna i szybka aktywacja receptorów AMPA (wiążą kwas
a-amino-3-hydroksy-5-metyło-4-izoksazolopropionowy) przez EAA po
woduje powstanie szybkich potencjałów synaptycznych i usunięcie jonów
Mg
2+
blokujących kanał jonowy związany z receptorem NMDA (pobudza
ny przez kwas A^-metylo-D-asparaginowy). Z kolei aktywacja receptora
NMDA przez aminokwasy pobudzające powoduje szybki przepływ jonów
Ca
2+
i Na
+
do wnętrza komórki, co prowadzi do dalszej depolaryzacji, zaś
pobudzenie przez SP receptorów neurokininowych (NK) powoduje depo
laryzację i wpływ jonów pozakomórkowego Ca
2+
przez kanał jonowy
związany z tym receptorem. Działanie SP na receptor NK oraz SP wraz
z EAA na receptor metabotropowy (mGluR) prowadzi do aktywacji fosfo-
lipazy C (PLC) i w następstwie do powstania trifosforanu inozytolu (IP
3
)
oraz diacyloglicerolu (DAG), które działają jako wewnątrzkomórkowe
przekaźniki II układu sygnałów. IP
3
uwalnia jony wapnia z siateczki śród-
plazmatycznej, które razem z jonami wapnia napływającymi przez kanały
jonowe związane z receptorami NMDA i NK powodują wzrost ekspresji
genowej (aktywacja III układu sygnałów). Z kolei DAG stymuluje translo-
kację i aktywację kinazy białkowej C (PKC), aktywowanej również przez
duży napływ jonów Ca
2+
do komórki. Kinaza ta, usuwając jony Mg
2+
blo
kujące kanał jonowy związany z receptorem NMDA, sama następnie akty-

Patomechanizm bólu ostrego
17
wuje napływ jonów wapnia do komórki przez kanał jonowy związany
z tym receptorem (dodatnie sprzężenie zwrotne) i razem z jonami Ca
;
~ na
sila ekspresję genów (III układ sygnałów).
W wyniku tego procesu może dochodzie do tworzenia, na matrycy ge
nowej kwasu rybonukleinowego, nowych cząsteczek białek i powstawania
nowych receptorów w błonie komórkowej. Zmienia to aktywność komórki
na dłuższy czas, mierzony w dniach, a w pewnych sytuacjach nawet
w sposób trwały. Postsynaptyczne mechanizmy w neuronach RT prowadzą
więc do rozwoju dodatniego sprzężenia zwrotnego, manifestującego się
narastającą nadwrażliwością neuronów. Ponadto wzrost stężenia jonów
Ca
2+
aktywuje syntazę tlenku azotu(II) (NOS) do produkcji tlenku azo-
tu(II) (NO), który - dyfundując swobodnie między neuronami, glejem
i wstecznie do zakończeń presynaptycznych - dodatkowo nasila ,.samona-
pędzający się" (wind-up) mechanizm aktywacji receptorów NMDA oraz
uwalnianie pronocyceptywnych neuroprzekaźników.
Dodatkowo wzrost stężenia jonów Ca
2
"*" aktywuje też fosfolipazę A-,
(PLA
2
), która, oddziałując na kwas arachidonowy (AA), inicjuje powsta
nie prostaglandyn (PG) w OUN - prostaglandyny odgrywają ważną rolę
w modulowaniu i percepcji informacji nocyceptywnej, gdyż uwalniane
w miejscu uszkodzenia tkanek i zapalenia obniżają próg pobudliwości dla
aktywacji neuronów czuciowych (w odpowiedzi na stymulację nocycep-
tywną).
Istotną grupą mediatorów uczestniczących w rozwoju procesu nocy-
cepcji są również cytokiny, które biorą aktywny udział w powstawaniu od
powiedzi organizmu na uraz. Są one wytwarzane głównie przez pobudzo
ne komórki układu odpornościowego, co przyczyniło się do powstania hi
potezy, iż są one pośrednikami między aktywowanym (w wyniku działania
bodźca uszkadzającego) układem immunologicznym a OUN. Udowodnio
no, że źródłem cytokin w OUN są m.in. astrocyty i neuroglej, aktywowane
przez EAA, SP oraz NO, uwalniane w strukturach rogu tylnego rdzenia
kręgowego w następstwie działania stymulacji nocyceptywnej. Należy też
podkreślić, że aktywowane komórki neurogleju oraz astrocyty mogą same
produkować szereg prozapałnych mediatorów (NO, EAA, IL-1, IL-6,
TNF-a, NGF, PG), a indukcja tych mediatorów, oddziałując na sąsiednie
komórki struktur rdzenia kręgowego, powoduje rozszerzenie procesu akty
wacji i zmianę właściwości przyległych neuronów. Tworzą się dodatnie
sprzężenia zwrotne między mikroglejem i astrocytami oraz między tymi
komórkami a komórkami nerwowymi, doprowadzające do rozwoju zmian
objawiających się klinicznie jako hiperalgezja i alodynia.
Opisany powyżej proces ośrodkowej sensytyzacji (patrz ryc. 1.2) jest
prawdopodobnie przyczyną powstawania hiperalgezji wtórnej, bólu rzuto-

18
Mechanizmy powstawania bólu
wanego i tzw. pamięci bólowej, związanej z nadpobudliwością komórek
układu nocyceptywnego i neuronów typu WDR (ang. wide dynamie ran
gę).
Z rogu tylnego rdzenia kręgowego informacja nocyceptywna jest prze
kazywana do wyższych pięter OUN, przede wszystkim drogami zlokalizo
wanymi (boczną drogą rdzeniowo-wzgórzową, przyśrodkową drogą rdze-
niowo-wzgórzową, drogą rdzeniowo-śródmózgowiową i rdzeniowo-siat-
kowatą) w przednio-bocznym kwadrancie istoty białej rdzenia kręgowego
oraz w sznurach tylnych.
Końcowym etapem procesu nocycepcji jest percepcja, mająca miejsce
w mózgowiu, które odgrywa rolę poznawczą i jest odpowiedzialne za
uświadomienie działania stymulacji bólowej, jej ocenę oraz za reakcje
afektywne i emocjonalne (tu powstaje lęk, agresja, gniew oraz kształtowa
ne są modele zachowań związanych z zapamiętanym bólem).
Należy jednak podkreślić, że około 30% populacji nie odczuwa bólu
przez minuty lub nawet godziny po zadziałaniu urazu - zjawisko to nosi
nazwę „analgezji wywołanej przez stres" i jest wynikiem aktywacji endo
gennych układów antynocyceptywnych, bowiem w komórkach rogów
tylnych oraz całym układzie rdzeniowo-wzgórzowym dochodzi do równo
czesnego modulowania (hamowania lub torowania) przewodzonych im
pulsów, którego rezultatem jest zahamowanie uwalniania neuroprzekaźni-
ków z ośrodkowych zakończeń pierwotnych aferentnych neuronów nocy-
ceptywnych lub też modulowanie aktywności neuronów rogu tylnego.
Stymulacja nocyceptywna przewodzona z obwodu do kory mózgowej
jest więc poddawana modyfikacji, w której biorą udział m.in. endogenne
układy opioidowe, układ noradrenergiczny, cholinergiczny, serotoniner-
giczny oraz GABAergiczny.
Poznanie przedstawionych powyżej mechanizmów neurofizjologicz
nych pozwoliło na rozwój badań nad zastosowaniem w postępowaniu
przeciwbólowym całego szeregu nowych leków, bowiem jego celem jest
nie tylko uśmierzenie bólu, ale także zapobieganie rozwojowi sensytyzacji
i jej następstw.
PATOMECHANIZM BÓLU PRZEWLEKŁEGO
Ból powstaje najczęściej w wyniku podrażnienia zakończeń nerwowych
układu nocyceptywnego przez silne bodźce (uraz, choroba), które mogą
spowodować uszkodzenie tkanek. Ten rodzaj bólu nazywany jest bólem
receptorowym lub normalnym, ponieważ jest doznaniem, które poznaje

Patomechanizm bólu przewlekłego
19
w ciągu swojego życia prawie każdy człowiek. Jeżeli dolegliwości bólowe
trwają dłużej niż 3 miesiące lub utrzymują się po wygojeniu uszkodzonych
tkanek, to mówi się o bólu przewlekłym.
Znacznie rzadziej powstaje ból niereceptorowy, czyli patologiczny,
w którym wyróżnia się ból neuropatyczny oraz ból psychogenny, związany
z procesem myślenia, stanem emocjonalnym lub osobowością, występują
cy bez uszkodzenia tkanek, chociaż opisywany w takich kategoriach.
U wszystkich chorych cierpiących z powodu przewlekłego bólu wystę
pują podobne mechanizmy powodujące obniżenie jakości życia: zaburze
nia fizjologiczne, psychologiczne i społeczne. Zależą one od czasu trwania
bólu i stopnia jego natężenia, nie zaś od przyczyny powstania bólu. Nowo
czesne techniki obrazowania, np. pozytonowa lub fotonowa tomografia
komputerowa, wskazują na neurofizjologiczne przyczyny różnic między
bólem ostrym i przewlekłym - w badaniach tych obserwuje się zmniejszo
ny przepływ krwi przez znaczne obszary wzgórza u chorych z bólem prze
wlekłym, natomiast w bólu ostrym przepływ w tych rejonach mózgowia
jest większy. Ponadto u chorych z bólem przewlekłym nie obserwuje się,
charakterystycznego dla bólu ostrego, pobudzenia układu współczulnego
i wewnątrzwydzielniczego. W miejsce pobudzenia psychicznego i niepo
koju pojawia się depresyjny nastrój, nadmierna drażliwość i zdenerwowa
nie. Opisano także charakterystyczne dla chorych z tym bólem zaburzenia
snu, obniżone libido i aktywność seksualną, ociężałość psychoruchową
oraz obniżony próg bólu. Ból przewlekły jest również przyczyną zmiany
wzorca zachowań związanego z jedzeniem. Możliwa jest utrata łaknienia
i spadek masy ciała. U części chorych pojawia się jednak niepokój i nad
mierna chęć jedzenia, co przy niedostatecznej, spowodowanej bólem ak
tywności ruchowej jest przyczyną otyłości, dodatkowo upośledzającej ak
tywność fizyczną. Większość pacjentów nie jest w stanie pracować zawo
dowo, co znacznie obniża ich dochody, standard życia oraz pozycję
w rodzinie. Chorzy postrzegają swoją sytuację jako beznadziejną. Zdespe
rowani domagają się coraz to nowych zabiegów operacyjnych, szukają po
mocy u znachorów, bioenergoterapeutów lub - co zdarza się najczęściej
- domagają się przepisywania wielu preparatów mających zmniejszyć ból
i cierpienie. Nadużywanie leków może prowadzić do zatrać i uzależnień,
ponadto obniża aktywność fizyczną i niekorzystnie wpływa na procesy
myślowe i psychikę chorego. Wielu badaczy określa ból przewlekły jako
chorobę samą w sobie, wymagającą wielokierunkowego postępowania
(w praktyce lekarza rodzinnego w przychodni podstawowej opieki zdro
wotnej, a także w poradni leczenia bólu często spotyka się on z przewle
kłym bólem receptorowym).

20
Mechanizmy powstawania bólu
BÓL NEUROPATYCZNY
Ból neuropatyczny jest rodzajem bólu patologicznego. Jest on zainicjo
wany lub spowodowany pierwotnym uszkodzeniem układu nerwowego.
Definicja obejmuje różnorodne zespoły bólowe, które nie mają wspólnej
etiologii ani umiejscowienia. Wiele zespołów bólu neuropatycznego posia
da jednak wspólne cechy kliniczne, co może sugerować podobny mecha
nizm powstawania, wynikający z nadpobudliwości neuronów zarówno ob
wodowych, jak i ośrodkowych. Dodatkowo mechanizm powstawania
w niektórych zespołach bólu neuropatycznego może mieć komponent re
ceptorowy, wynikający z obniżenia progu pobudliwości zakończeń nerwo
wych nervi nemorum. Ból może być także zależny, przynajmniej
w pewnym okresie jego trwania, od pobudzenia układu współczulnego.
Pomimo wspólnej przyczyny mechanizmy powstawania bólu neuropatycz
nego różnią się w bardzo istotny sposób i jest to jeden z powodów niesku
teczności zastosowania leczenia przepisanego w związku z etiologią po
wstawania bólu. Nadpobudliwość neuronów nie jest zatem przejawem jed
nego mechanizmu, ale wynika z kombinacji czynników, które sumując się,
określają stopień i typ nadpobudliwości u poszczególnych pacjentów
i w poszczególnych zespołach bólowych.
Częstość występowania bólu neuropatycznego wynosi 0,5-0,8%
wszystkich chorych z bólem przewlekłym. U pacjentów z cukrzycą do po
wstania neuropatii dochodzi w 2-3,5% przypadków, po półpaścu prawdo
podobieństwo powstania przewlekłej neuralgii wynosi 10%, zaś ból fanto-
mowy po dwóch latach od amputacji kończyny występuje u 4% chorych.
Ból neuropatyczny nie jest zatem nieodłączną konsekwencją uszkodzenia
nerwu. Stwierdza się dużą różnorodność zależną od czasu powstawania,
miejsca uszkodzenia, przyczyny wywołującej, współistnienia innych pro
cesów patologicznych, a także czynników psychicznych, wieku oraz
skłonności osobniczej. Bardzo podobne uszkodzenie u jednych osobników
prowadzi do powstania bólu neuropatycznego, natomiat u innych nie - po
wstanie bólu zależy bowiem od upośledzenia lub wyczerpania adaptacyj
nych procesów neuronalnej plastyczności, czyli zespołu czynników neuro
fizjologicznych i neurochemicznych, które w większości przypadków
uszkodzeń układu nerwowego są w stanie zapobiec powstawaniu przewle
kłego bólu neuropatycznego.
Przyczyny powstawania bólu neuropatycznego w zależności od miej
sca (poziomu) uszkodzenia struktur układu nerwowego:
1. Nerw
• nerwiak (amputacja, przecięcie nerwu),

Patomechanizm bólu przewlekłego
21
• ucisk (zespoły cieśni, guzy),
• zmiażdżenie, rozciągnięcie, niecałkowite przecięcie (uraz),
• mononeuropatie (cukrzyca, napromienianie, niedokrwienie),
• polineuropatie (cukrzyca, amyloidoza. zatrucia).
2. Zwój rdzeniowy
• ucisk (krążek międzykręgowy, guz, blizna tkankowa),
• wyrwanie (awulsja) korzeni,
• infekcja (neuralgia popółpaścowa).
3. Rdzeń kręgowy
• stłuczenie,
• guz,
• niecałkowite przecięcie.
4. Pień mózgu, wzgórze, półkule mózgu
• zaburzenia ukrwienia (zawał, zator),
• guz,
• uraz.
Najczęściej występujące zespoły bólu neuropatycznego u ludzi po
wstają w wyniku uszkodzenia obwodowego układu nerwowego i są spo
wodowane urazem lub dwoma schorzeniami: półpaścem i cukrzycą.
W warunkach prawidłowych zablokowanie przewodnictwa w nerwie
obwodowym, np. przez środki znieczulenia miejscowego, przejawia się
czasowym porażeniem ruchowym mięśni w obszarze unerwianym przez
zblokowany nerw, zniesieniem czucia dotyku, bólu i temperatury oraz roz
szerzeniem naczyń, zmniejszoną potliwością i wzrostem temperatury skó
ry, co jest wyrazem przerwania przewodnictwa we włóknach zarówno afe-
rentnych, jak i eferentnych A, B i C.
W przypadku uszkodzenia nerwu obserwuje się podobne objawy, jed
nak po kilku godzinach sytuacja zmienia się w sposób paradoksalny -
osłabione czucie bólu zmienia się w patologiczny zespół bólowy, zaś ob
serwowane początkowo odnerwienie we włóknach współczulnych może
ulec zmianie w nadpobudliwość wyrażającą się skurczem naczyń, zwięk
szoną potliwością czy piloerekcją. Występuje zespół objawów charaktery
stycznych dla bólu neuropatycznego, m.in. deficyt ruchowy, ból samoist
ny, hiperalgezja, hiperpatia, alodynia i zaburzenia wegetatywne. Wyniki
badań doświadczalnych wskazują na prawdopodobnie łączne działanie co
najmniej trzech komponentów w powstawaniu bólu neuropatycznego:
• Pierwszy dotyczy zmian pobudliwości elektrycznej błon komórko
wych uszkodzonego aksonu oraz zwoju rdzeniowego pierwszego
neuronu aferentnego.

22
Mechanizmy powstawania bólu
• Drugi odnosi się do zmian przetwarzania otrzymanych sygnałów
w rogu tylnym rdzenia kręgowego.
• Po trzecie wiele wskazuje na to, że w wyższych piętrach OUN do
chodzi do dezintegracji zaprogramowanych i skoordynowanych od
powiedzi na sytuacje naruszające integralność organizmu w obsza
rze różnych poziomów układu nerwowego.
Zmiany w zakresie pierwszego neuronu
Uszkodzenie nerwu, a w szczególności jego całkowite lub częściowe prze
cięcie, powoduje bombardowanie impulsami nerwowymi o wysokiej czę
stotliwości ciała macierzystego neuronu w zwoju rdzeniowym (DRG).
W wyniku informacji o uszkodzeniu nerwu (afferent barrage), w części
macierzystej komórki, która znajduje się w DRG, dochodzi do ekspresji
genowych i produkcji drobin białek transportowanych śródaksonalnie do
tworzącego się nerwiaka. Jednak część drobin pozostaje w centralnej czę
ści komórki w DRG lub na przebiegu włókna nerwowego. Drobiny białek
zostały zidentyfikowane jako oporne na tetrodotoksynę (TTX) kanały so
dowe, które posiadają (patologiczną w nerwie obwodowym) zdolność
transdukcji bodźców mechanicznych na impuls elektryczny. W wyniku
ekspresji genowych dochodzi także do powstawania receptorów a
2
, które
mogą być zlokalizowane w zakończeniach nerwowych regenerującego się
nerwu, w ektopowych rozrusznikach nerwu oraz w DRG. Powstałe de no-
vo
receptory stanowią źródło samoistnych pobudzeń, mają zdolność trans
dukcji słabych bodźców mechanicznych i termicznych, a ponadto są wraż
liwe na działanie katecholamin (powstanie ognisk samoistnych pobudzeń
dokonuje się w ciągu kilkudziesięciu godzin od uszkodzenia nerwu).
Następstwem zachodzących zmian jest powstanie bólu samoistnego
oraz napadowego, wynikającego z podrażnienia bodźcami mechaniczny
mi, termicznymi lub chemicznymi, a źródłem ich powstawania jest zarów
no nerwiak, jak i włókno nerwowe oraz DRG (ryc. 1.3).
Powstawanie patologicznych połączeń między
włóknami pnia nerwu
Uszkodzenie nerwu prowadzi do powstania patologicznej interakcji ukła
du nocyceptywnego i autonomicznego. Przyczyną tego zjawiska jest po
wstawanie patologicznych połączeń - efaps - między aferentnymi włókna
mi nocyceptywnymi i eferentnymi włóknami współczulnymi, zarówno
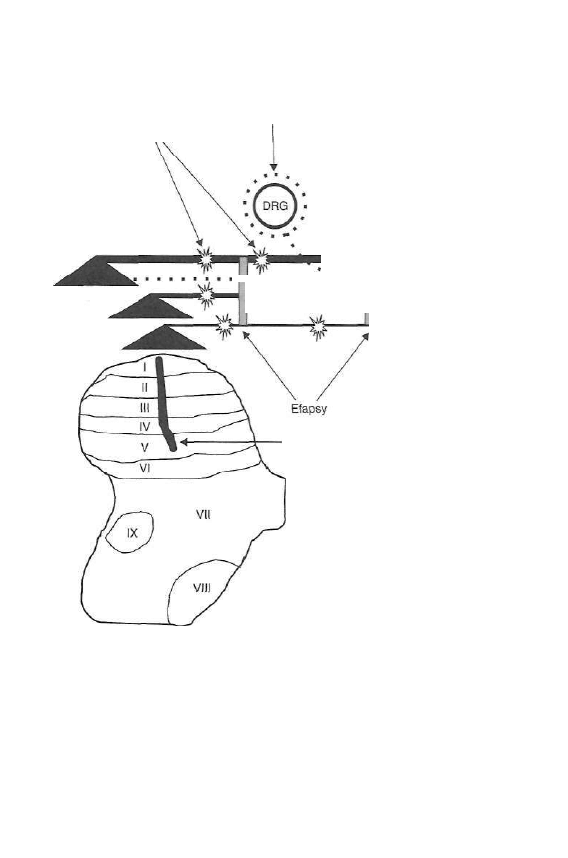
Patomechanizm bólu przewlekłego
23
„Pączkowanie" włókien współczulnych
wokół zwoju rdzeniowego
Ektopowe rozruszniki nerwu
Włókna B
Włókna Ap
. . . . . . . * . .
Włókna A5
Włókna C
Wstęga patologicznych połączeń
między warstwami tylnego rogu
rdzenia kręgowego
Rycina 1.3. Patomechanizm bólu neuropatycznego.
wzdłuż nerwu, jak i w nerwiaku. Wzajemne pobudzenie może dokonywać
się bezpośrednio, przez przeniesienie bodźca elektrofizjołogicznego, lub
pośrednio, poprzez produkowane endogennie katecholaminy. Zmiany
w proksymalnej części uszkodzonego nerwu powodują, że pobudzenie
układu współczulnego lub wstrzyknięcie noradrenaliny prowadzi do po
wstania patologicznej aktywności we włóknach aferentnych, a w konse-

24
Mechanizmy powstawania bólu
kwencji zmiany te są jedną z przyczyn powstawania bólu zależnego od
układu współczulnego. Dlatego też sympatektomia czy blokada układu
współczulnego powoduje zmniejszenie powstawania oraz natężenia bólu
zarówno w warunkach eksperymentalnych, jak i klinicznych.
Zmiany fenotypu w zakresie pierwszego neuronu
aferentnego
W warunkach fizjologicznych fenotyp neuronu nocyceptorowego regulo
wany jest przez czynniki wzrostowe nerwu transportowane do ciała macie
rzystego komórki, co powoduje uwalnianie wielu neuropeptydów, m.in.
SP, CGRP (ang. calcitonin gene related peptide) i somatostatyny. Po prze
cięciu nerwu w małych nocyceptorowych neuronach ekspresja tych neuro
peptydów zmniejsza się, natomiast rośnie stężenie VIP (ang. vasoactive
intestinal peptide), PACAP (ang. pituitary adenylate cyclase-activating
peptide) i galaniny. Zaburzenie równowagi i regulacji fenotypu po uszko
dzeniu nerwu jest również uznawane za jedną z przyczyn powstawania pa
tologicznego bólu po uszkodzeniu nerwu.
Przerwanie ukrwienia i osłonek nerwu
W wyniku uszkodzenia nerwu zostaje zaburzony stan równowagi między
neuronami i ich otoczeniem, co także jest przyczyną zmian wrażliwości,
pobudliwości, przewodnictwa oraz metabolizmu i może doprowadzać do
obumarcia komórki, a w przypadku przeżycia powoduje głębokie zmiany
funkcji neuronu i powstanie patologicznego bólu.
Obrzęk neurogenny i odczyn zapalny nerwu
W powstawaniu bólu neuropatycznego rolę odgrywa nie tylko strukturalne
uszkodzenie nerwu, lecz również towarzyszące mu procesy zapalne.
W miejscu uszkodzenia dochodzi bowiem do uwalniania z tkanek i naczyń
np. BK, 5-HT, jonów wodorowych, prostanoidów, NGF, cytokin oraz wol
nych rodników, co prowadzi do napływu komórek układu immunologicz
nego, przesięku surowicy i obniżenia progu pobudliwości zakończeń ner
wowych nerwów unerwiających pnie nerwu (nervi neryorum).
Procesy zapalne mogą doprowadzić do powstania bólu neuropatyczne
go również bez strukturalnego uszkodzenia nerwu. W ostatnich latach

Patomechanizm bólu przewlekłego
25
podkreśla się rolę pobudzenia komórek gleju i zwiększonego uwalniania
prozapalnych cytokin w powstawaniu przewlekłych zespołów bólowych
zarówno po uszkodzeniu rdzenia, jak i nerwów obwodowych - postuluje
się, że pobudzenie komórek gleju jest przyczyną powstawania bólu odle
głego od miejsca uszkodzenia oraz pojawienia się objawów „lustrzanego
odbicia" bólu, zaś produkcja cytokin prozapalnych może być przynajmniej
częściowo odpowiedzialna za powstawanie oporności na opioidy.
Ośrodkowa sensytyzacja
Jednym z mechanizmów odpowiedzialnych za nasilanie doznań bólowych,
rozszerzenie miejsca odczuwania bólu i jego przewlekły charakter jest po
wstanie nadwrażliwości neuronów rdzenia kręgowego i wyższych pięter
OUN, tzw. ośrodkowej sensytyzacji (szczegółowe omówienie tego za
gadnienia przedstawiono na początku tego rozdziału).
Zmiany morfologiczne w neuronach
rdzenia kręgowego
Po uszkodzeniu nerwu czynnikami wywołującymi powstanie zjawiska
ośrodkowej sensytyzacji są w początkowym okresie ektopowe rozruszniki
nerwu, jednak po pewnym czasie (mierzonym w tygodniach) w neuronach
rogów tylnych rdzenia kręgowego dochodzi do powstania zmian morfolo
gicznych. Dotyczy to w szczególności:
• powstawania dodatkowych wypustek nerwowych, penetrujących
nowe obszary rdzenia kręgowego,
• zwyrodnienia śródsynaptycznego powierzchniowych warstw rogu
tylnego rdzenia kręgowego,
• modulacji funkcji mikrogleju,
• neurochemicznych zmian właściwości komórki.
W warunkach prawidłowych ból przewodzony jest przez włókna afe-
rentne neuronów, których wypustki dośrodkowe dochodzą do warstwy I
i II rogu tylnego rdzenia kręgowego. Podrażnienie włókien AB nie ma
wpływu na powstawanie doznań bólowych, ponieważ włókna dośrodkowe
tych neuronów dochodzą do warstwy III rdzenia. Uszkodzenie nerwu ob
wodowego jest natomiast czynnikiem stymulującym tworzenie nowych
gałęzi pobocznych z neuronów warstwy II i w konsekwencji powstaje
wstęga włókien nerwowych łącząca neurony od warstwy II do V - to po-

26
Mechanizmy powstawania bólu
zwala wyjaśnić ośrodkowy mechanizm powstawania alodyni, a więc dla
czego podrażnienie neuronów Ap (przewodzących czucie dotyku) powo
duje powstanie doznań bólowych.
Zmiany ośrodkowe po uszkodzeniu nerwu dotyczą również ekspresji
genów kodujących białka receptorów błon komórkowych neuronów, co
w sposób trwały zmienia pobudliwość oraz może spowodować wytworze
nie zdolności do samoistnych wyładowań. Ból neuropatyczny w takich
przypadkach jest powodowany trwałymi zmianami w błonie komórkowej
neuronów i podobnie jak w padaczce występują samoistne wyładowania,
charakteryzujące się nie drgawkami czy utratą świadomości, lecz atakami
doznań bólowych.
Zaburzenie procesów hamowania
Modulacja przewodzenia bodźców nocyceptywnych obejmuje również
procesy hamowania zarówno w rdzeniu kręgowym, jak i poprzez zstępują
ce układy antynocyceptywne, w których impulsy hamujące przekazywane
są do neuronów rogów tylnych rdzenia kręgowego z wyższych pięter
OUN: substancji szarej okołowodociągowej w śródmózgowiu, substancji
szarej okołokomorowej w podwzgórzu, bocznych i grzbietowo-bocznych
adrenergicznych neuronów mostu i serotoninergicznych neuronów zlokali
zowanych w brzuszno-dogłowowej części rdzenia przedłużonego oraz
w jądrze wielkim szwu. Nasilenie się procesów hamowania powoduje
osłabienie aktywności neuronów rogów tylnych rdzenia kręgowego oraz
adaptacyjną modyfikację przewodzenia bodźców z obwodu do kory móz
gowej. Z neurochemicznego punktu widzenia najlepiej poznane spośród
wewnątrzrdzeniowych i zstępujących układów hamujących są: opioidowy,
GABAergiczny, serotoninergiczny, adrenergiczny i cholinergiczny. Po
uszkodzeniu nerwu, w wyniku różnych mechanizmów dochodzi do
zmniejszenia hamowania ośrodkowego i rdzeniowego neuronów rogów
tylnych rdzenia kręgowego. W wyniku ekscytotoksyczności część hamują
cych interneuronów blaszki II rogów tylnych rdzenia obumiera. To z kolei
powoduje zmniejszenie się stężenia GABA (kwasu y-aminomasłowego),
neuroprzekaźnika hamującego w interneuronach rdzenia kręgowego.
Zmniejsza się również gęstość presynaptycznych receptorów GABA oraz
opioidowych, wzrasta natomiast stężenie cholecystokininy, która jest inhi
bitorem receptorów opioidowych. W wyniku uszkodzenia nerwu wzrasta
też aktywność kinazy białkowej C oraz immunofilin, zaś zmniejsza fosfa
tazy kalmodulinowej - kalcyneuryny, co powoduje osłabienie wpływu en
dogennych opioidów na kanały wapniowe.

D
atomechanizm bólu przewlekłego
27
Po uszkodzeniu nerwu zmiany ośrodkowe obejmują nie tylko neurony
rogów tylnych rdzenia kręgowego, ale rozsiane są w całym układzie ner
wowym. Powstają układy wzajemnych pobudzeń i sprzężeń zwrotnych,
które jak fala obejmują coraz wyższe piętra OUN (zgodnie z zasadami cy
bernetyki pętle sprzężeń zwrotnych mają pewne granice wyznaczone przez
ich energię i umiejscowienie w czasie i jeżeli te granice zostaną przekro
czone, mechanizm „wymyka się" spod kontroli innych układów i oscyluje
jak gdyby bezsensownie, bez szans powrotu do stanu prawidłowego).
Wybrane piśmiennictwo:
1. Dobrogowski J., Wordliczek J. (red.): Medycyna bólu. Wyd. Lek. PZWL,
Warszawa 2004.
2. Loeser J. D. (red.): Bonica's management ofpain. Lippincott & Wilkins, Phila-
delphia 2001.
3. Pain 2005 - An Updated Review. IASP Press, Seattle 2005.
4. Rice A. i wsp. (red.): Clinical Pain Management. Arnold, London 2003.
Wyszukiwarka
Podobne podstrony:
3 Mechanizm powstawania odruchów warunkowych oraz metody ich badania
Kamien nazebny przyczyny i mechanizm powstawania
Odporność poszczepienna mechanizm powstawania
wiatry rodzaje i mechanizmy powstawania
Mechanizmy przewodzenia bolu Fi Nieznany
Melatonina mechanizm powstawania dziaania zastosowanie kliniczne, Biochemia, prace
Mechanizmy powstawania agresji i przemocy, Studia Administracja, LICENCJAT, Semestr IV, POLITYKA KAR
Przyczyna i mechanizm powstawania chorob zapalnych zatok i obocznych nosa nowe spojrzenie
Opisz mechanizm powstawania i rolę S, Biochemia, prace
9.11.2009-mechanizm powstawania głosu
MECHANIZM POWSTAWANIA I HISTOKLINIKA KRWIAKA PODTWARDÓWKOWEGO I NADTWARDÓWKOWEGO
zaparcia stresowe mechanizmy powstania i profilakyka
79 Wyjasnij mechanizm powstawania potencjalu czynnosciowego
Determinacja płci chromosomowej czyli mechanizm powstawania określonej płci chromosomowejx
Mechanizm powstawania moczu
Gorączka mechanizm powstawania gorączki
więcej podobnych podstron