
Krew obwodowa do badań DNA: krew obwodową pobrać w ilości 2-5 ml, stosując system
zamknięty, zawierający 10% wersenian sodowy (EDTA) jako antykoagulant. Po pobraniu
probówkę z krwią dokładnie wymieszać przez kilkukrotne odwracanie. Następnie umieścić
w temperaturze 2-8 st. C i przekazać do laboratorium. Czas przechowywania w temp. 2-8 st. C do
5 dniu, lub do 6 tygodni w temp. -20 st. C
Krew obwodowa do badań RNA: krew obwodową pobrać w ilości 2-5 ml, stosując system
zamknięty, zawierający 10% wersenian sodowy (EDTA) jako antykoagulant. Po pobraniu
probówkę z krwią dokładnie wymieszać przez kilkukrotne odwracanie. Następnie umieścić
w temperaturze 4 st. C i jak najszybciej przekazać do laboratorium. Jeżeli istnieje konieczność
dłuższego przechowywania materiału: do 3 dni w temp. Pokojowej lub w temp. -20 st. C
z użyciem odczynnika stabilizującego.
Krew obwodowa do badań cytogenetycznych: krew obwodową pobrać w ilości 2-5ml
stosując system zamknięty, zawierający heparynę jako antykoagulant. Po pobraniu probówkę
z krwią dokładnie wymieszać i niezwłocznie przekazać do laboratorium. Jeżeli istnieje
konieczność dłuższego przechowywania materiału: do 2 dni w temp. 2-8 st. C Krwi nie wolno
zamrażać.
Fragmenty tkanek do badań DNA: fragment tkanki pobrany wg zaleceń lekarza, umieścić
w sterylnym naczyniu, utrzymując temp. 4 st. C i jak najszybciej przekazać do laboratorium.
Jeżeli istnieje konieczność dłuższego przechowywania materiału: do 6 tygodni w temp. -20 st. C
lub w temp. -70 st. C przez dłuższy czas. Unikać kilkukrotnego rozmrażania i zamrażania próbki.

Amplifikacja klasyczna
Reakcja PCR (ang. polymerase chain reaction, pol. łańcuchowa reakcja polimerazy)
opiera się na metodzie powielania specyficznych fragmentów DNA. Odkryta została przez
Kary’ego Mullis’a w 1980 roku. Standardowy test PCR polega na powieleniu in vitro
określonych sekwencji wyjściowego DNA, co możliwe jest nawet w przypadku
dysponowania niewielkimi ilościami materiału. Dzięki tej metodzie w łatwy sposób można
uzyskać dużą ilość kopii pojedynczego fragmentu DNA. Proces amplifikacji można
przeprowadzić poddając mieszaninę enzymów, substratów i określonego odcinka matrycy
wielokrotnej reakcji zmieniającej się cyklicznie temperatury, zgodnie ze schematem:
Termiczna denaturacja matrycy (94-98º C, 15-60 sek.)
Wiązanie starterów w określonym miejscu powielanego DNA (40-70º C, 15-60 sek.)
Elongacja nowych nici przez termo stabilną polimerazę DNA (na odcinkach
ograniczonych starterami) (72ºC, 5-60 sek.)
Reakcja prowadzona jest w cyklach powtarzających się 20-40 razy. W każdym cyklu
następuje etap denaturacji cząsteczki DNA, przyłączenie starterów i syntezy nowej nici.
Każdy etap zachodzi w innej temperaturze. Reakcja prowadzona jest w probówce
umieszczonej w bloku grzejnym (termocykler), który umożliwia szybką i precyzyjną zmianę
temperatury, właściwej dla poszczególnych etapów.
Schemat łańcuchowej reakcji polimerazy.

Mieszanina reakcyjna składa się z kilku podstawowych elementów:
1. Matrycy DNA.
2. Starterów reakcji PCR – krótkie sztucznie syntetyzowane odcinki jednoniciowego
DNA, komplementarne do końców badanego fragmentu matrycy.
3. Trifosforanów deoksynukleotydów (dATP, dGTP, dCTP, dTTP), służących do
syntezy nowych łańcuchów DNA.
4. Polimerazy DNA, katalizującej reakcję syntezy nowych nici.
5. Odpowiedniego buforu reakcji, zapewniającego optymalne środowisko do
przyłączania starterów i prawidłowego działania polimerazy DNA.
6. Jony dwuwartościowe (najczęściej Mg2+) jako kofaktor aktywności enzymatycznej
polimerazy.
PCR jest reakcją łańcuchową, ponieważ nowo zsyntetyzowana nić DNA służy jako
matryca do dalszej syntezy. Powtarzanie cykli amplifikacji prowadzi do wzrostu
odpowiednich fragmentów DNA w postępie wykładniczym. Teoretycznie po n cyklach
badana sekwencja zostaje powielona 2
n
-krotnie, jednak w trakcie procesu substraty ulegają
zużyciu, spada też aktywność polimerazy i w efekcie wydajność reakcji jest niższa.
Tradycyjne metody detekcji powielanego DNA polegają na elektroforezie kwasów
nukleinowych w obecności bromku etydyny. Równocześnie z badanym fragmentem rozdziela
się w żelu standardy masowe (fragmenty DNA o znanych masach cząstek wyrażonych
w ilości par zasad). Pomiar polega na wizualnej lub densytometrycznej analizie prążków,
widocznych po naświetleniu żelu światłem ultrafioletowym. Na podstawie położenia
badanego materiału w stosunku do standardów masowych określana jest jego wielkość
Na efektywność reakcji PCR w istotny sposób wpływają różne czynniki:
Typ termocyklera
Temperatura i czas poszczególnych etapów oraz ilość przeprowadzonych cykli
Jakość enzymu, jego stężenie
Jakość buforu reakcyjnego
Primery
Stężenie dNTP
Temperatura denaturacji i czas jej trwania zależy od matrycowego DNA używanego
do reakcji. Niekompletna denaturacja może obniżyć wydajność reakcji, z kolei za długa
denaturacja doprowadza do zmniejszenia aktywności enzymu.

Temperatura hybrydyzacji jest najważniejszym czynnikiem wpływającym na
specyficzność reakcji. Jeżeli temperatura jest za wysoka przyłączenie primerów jest
niemożliwe, z kolei przy niskiej temperaturze zachodzi niespecyficzne przyłączanie starterów
do wielu miejsc na matrycy, co prowadzi do powstawania wielu niespecyficznych
fragmentów o nieznanej sekwencji. Temperaturę przyłączania należy ustalać oddzielnie dla
każdego startera.
Temperatura i czas trwania elongacji zależy od używanej polimerazy. Optymalny
zakres działania wynosi zwykle 68-72ºC.
Ilość przeprowadzanych cykli zależy głównie od początkowego stężenia DNA,
optymalna ilość to zazwyczaj 25-35 cykli. Zmniejszona liczba cykli powoduje małą
wydajność reakcji, z kolei zwiększona liczba cykli prowadzi do wzrostu ilości
niespecyficznych produktów.
Stosowanie odpowiedniej polimerazy znacznie zwiększa specyficzność i wydajność
reakcji. Nadmiar enzymu prowadzi do powstawania niespecyficznych produktów, podczas
gdy jego zmniejszone stężenie może spowodować powstawanie zbyt małych ilości produktu.
Bufor zapewnia odpowiednie warunki dla działania polimerazy. Ważnym składnikiem
buforu są jony Mg
2+
, wpływają one na aktywność enzymu. Zbyt wysokie stężenie jonów
magnezowych powoduje powstawanie niespecyficznych produktów reakcji, natomiast niskie
stężenia są przyczyną słabszego sygnału amplifikacji.
Projektowanie optymalnych starterów dla amplifikacji. Długość primerów powinna
wynosić około 20 nukleotydów, zawierających 50-60% par G-C. Startery nie powinny
tworzyć dimerów lub innych struktur wyższego rzędu. Końce 3’ starterów nie mogą być
komplementarne do siebie. Ważne jest odpowiednie dobranie temperatury wiązania starterów.
Zbyt wysokie stężenie primerów może prowadzić do akumulacji niespecyficznych produktów
i zwiększa ryzyko tworzenia dimerów.
Optymalne stężenie nukleotydów. Nierówne ilości czterech dNTP redukują wydajność
amplifikacji. Końcowe stężenie każdego nukleotydu w roztworze powinno wynosić 200μM.
Istotnymi czynnikami wpływającymi na powodzenie reakcji są także: unikanie błędów
laboratoryjnych, stosowanie odczynników o najwyższej jakości. Powielany fragment DNA
nie może być zdegradowany ani zanieczyszczony. Szczególnym nadzorem należy otoczyć
etapy pobrania materiału, jego prawidłowego oznaczenia, odpowiedniego transportu oraz
przechowywania. Izolacji materiału należy dokonywać za pomocą technik gwarantujących
wysoką wydajność izolacji i wysoki stopień czystości DNA.

PCR jest metodą szeroko stosowaną, ma ogromne znaczenie dla biologii molekularnej
i znajduje liczne zastosowania w takich dziedzinach jak klonowanie, sekwencjonowanie,
wprowadzanie specyficznych mutacji, diagnostyka medyczna (onkologia, transplantologia,
mikrobiologia) oraz medycyna sądowa.
Na bazie klasycznej metody PCR powstało wiele wariantów PCR będących
modyfikacjami wyjściowego mechanizmu.
Modyfikacje PCR
RT-PCR (ang. Reverse Transcriptase PCR)
Głównym ograniczeniem enzymu polimerazy DNA jest to, że jako matrycy do syntezy
może używać jedynie DNA. Z użyciem tego enzymu nie można amplifikować RNA.
Rozwiązaniem tego problemu jest RT-PCR.
RT-PCR jest techniką, która łączy w sobie reakcję odwrotnej transkrypcji oraz reakcję
PCR. Sprowadza się ona do amplifikacji specyficznego fragmentu RNA. Dzięki tej metodzie
możliwe jest zbadanie aktywności genu, a nie jedynie jego obecności w genomie.
Proces PCR jest poprzedzony przepisaniem sekwencji mRNA na cDNA w reakcji
odwrotnej transkrypcji. Jako matrycowy RNA może być stosowany całkowity RNA
komórkowy jak i wstępnie frakcjonowany mRNA. Aby reakcja odwrotnej transkrypcji miała
dobrą wydajność wyizolowany RNA musi być wolny od zanieczyszczeń. Jakość oraz ilość
RNA można ocenić elektroforetycznie lub spektrofotometrycznie.
Reakcję syntezy cDNA (ang. Complementary DNA) na matrycy RNA przeprowadza
się za pomocą enzymu odwrotnej transkryptazy. Reakcja ta może być wykonana trzema
metodami: przy użyciu heksametrów, oligo(dT) lub startera specyficznego dla analizowanego
genu. Primer oligo(dT) jest komplementarny do fragmentu poliA na końcu 3’cząstek mRNA.
Zastosowanie tego primera pozwala na uzyskanie puli cDNA odpowiadającej całemu mRNA
komórki. Przypadkowe promery heksametrowe, dają jednakową reprezentację całego RNA
komórki.
W celu oceny jakości uzyskanego cDNA należy przeprowadzić reakcję kontrolną PCR
z użyciem primerów specyficznych do genu eksprymowanego konstytutywnie. Wydajność
reakcji RT jest bardzo ważna w przypadku późniejszego zastosowania uzyskanego cDNA do
analizy ekspresji genów.
Po reakcji odwrotnej transkrypcji następuje właściwa reakcja PCR. RT-PCR jest
stosowany do określania wpływu różnych czynników na poziom ekspresji genów,
wykrywania markerów nowotworowych, badania alternatywnych form splicingowych.

Real- time PCR
Łańcuchowa reakcja polimerazy z analizą w czasie rzeczywistym jest czułą metodą
analityczną pozwalającą na amplifikowanie wybranego odcinka DNA przy jednoczesnym
pomiarze ilości produktu, dzięki wykorzystaniu technik fluorescencyjnych.
W przeciwieństwie do klasycznej reakcji PCR, gdzie oznaczanie produktu zachodziło
w końcowym etapie, system ten pozwala na monitorowanie reakcji w czasie w którym ona
rzeczywiście zachodzi. W tym celu znakuje się startery, sondy lub powielane fragmenty
fluorochromami. Następnie mierzy się poziom emitowanej przez nie fluorescencji za pomocą
fluorymetru sprzężonego z termocyklerem. Im silniejsza fluorescencja tym więcej kopii
amplifikowanego fragmentu. Do analizy przyrostu ilości produktu w czasie rzeczywistym
opracowano różne interkalujące barwniki (bromek etydyny, SYBR Green I) oraz
hybrydyzujące sondy (TaqMan, Molecular Beacons, Fluorescence Resonance Energy
Transfer, Scorpions, TaqMan Minor Groove Binder)
Najpopularniejszym barwnikiem stosowanym w rt-PCR jest SYBR Green I. Jego
działanie polega na emisji fluorescencji po wbudowaniu się w mniejszą bruzdę dwuniciowej
cząsteczki DNA. Wzrastająca ilość amplifikowanego fragmentu powoduje wzrost
fluorescencji. Analizy z zastosowaniem barwników są mniej swoiste niż w przypadku
wykorzystania sond DNA specyficznie znakowanych flurochromem (komplementarnych do
badanego fragmentu). Wyróżnia się kilka rodzajów sond: hydrolizujące typu TaqMan,
Molecular Beacon, Scorpion, hybrydyzujące. Sondy te różnią się między innymi działaniem
oraz budową
W analizie z zastosowaniem Real-time PCR wykorzystuje się zasadę: im wyższa jest
początkowa liczba cząstek badanego materiału tym mniejsza ilość cykli jest potrzebna do
uzyskania określonej ilości produktu. Aby określić wyjściową ilość transkryptów można
zastosować metodę bezpośrednią, wyznaczając dokładną ilość badanego materiału lub metodę
względną pozwalającą określić ilość badanego materiału w stosunku do wzorca. Standardem
może być sekwencja występująca w analizowanym materiale np. geny metabolizmu
podstawowego. Z założenia ulegają one stałej ekspresji w komórkach organizmu i stanowią
punkt odniesienia do badanych genów. W tym przypadku poziom ekspresji ocenianego genu
wyrażony jest w stosunku do poziomu ekspresji genu referencyjnego. Stosuje się także
standardy zewnętrzne, będące cząsteczkami RNA lub DNA, które dodawane są w znanej
ilości do reakcji. Na podstawie liczby cząstek sekwencji standardowej określa się ilość
badanego materiału

Wyznacznikiem początkowej ilości badanej matrycy jest wartość C
T
. Jest to wartość
progowa fluorescencji, numer cyklu, w którym fluorescencja przekracza poziom tła,
przechodzi w fazę logarytmicznego wzrostu. Im niższa jest wartość C
T
tym większa była
liczba badanych cząstek na początku reakcji
Metoda ta cieszy się ogromną popularnością między innymi dzięki temu, że jest mniej
pracochłonna, bardziej czuła i wiarygodna, a ryzyko zanieczyszczenia wysoce
zminimalizowane. Wielkim udogodnieniem jest znaczne skrócenie czasu analizy oraz
wydania wyniku
Real-time PCR jest metodą pozwalającą na ilościową ocenę ekspresji genów,
stosowany jest w wielu dziedzinach. Technikę tę można wykorzystać między innymi do
potwierdzenia wyników analiz mikromacierzy oligonukleotydowych, monitorowania terapii
oraz analizy mutacji. Stosowana jest też w diagnostyce mikrobiologicznej do wykrywania
patogenów
Cytogenetyka
Cytogenetyka kliniczna to dział genetyki, którego przedmiotem zainteresowania są
chromosomy w komórce organizmu ludzkiego. Dziedzina ta znalazła ścisłe zastosowanie
diagnostyczne w medycynie. Zajmuje się badaniem liczby, budowy morfologicznej oraz
nieprawidłowości (aberracji) w strukturze chromosomów.
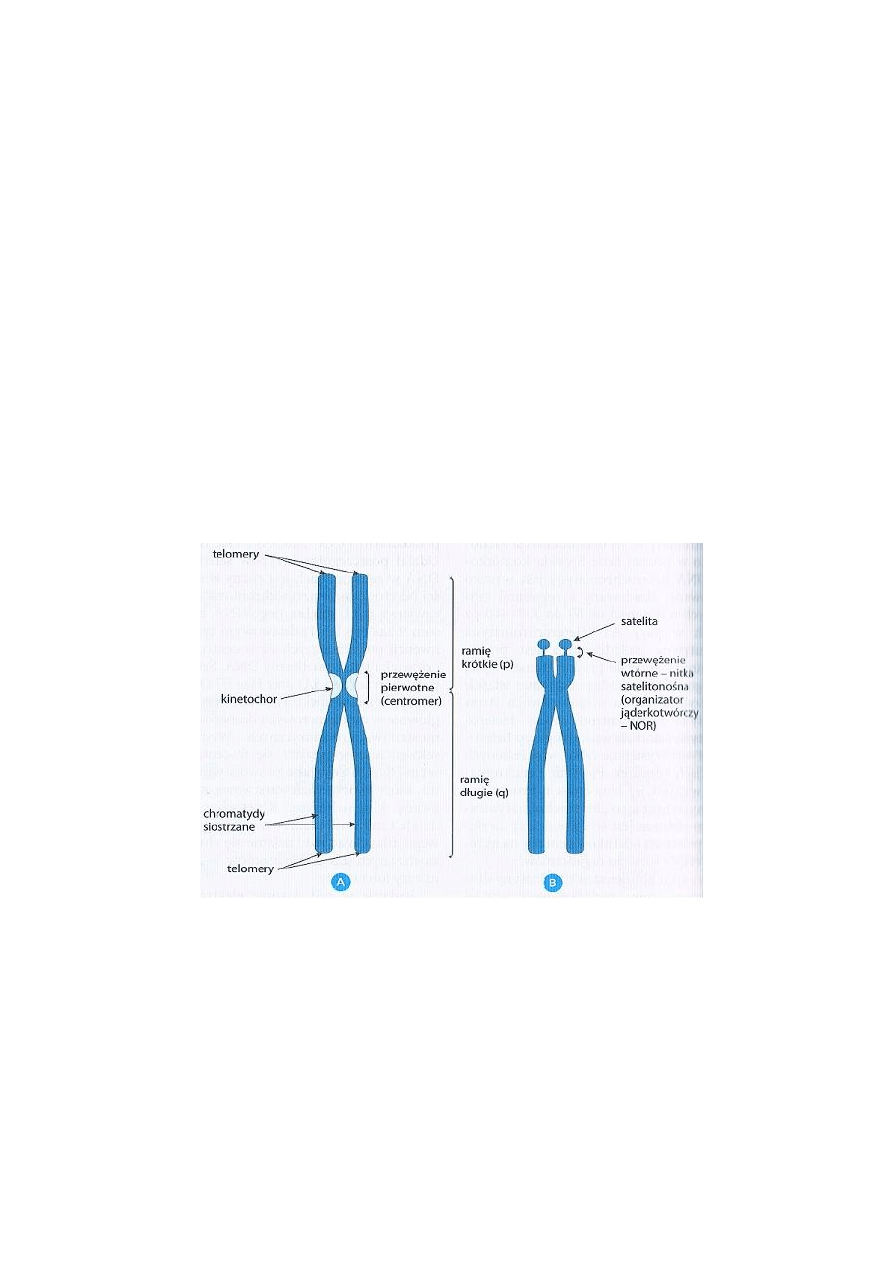
Budowa chromosomu metafazowego
Nośnikiem informacji genetycznej jest kwas deoksyrybonukleinowy (DNA), który w powiązaniu z
białkami histonowymi tworzy kompleks kwasy nukleinowe-białka inaczej nazywany chromatyną
- zlokalizowaną w jądrze komórkowym. Nić chromatynowa w zależności od fazy cyklu
komórkowego ulega kondensacji lub dekondensacji.
Chromosomy jako indywidualne struktury widoczne są tylko podczas podziałów komórkowych.
Najbardziej skondensowaną formą chromatyny jądrowej są chromosomy metafazowe.
charakterystyczny kształt pozwolił na wyróżnienie kilku elementów w ich morfologii (ryc.1):
- ramię krótkie p i długie q
- przewężenia pierwotnego czyli centromeru, przez który ramię krótkie łączy się z długim
- telomerów
- organizatorów jąderka (NOR), nazywanych przewężeniami wtórnymi
Ryc. 1 Schemat budowy chromosomu metafazowego. A – submetacentryczny, B – akrocentryczny
(Genetyka Medyczna, G. Drewa, T. Ferenc)
Na podstawie położenia centromeru chromosomy człowieka dzieli się na trzy grupy (ryc.2):
- chromosomy metacentryczne o równych ramionach pi q
- chromosomy submetacentryczne o ramionach p nieco krótszych niż q
- chromosomy akrocentryczne gdy centromer jest blisko jednego końca i ramiona górne p są bardzo
krótkie.
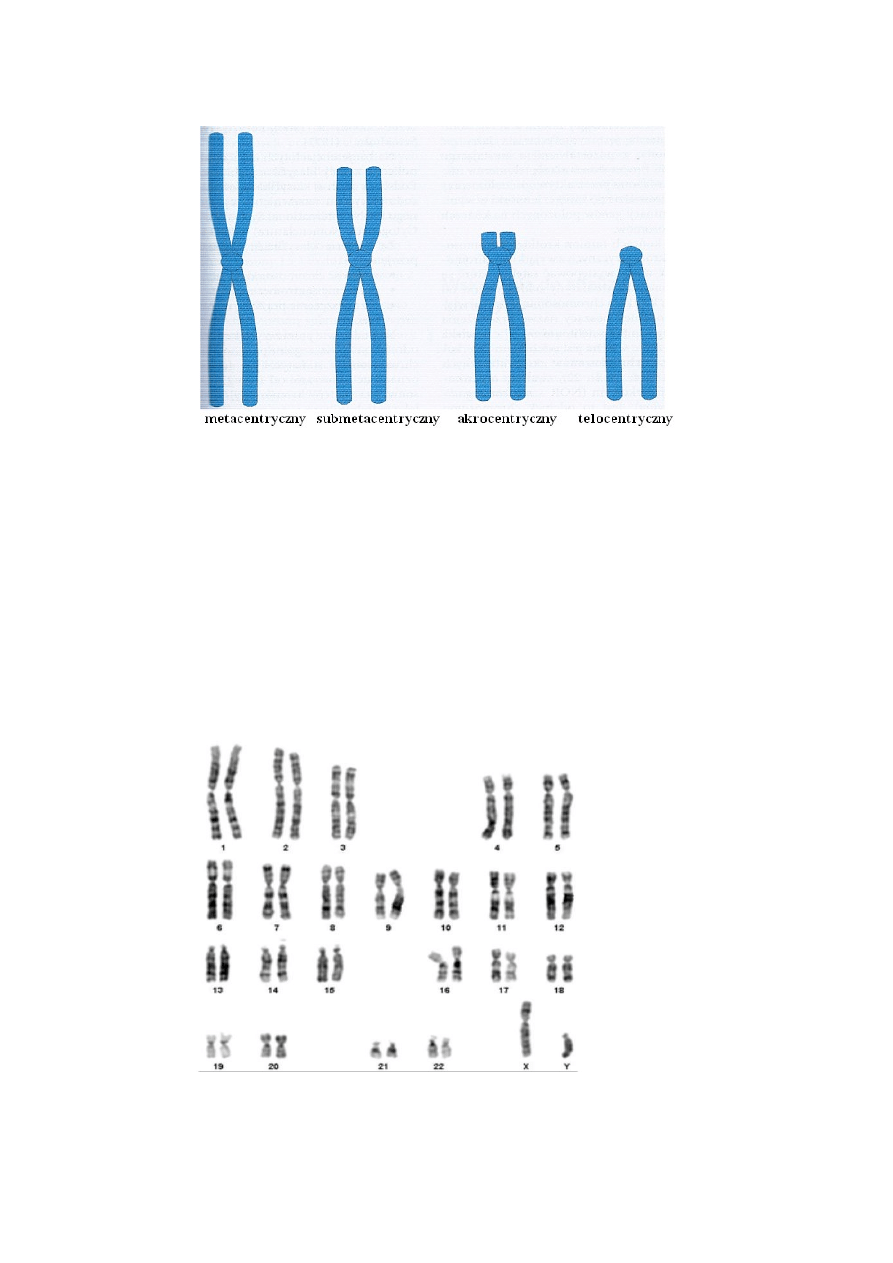
Ryc. 2 Typy budowy chromosomów metafazowych. (Genetyka Medyczna, G. Drewa, T. Ferenc)
Kariotyp i kariogram
Zestaw chromosomów występujący w komórce somatycznej charakterystyczny dla danego
organizmu nazywany jest kariotypem. Chromosomy człowieka sklasyfikowano zgodnie z zasadami
międzynarodowego systemu nazewnictwa cytogenetycznego ISCN (International System for
Human Cytogenetic Nomenclature). Za kryteria klasyfikacji przyjęto wielkość chromosomów,
położenie centromeru oraz pozycję danych prążków na ramionach p i q.
W normalnej komórce somatycznej człowieka znajdują się 22 pary homologiczne chromosomów
somatycznych (autosomy) oraz jedna para chromosomów płciowych (heterochromosomy XX lub
XY). Autosomy przyporządkowano do 7 grup (A-G). Chromosom X najbardziej podobny do 6 pary
zaliczono do grupy C, a chromosom Y zaliczono do grupy G.
Kariogram człowieka to inaczej kariotyp przedstawiony w postaci graficznej (Ryc. 3).
Ryc. 3 Kariogram człowieka – 46, XY (prążki GTG) (oryg. z archiwum Zakładu Genetyki
Klinicznej UM w Lublinie).

Ryc. 4 Podział chromosomów człowieka
Grup
a
Para
A
1-3
B
4-5
C
6-12 i chromosom X
D
13-15
E
16-18
F
19-20
G
21-22 i chromosom Y
−
do grupy A należą pary 1-3 gdzie chromosomy 1 i 3 to chromosomy metacentryczne,
a chromosom 2 jest submetacentryczny (Ryc. 4)
−
do grupy B należą submetacentryczne pary chromosomów 4 i 5
−
do grupy C zaliczono średniej wielkości submetacentryczne pary chromosomów 6-12 oraz
zbliżony wielkością chromosom X
−
grupę D tworzą akrocentryczne pary chromosomów 13-15
−
grupę E tworzą pary 16-18 gdzie chromosom 16 jest metacentryczny, a 17 i 18 to małe
chromosomy submetacentryczne
−
do grupy F należą najmniejsze metacentryczne pary chromosomów 19 i 20
−
grupę G tworzą małe akrocentryczne pary chromosomów 21-22 oraz zbliżony wielkością
chromosom Y
Wskazania do badania cytogenetycznego - analizy kariotypu
Badanie cytogenetyczne w celu oznaczenia kariotypu wykonuje się u pacjentów u których po
uprzednim kontakcie z lekarzem poradni genetycznej stwierdza się, że obraz kliniczny choroby
może być spowodowany obecnością aberracji chromosomowej. Do najczęstszych wskazań
określenia kariotypu należą:
−
występowanie zespołu wad rozwojowych współistniejący z opóźnieniem rozwoju
psychoruchowego lub umysłowego
−
występowanie cech fenotypowych charakterystycznych dla danego zespołu
chromosomowego
−
nieprawidłowa budowa narządów płciowych
−
niepowodzenia rozrodu
−
brak cech dojrzewania płciowego
−
kliniczne zaburzenia wzrostu
−
pierwotny lub wtórny brak miesiączki
−
występowanie znanej aberracji strukturalnej w rodzinie
Przebieg badania cytogenetycznego
Badanie cytogenetyczne jest procesem wieloetapowym i długotrwałym. Czas wykonania zależy od
typu hodowli komórkowej, od konieczności zastosowania metod molekularnych czy też wykonania
dodatkowego barwienia.

Pobranie materiału
Pierwszym etapem przebiegu badania cytogenetycznego jest pobranie materiału biologicznego,
którym jest krew, płyn owodniowy, kosmówka, fibrobalsty skóry, tkanki płodu po poronieniu.
W celu określenia struktury i liczby chromosomów najczęściej bada się limfocyty krwi obwodowej
i komórki szpiku kostnego.
Materiał pobierany jest przez wykwalifikowany personel w poradniach lub na oddziałach
szpitalnych w warunkach aseptycznych do sterylnych probówek na odpowiedni koagulant lub
pożywkę komórkową. Zakażenie bakteryjne lub grzybiczne uniemożliwia dalsze przeprowadzenie
badania. W chwili otrzymania materiału pracownik laboratoryjny sprawdza jego jakość, ilość, czy
krew pobrana jest na heparynę czy zawiera skrzepy, czy tkanki pobrane są na odpowiednie podłoże
komórkowe, czy zawierają substancje zapobiegające infekcji materiału i przede wszystkim czy
materiał przeznaczony do dalszych badań jest opisany imieniem i nazwiskiem pacjenta oraz jego
datą urodzenia.
Hodowla komórkowa
Hodowla komórek in vitro jest procesem namnażania się komórek w warunkach sztucznych poza
organizmem. Brak wzrostu hodowli uniemożliwia wykonanie badania ponieważ chromosomy
można analizować tylko podczas prometafazy lub metafazy podczas podziału komórkowego.
Najczęstszą metodą pozwalającą otrzymać dużą liczbę komórek do hodowli jest mikro lub
makrohodowla limfocytów krwi obwodowej.
Izolacja limfocytów
W metodzie tej wykorzystuje się różnice w wielkości i ciężarze właściwym poszczególnych
komórek. Stosuje się mieszaniny rozdzielające: Ficoll (Percoll), Isopaque (Uropolina) [metoda
Boyum'a], Gradisol (mieszanina Uropoliny i dekstranu), Lymphoprep – o różnej gęstości.
Krew wymieszaną z PBS i pobraną na antykoagulant nawarstwia się na odpowiedni gradient
i wiruje . Po wirowaniu widoczne są 3 warstwy:
−
na dnie – erytrocyty i i granulocyty
−
warstwa mieszaniny rozdzielającej z utworzona interfazą MNC (Mono Nuclear Cells)
−
górna warstwa – rozcieńczone osocze
Po wirowaniu komórki z powstałej interfazy są zbierane płukane w PBS, następnie zamrażane lub
wykorzystywane do hodowli komórkowej.
Izolacja limfocytów
Potrzebne urządzenia:
−
pipeta 1000-5000 µL
−
pipeta 20-200 µL
−
probówka typu Falcon (15 ml)
−
wirówka
−
probówki typu Eppendorf
Potrzebne odczynniki:
−
PBS
−
Gradisol lub Lymphoprep

Wykonanie:
−
pobraną krew rozcieńczamy dwukrotnie w PBS (np. 3 ml krwi i 3 ml PBS)
−
do probówek typu Falcon (15 ml) pipetą dodać 3ml Gradisolu lub Lymphoprepu
−
rozcieńczoną w PBS krew delikatnie nawarstwiamy pipetą po ściance do probówki
zawierającej Gradisol L
−
probówki wirujemy z prędkością 2700 rpm przez okres 30 min w temperaturze pokojowej
−
po wirowaniu widoczne są trzy warstwy
−
zebrać pipetą utworzona interfazę MNC do probówki o pojemności 15 ml, a następnie
dodać PBS do 15 ml
−
wstawić probówki do wirówki i wirować z prędkością 3000 rpm przez okres 10 min
−
używając pipety odciągamy supernatant tak by nie naruszyć osadu
−
do osadu komórkowego dodać za pomocą pipety PBS do 15 ml i wirować z prędkością
3000 rpm przez okres 10 min. Tą czynność powtarzamy dwukrotnie.
−
używając pipety odciągamy supernatant tak by nie naruszyć osadu
−
dodać 2 ml PBS i całość przenieść do probówki Eppendorf
−
wstawić probówki do wirówki i wirować z prędkością 800 rpm przez okres 3 min
−
używając pipety odciągamy supernatant tak by nie naruszyć osadu
−
osad komórek zamrozić w temp - 80°C lub wykorzystać do hodowli komórkowej
Zakładanie hodowli
Zakładanie hodowli in vitro odbywa się w aseptycznych warunkach przy użyciu komór
z przepływem laminarnym. Wyróżnia się dwa rodzaje hodowli komórkowych:
−
hodowle w zawiesinie gdzie komórki do prawidłowego wzrostu nie muszą wykazywać
adherencji (przyklejania do podłoża stałego). Prawidłowy rozwój zapewniony jest przez
utrzymanie ruchu pożywki hodowlanej. Komórki inkubuje się w specjalnych naczyniach z
wbudowanym mieszadłem magnetycznym i wiosełkami poruszającymi zawiesinę
komórkową (Ryc. 5). Do hodowli stosuje się specjalne pożywki tzw. płyny spinerowe, które
nie zawierają jonów magnezu i wapnia sprzyjających przyklejaniu komórek do podłoża.
−
hodowle adhezyjne, jednowarstwowe. Komórki przyklejają się do powierzchni naczynia,
dzielą się i rosną w postaci pojedynczej warstwy (ang. monolayer). Warunkiem niezbędnym
do rozwoju prawidłowej hodowli jednowarstwowej jest zdolność komórek do przylegania
do podłoża. Jeżeli hodowlę przeprowadza się na szkiełku mikroskopowym jest to tzw.
hodowla in situ.
butelka do hodowli komórkowych
naczynia do hodowli adhezyjnych
zawieszonych w płynie
Ryc. 5 Naczynia do hodowli komórkowych

Do każdego rodzaju materiału dobiera się odpowiedni typ pożywki i substancje stymulujące
podziały komórkowe w warunkach sztucznych. W celu zmniejszenia ryzyka kontaminacji do
podłoża dodawane są antybiotyki i substancje przeciwgrzybiczne (Ryc. 7). Hodowle inkubowane są
najczęściej w temperaturze 37°C, atmosferze 5% CO
2
i przy 95% wilgotności. Czas trwania
hodowli jest zależny od rodzaju materiału do badań (Ryc. 6).
Typ hodowli
Czas trwania hodowli
limfocyty
72 h
komórki płynu owodniowego
7-10 dni
kosmówki
STC (short term culture) 24 godziny
LTC (long term culture) 72 godziny
fibroblasty
1 miesiąc
Ryc. 6 Czas trwania hodowli
Komórki człowieka mają ograniczoną zdolność do wzrostu i podziałów, zbyt długo hodowane
obumierają jednak żywotną hodowlę komórek można przechowywać w pożywce komórkowej z
dodatkiem DMSO (sulfotlenku dimetylu) w atmosferze ciekłego azotu.
Utrwalanie hodowli komórkowej
Ogromną rolę w hodowli komórkowej odgrywa synchronizacja cyklu komórkowego, potrzebna ze
względu na uzyskanie wysokiego indeksu mitotycznego, a także chromosomów o odpowiedniej
długości i rozdzielczości.
Na etapie fazy S cyklu komórkowego hodowle komórkowe są wstrzymywane (kolchicyna,
kolcemid), następnie uwalniany czynnik hamujący powoduje, że wszystkie nagromadzone komórki
równocześnie wchodzą w fazę G2 i mitozę. Synchronizacja cyklu komórkowego ułatwia wybór
momentu utrwalania kiedy wiele komórek jest w stadium prometafazy. Przed procesem utrwalania
hodowlę komórkową traktuje się również czynnikami niszczącymi wrzeciono kariokinetyczne
(odpowiadające za rozejście się chromosomów do komórek potomnych), a także poddaje się
działaniu roztworu hipotonicznego (KCl), dzięki któremu cytoplazma staje się rzadsza,
a wykonanie preparatu staje się łatwiejsze.
Do utrwalania DNA chromosomów stosuję się mieszaninę stężonego kwasu octowego i metanolu
(w stosunku 1:3). Cały proces przeprowadza się według odpowiedniej dla każdego laboratorium
procedury w taki sposób, aby uzyskać jak największą ilość metafaz oraz jak najdłuższe
chromosomy.
Odczynnik
Opis
Fitohemaglutynina (PHA)
Mitogen limfocytów T, umożliwiający hodowlę komórek
w warunkach sztucznych
Glutamina L
Aminokwas, suplement do podłoża komórkowego
FBS
Surowica używana jako suplement do podłoży komórkowych
Penicylina, streptomycyna, gentamycyna
Antybiotyki, suplementy do podłoży komórkowych
RPMI 1640
Podłoże komórkowe, stosowane najczęściej
do hodowli limfocytów
Kolcemid, kolchicyna, winblastyna
Odczynniki stosowane do zatrzymania podziałów
komórkowych
MTX, FdU, BrdU, tymidyna, urydyna
Odczynniki używane do synchronizacji hodowli
komórkowych
DMSO
Suplement
podłoża
komórkowego
stosowany
do krioprzechowywania żywych komórek
Ryc. 7 Odczynniki pomocnicze stosowane w hodowli in vitro.

Zakładanie hodowli komórkowych:
Potrzebne urządzenia:
−
pipeta 100-1000 µL
−
pipeta 1000-5000 µL
−
strzykawka 10 ml
−
falkon do zakładania hodowli
−
probówka 50ml, do utworzenia podłoża komórkowego
−
komora laminarna
−
inkubator
Potrzebne odczynniki:
−
FBS
−
antybiotyki (penicylina, streptomycyna)
−
fitohemaglutynia (PHA)
−
RPMI 1640
Wykonanie:
−
sprawdzić czy krew została pobrana do probówki na heparynę sodową
−
sprawdzić czy probówka została podpisana
−
podpisać falkon w którym będzie przeprowadzona hodowla komórkowa. Na falkonie
powinno znaleźć się imię i nazwisko pacjenta, data i godzina założenia hodowli
−
do probówki przeznaczonej na utworzenie 20ml podłoża komórkowego pipetą dodajemy:
* 2 ml surowicę FBS
* 0,2 ml penicyliny i streptomycyny
* 0,6 ml PHA
* 17,2 ml podłoża RPMI
−
do podpisanego falkonu strzykawką dodać 10 ml utworzonego podłoża komórkowego,
następnie pipetą dodajemy 0,8 ml krwi pełnej dobrze wymieszanej (od dorosłych) lub 0,6 ml
krwi pełnej od dzieci
−
falkon z hodowlą komórkową wstawiamy do inkubatora do temp. 37°C na czas 72 h
Traktowanie i utrwalanie hodowli komórkowej
Potrzebne urządzenia:
−
pipeta 20 – 200 µL
−
pipety Pasteura
−
probówka 15 ml
−
inkubator
−
wirówka
−
wortex
−
komora laminarna
Potrzebne odczynniki:
−
kolcemid
−
0,075M KCl
−
utrwalacz (kwas octowy i metanol w stosunku 1:3)

Wykonanie:
−
po 72 h wyjąć hodowle komórkowe z inkubatora, dokładnie wymieszać, postawić w pozycji
pionowej
−
pipetą dodać 80 µL kolcemidu i ponownie wymieszać
−
wstawić hodowle komórkowe do inkubatora do temp. 37°C na czas 20 min
−
wyjąć falkon z inkubatora i przelać hodowle komórkowe do probówek (poj. 15 ml)
−
wstawić do wirówki i wirować z prędkością 1500 obr/min przez czas 10 min w temp. 23°C
−
używając pipety pasteura odciągamy supernatant tak by nie naruszyć osadu
−
osad mieszamy na worteksie i równocześnie korzystając z pipet powoli dodajemy 8 ml
0,075M KCl
−
probówki z KCl inkubujemy w temp. 37°C przez czas 20 min
−
wstawiamy próbki do wirówki i wirujemy z prędkością 1500 obr/min przez czas 20 min
w temp. 23°C
−
używając pipety pasteura odciągamy supernatant tak by nie naruszyć osadu następnie
pozostały osad mieszamy na worteksie i równocześnie korzystając z pipet powoli dodajemy
8 ml utrwalacza. Próbki wstawiamy do wirówki i wirujemy z prędkością 1500 obr./min
przez okres 10 min. Tą czynność wykonujemy trzykrotnie
−
po trzecim utrwaleniu wkładamy probówki do zamrażalki do temp. -20°C
Sporządzanie preparatów
Po utrwaleniu hodowli komórkowej przygotowuje się preparaty cytogenetyczne. Na mikroskopowe
szkiełka podstawowe nakrapiane są zawiesiny utrwalonych komórek, które następnie podlegają
procesowi suszenia. Etap ten odbywa się w odpowiedniej dla danej tkanki temperaturze
i wilgotności co powoduje rozciągnięcie i pęknięcie komórek, a w konsekwencji powstanie
preparatu nadającego się do dalszej analizy.
Sporządzanie preparatów cytogenetycznych przygotowuje się metodą ręczną lub automatycznie
z wykorzystaniem odpowiedniej aparatury.
Analiza mikroskopowa i zapisywanie wyników cytogenetycznych
Analiza chromosomów od danego pacjenta przeprowadzana jest za pomocą mikroskopu oraz
systemu komputerowego umożliwiającego ułożenie kariogramu. Według obowiązującego systemu
ISCN (Ryc. 8) zapis cytogenetyczny rozpoczyna się od podania liczby chromosomów, a po
przecinku liter oznaczających chromosomy płciowe (46, XX kobieta; 46, XY mężczyzna). Jeżeli
w kariotypie występują aberracje to zapisywane są po przecinku i dodatkowo słownie opisane
w epikryzie.
Ryc. 8. Wykaz symboli i skrótów stosowanych w zapisie kariotypu (według ISCN)
Skrót
Objaśnienie
+
nadmiar, dodatek
-
niedobór, ubytek
:
pęknięcie
::
pęknięcie i połączenie
,
Oddziela liczbę chromosomów, chromosomy płci i ewentualne
aberracje
;
Rozdziela zapisy dotyczące różnych chromosomów

Skrót
Objaśnienie
.
Oddziela zapisy subprążków od cyfry określającej prążek
→
określa zakres regionu chromosomowego
()
nawiasy obejmujące uszkodzony chromosom oraz punkty pęknięć
/
oddziela różne linie komórkowe w mozaikach
cen
centromer
chi
chimera
del
delecja (wypadnięcie fragmentu chromosomu)
der
chromosom pochodny
dic
chromosom dicentryczny (posiadający dwa centromery
dup
duplikacja
fra
miejsce kruche
h
heterochromatyna konstytutywna
i
izochromosom (posiadający oba ramiona takie same
ins
insercja (wstawianie fragmentu)
inv
inwersja (obrócenie fragmentu)
kpz
kilo par zasad
mar
chromosom markerowy, często pochodzący od chromosomu
akrocentrycznego
mat
pochodzenie matczyne
mos
mozaikowość (kariotyp złożony z kilku linii komórkowych)
NOR
organizator jąderkowy
p
krótkie ramię chromosomu
pat
materiał pochodzący od ojca
PAR
region pseudoautosomalny
q
długie ramię chromosomu
r
chromosom pierścieniowy
s
satelity
stk
nitki satelitonośne
t
translokacja
tel
telomer
ter
koniec chromosomu
UPD
jednorodzicielska disomia
UPHD
jednorodzicielska heterodisomia
UPID
jednorodzicielska izodisomia

Przykłady zapisów aberracji liczbowych
–
45, X (monosomia X) – kariotyp w którym brakuje jednego chromosomu X u płci żeńskiej
–
47, XYY (polisomia Y) – kariotyp z jednym chromosomem X i dwoma chromosomami Y
–
47, XXX (polisomia X) – kariotyp z 3 chromosomami X
–
47, XY, +21 (trisomia 21) – kariotyp z dodatkowym chromosomem pary 21
–
46, XY, upd(15) mat – męski kariotyp wykazujący jednorodzicielską disomię chromosomu
15 pochodzenia matczynego
–
mos 47, XX,+21[20]/46,XX,upd(21) pat[8] – żeński kariotyp mozaikowy składający się z
dwóch linii komórkowych, jednej z jednorodzicielską disomią chromosomu 21 pochodzenia
ojcowskiego stwierdzoną w 8 komórkach i drugiej linii z trisomią chromosomu 21
stwierdzonej w 20 komórkach
Przykłady zapisów aberracji strukturalnych
–
46, XY,del(5)(pter→q13) – delecja dotycząca końcowego fragmentu ramion długich
chromosomu pary 5 od prążka q13
–
46, XX,del(5)(pter→q13::q33→qter) – delecja oznaczająca utratę wewnętrznego fragmentu
ramienia długiego chromosomu pary 5 między prążkami q13 i q33
–
45, XX,der(13;21)(q10;10) – pęknięcie w prążku q10 chromosomu pary 13 i 21.
–
46, XX,dup(1)(q22q25) – podwojenie fragmentu chromosomu pary 1 zawartego pomiędzy
prążkami q22 i q25
–
45, XX,dic(13;15)(q22;q24) – chromosom dicentryczny złożony z fragmentów zwierających
centromery chromosomów pary 13 i 15
Wyszukiwarka
Podobne podstrony:
materiał do badań, PCR, Cytogenetyka
POBIERANIE I PRZECHOWYWANIE MATERIAŁÓW DO BADAŃ wiRUSOLOGICZNYCH prezentacja
pobieranie i przesyłanie materiałów do badań mikrobiologicznych, mikrobiologia
Pobieranie materiałów do badań
POBIERANIE MATERIALOW DO BADAN MIKROBIOLOGICZNYCH ppt
genetyka cw 9 materiał do badan
pobieranie materiału d do badań
pobieranie materiału do badań
technika pobierania materiału do badań
Pobieranie i przesyłanie materiału do badań, UM Wrocław - Stomatologia, Mikrobiologia i mikrobiolog
materiały do badań, Medycyna, Choroby zakaźne
Pobieranie materiału do badań biologicznych, Ratownicto Medyczne, MIKROBIOLOGIA
Materiał do badań hematologicznych oraz interpretacja wyników badań układ czerwonokrwinkowy ppt
Procedura Przyjmowanie materiału do badań laboratoryjnych
pobieranie materialu do badań morfologicznych wyklady
07 Procedury zabiegowe służące do uzyskania materiału do badań hist patid 6938 ppt
POBIERANIE I PRZECHOWYWANIE MATERIAŁÓW DO BADAŃ wiRUSOLOGICZNYCH prezentacja
Pobieranie materiałów do badań
więcej podobnych podstron