Materiały z chemii organicznej
kg
- 1 -
Elektroujemność niektórych grup:
-CH
3
2,30
-CJ
3
2,50
-CH
2
Cl
2,47
-CBr
3
2,57
-CHCl
2
2,63
-CF
3
3,29
-CCl
3
2,79
Elektroujemność węgla w zależności od hybrydyzacji:
C
sp3
< C
sp2
< C
sp
Efekt indukcyjny różnych grup odniesiony do wodoru:
+I
-I
-O
-
-NR
3
+
-COOH
-OR
-COO
-
-SR
2
+
-F
-COR
-CR
3
-NH
3
+
-Cl
-SH
-CHR
2
-NO
2
-Br
-SR
-CH
2
R
-SO
2
R
-I
-OH
-CH
3
-CN
-OAr
-C
≡CR
-SO
2
Ar
-COOR
Ar
Efekt mezomeryczny najczęściej spotykanych grup:
+M
-M
-O
-
-NH-COR
-NO
2
-CO-H
-S
-
-O-COR
-CN
-CO-R
-NR
2
-SR
-SO
3
H
-NO
-NHR
-SH
-SO
2
-OR
-Ar
-NH
2
-F > -Cl > -Br > -I
-COOH
-OR
-CH
3
(hiperkonjugacja)
-COOR
-OH
-CONH
2
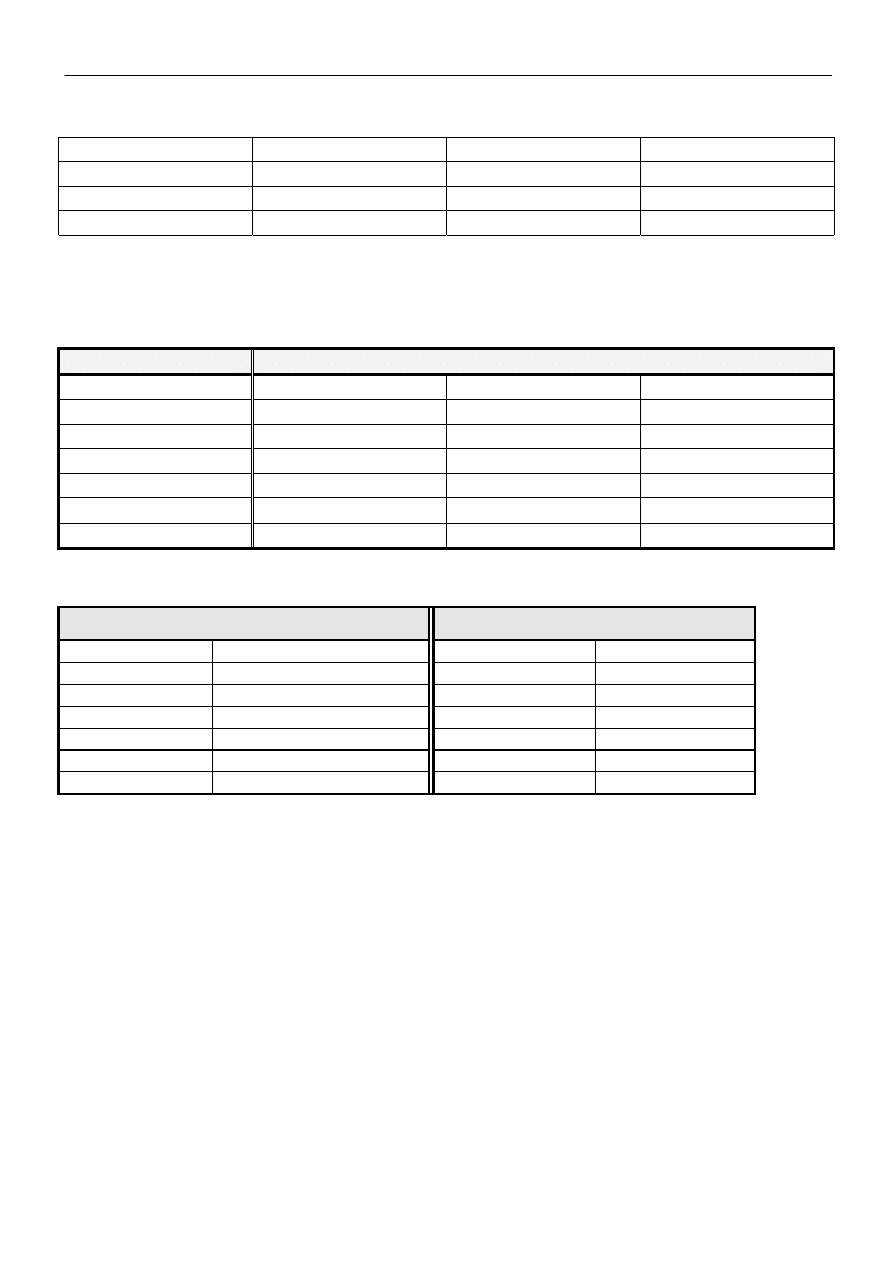
Reguły stosowane przy rysowaniu struktur granicznych:
•
Struktury graniczne mogą różnić się tylko rozmieszczeniem elektronów, lecz nie położeniem jąder
atomowych. Jeżeli położenia jąder nie są takie same, to mamy do czynienia z izomerią albo tautomeria, a nie
mezomerią.
•
W mezomerii uczestniczą tylko takie struktury, które mają taką samą ilość par elektronów. Jeżeli rozpatruje
się mezomerię rodników, to ilość niesparowanych elektronów musi być taka sama we wszystkich strukturach
granicznych.
•
Struktury graniczne powinny mieć taką samą, albo porównywalną energię. Te struktury, których wzory
pozwalają przypuszczać, że odpowiadają im o wiele większe zawartości energii niż dla pozostałych struktur, nie
biorą udziału w mezomerii lub ich udział w rzeczywistej budowie cząsteczki jest znikomy.
•
Mezomeria występuje tylko w tych cząsteczkach, lub w częściach cząsteczek, które mają budowę płaską. W
mezomerii biorą udział tylko elektrony należące do tych atomów, które leżą w jednej płaszczyźnie. Znaczne
wychylenie atomów z płaszczyzny utrudnia nakładanie się orbitali p.
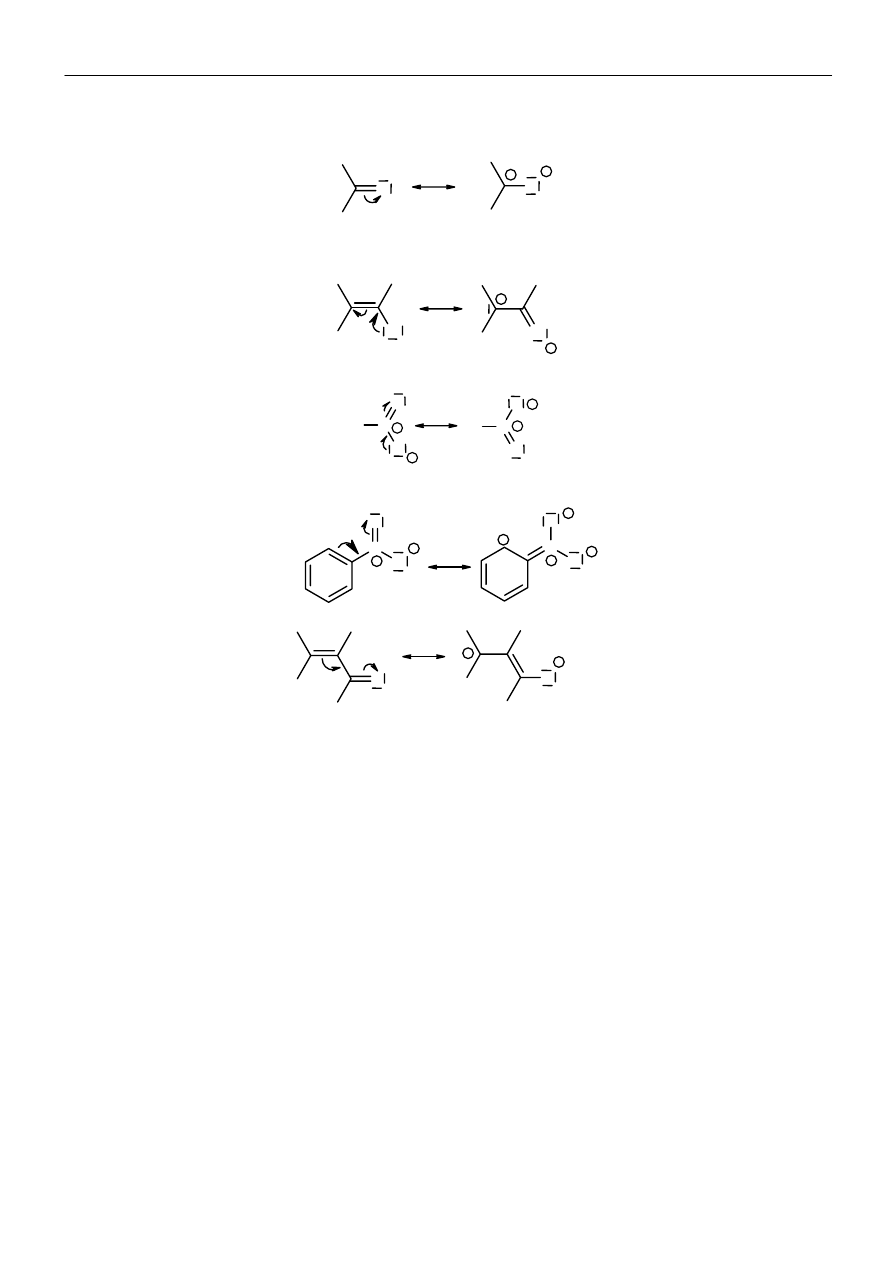
Materiały z chemii organicznej
kg
- 2 -
Gdy dwa atomy różniące się elektroujemnością połączone są wiązaniem
π, atom o większej elektroujemności może ściągnąć do
siebie ruchliwe elektrony
π:
O
O
+
-
Wolna para elektronowa atomu związanego wiązaniem
σ z innym atomem o hybrydyzacji sp lub sp
2
jest zdelokalizowana:
Cl
Cl
+
-
R N
O
O
+
-
R N
O
O
+
-
Wiązania
π rozdzielone jednym wiązaniem σ są ze sobą sprzężone i ulegają delokalizacji:
N
O
O
O
N
O
O
O
+
-
+
-
-
-
+
+
W chemii organicznej zdecydowana większość reakcji przebiega z udziałem atomu węgla (na atomie węgla). Pojęcie
kwasów i zasad wg Bröensteda, Lewisa czy Pearsona nie było zbyt dogodne, ponieważ w żaden sposób moc zasad (kwasów) nie
przekładała się na szybkość reakcji. Wprowadzono więc podział substancji reagujących na nukleofile i elektrofile.
Nukleofilem
- nazywa się jony ujemne, cząsteczki obojętne, które dysponują wolnymi parami elektronów i podczas reakcji
ze związkami organicznymi atakują te atomy węgla, przy których występuje zmniejszona gęstość elektronów. Odczynniki
nukleofilowe są zasadami zarówno według definicji Bröensteda, jak i Lewisa.
Elektrofilem
- nazywa się jony dodatnie i obojętne cząsteczki, które są zdolne do przyłączenia pary elektronów i podczas
reakcji ze związkami organicznymi atakują te atomy węgla, przy których występuje zwiększona gęstość elektronów.
Odczynniki elektrofilowe są kwasami Lewisa.
Miarą nukleofilowości odczynnika są stałe szybkości ich reakcji z cząsteczką substratu. O nukleofilowości
odczynnika decydują dwa czynniki: zasadowość i polaryzowalność.
•
Nukleofilowość wzrasta wraz z zasadowością np.: R-O
-
>OH
-
>C
6
H
5
-O
-
>CH
3
COO
-
>H
2
O
Reguła ta spełniona jest również w wielu jonach, w których ładunek ujemny znajduje się na atomie
tego samego okresu: R
3
C
-
>R
2
N
-
>RO
-
>F
-
•
W śród chlorowców charakter nukleofilowy wzrasta od jonu fluorkowego do jonu jodkowego, a więc odwrotnie do
zasadowości. Nukleofilowość zależy tu bardziej od polaryzowalności: I
-
>Br
-
>Cl
-
>F
-
Materiały z chemii organicznej
kg
- 3 -
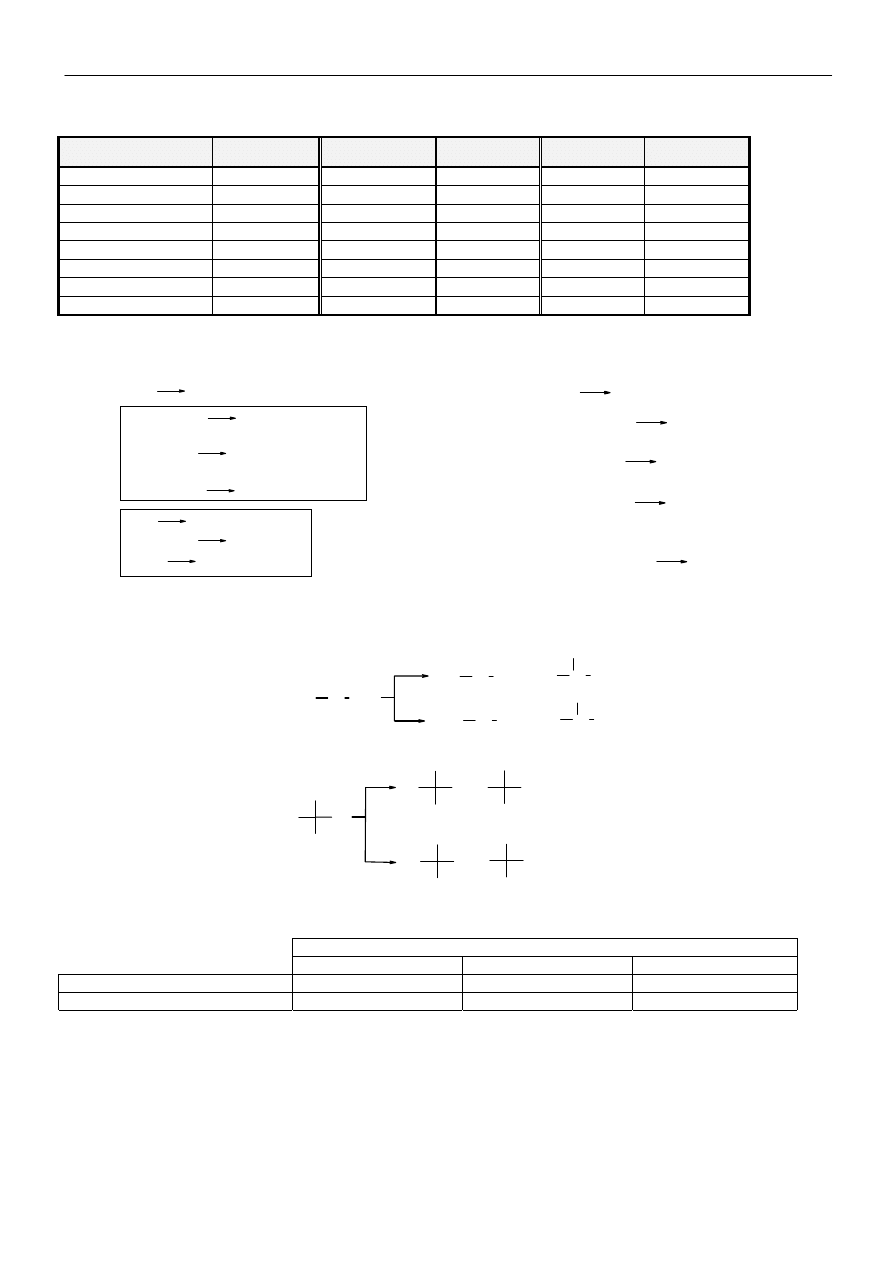
Energia dysocjacji niektórych wiązań (rozpad homolityczny):
Wiązanie
Energia dysocjacji
KJ/mol
Wiązanie
Energia dysocjacji
KJ/mol
Wiązanie
Energia dysocjacji
KJ/mol
CH
3
-H
436
CH
3
-F
453
F-F
126
CH
3
-CH
2
-H
411
CH
3
-Cl
352
Cl-Cl
243
(CH
3
)
2
CH-H
394
CH
3
-Br
293
Br-Br
193
(CH
3
)
3
C-H
381
CH
3
-I
235
I-I
151
CH
2
=CH-H
453
CH
3
-CH
2
-Cl
348
H-F
570
CH
2
=CH
2
-CH
2
-H
323
(CH
3
)
2
CH-Cl
306 ?
H-Cl
432
C
6
H
5
-CH
2
-H
327
(CH
3
)
3
C-Cl
314
H-Br
369
CH
2
=CH-Cl
436
H-I
298
Mechanizm i entalpia reakcji chlorowania alkanów
Mechanizm i entalpia reakcji jodowania
alkanów
Cl
2
2Cl
.
h
ν
zapoczątkowanie reakcji
Cl
.
+
CH
3
-H
CH
3
.
+ HCl
∆H=436 KJ/mol -432 KJ/mol = 4KJ/mol
CH
3
.
+ Cl
2
Cl
.
+ CH
3
Cl
∆H=243 KJ/mol -352 KJ/mol = -109KJ/mol
Cl
.
+
CH
3
-H
CH
3
.
+ HCl
∆H reakcji =-105KJ/mol
zakończenie łańcucha
propagacja,
wzrost łańcucha
Cl
2
2Cl
.
Cl
.
+ CH
3
.
CH
3
Cl
2CH
3
.
C
2
H
6
I
2
2I
.
h
ν
I
.
+
CH3-H
CH3
.
+ HI
∆H=436 KJ/mol -298 KJ/mol = 138KJ/mol
CH
3
.
+ I
2
I
.
+ CH
3
I
∆H=151 KJ/mol -235 KJ/mol = -84KJ/mol
I
.
+
CH
3
-H
CH
3
.
+ HI
∆H reakcji = +54KJ/mol
CH
3
I
+
HI
CH
4
+ I
2
∆H reakcji = - 54KJ/mol
Obliczanie składu mieszaniny poreakcyjnej w reakcji halogenowania alkanów:
C
H
3
CH
2
CH
3
C
H
3
CH
2
CH
2
Cl
C
H
3
CH CH
3
Cl
+
C
H
3
CH
2
CH
2
Br
C
H
3
CH CH
3
Br
+
CH
3
C
H
3
CH
3
H
CH
3
C
H
3
CH
2
Cl
H
CH
3
C
H
3
CH
3
Cl
+
CH
3
C
H
3
CH
2
Br
H
CH
3
C
H
3
CH
3
Br
+
45%
55%
3%
97%
64%
36%
1%
99%
Względne reaktywności wiązań C-H w reakcji:
rzędowość atomu wegla
I
o
II
o
III
o
chlorowania (25
o
C)
1
3,7
5
bromowania (130
o
C)
1
80
1600
Obliczony skład mieszaniny poreakcyjnej dla powyższych reakcji:
Chlorowanie propanu: 6
I
o
H, reaktywność 6*1=6;
2 II
o
H, reaktywność 2*3,7=7,4; skład 45%, 55%
Bromowanie propanu: 6
I
o
H, reaktywność 6*1=6;
2 II
o
H, reaktywność 2*80=160; skład 3,6%, 96,4%
Chlorowanie izobutanu: 9 I
o
H, reaktywność 9*1=9;
1 III
o
H, reaktywność 1*5=5; skład 64%, 36%
Bromowanie izobutanu: 9 I
o
H, reaktywność 9*1=9;
1 III
o
H, reaktywność 1*1600=1600; skład 0,6%, 99,4%

Materiały z chemii organicznej
kg
- 4 -
CH
3
CH
3
C
H
3
CH
3
C
H
3
CH
3
CH
3
CH
3
C
H
3
CH
3
C
H
3
CH
3
CH
3
C
H
3
CH
3
CH
3
C
H
3
CH
3
C
H
3
CH
3
C
H
3
C
H
3
CH
3
CH
3
C
H
3
CH
3
C
H
3
C
H
3
CH
3
CH
3
CH
3
C
H
3
C
H
3
C
H
3
CH
3
CH
3
C
H
3
CH
3
C
H
3
C
H
3
CH
3
CH
3
CH
3
C
H
3
CH
3
C
H
3
CH
3
CH
3
CH
3
C
H
3
CH
3
C
H
3
CH
3
CH
3
Wzory perespektywistyczne
Kozłowe
Wzory projekcyjne (rzutowy) Newmana
Wzór projekcyjny (rzutowy)Fischera
Przestrzenne
CH
3
C
H
3
CH
3
C
H
3
CH
3
CH
3
CH
3
CH
3
CH
3
C
H
3
C
H
3
CH
3
Konstytucja związku o danym wzorze cząsteczkowym
określa sposób i kolejność powiązania atomów.
Konfiguracja cząsteczki o określonej konstytucji jest to
rozmieszczenie w przestrzeni jej atomów, bez
uwzględnienia różnych położeń atomów wynikających z
rotacji wewnętrznej wokół jednego lub kilku wiązań
pojedynczych.
Stereoizomerami są takie izomery, które mają
identyczną konstytucję, a różnią się jedynie
przestrzennym rozmieszczeniem atomów.
Chiralność jest to właściwość przedmiotu polegająca na
tym, że jest on nieidentyczny ze swym odbiciem
lustrzanym. Dotyczy tylko indywidualnych cząsteczek, a
ściślej ich modeli. Jeżeli cząsteczka związku nie posiada
takich elementów symetrii jak: płaszczyzna symetrii (m),
środek symetrii (i) (może posiadać oś dwukrotną), to jest
chiralna.
Enancjomery są to cząsteczki stanowiące nawzajem
odbicie lustrzane.
Asymetria oznacza brak elementów symetrii. Każda
cząsteczka asymetryczna jest chiralna.
Diastereoizomery
- stereoizomery nie będące
enancjomerami.
Konformacja cząsteczki o określonej strukturze jest to
różne rozmieszczenie jej atomów w przestrzeni,
wynikające z rotacji wewnętrznej wokół wiązań
pojedynczych.
Reguły pierwszeństwa:
1
Atomy o wyższych liczbach atomowych mają pierwszeństwo przed atomami których liczby atomowe są niższe.
W przypadku izotopów o pierwszeństwie decyduje liczba masowa.
2
Jeżeli zastosowanie reguły 1 dla atomów bezpośrednio związanych z centrum asymetrii nie pozwala jednoznacznie
uporządkować podstawników wg pierwszeństwa, to rozpatruje się następne z kolei atomy, nie połączone bezpośrednio z centrum
asymetrii.
3
Sposób postępowania wobec grup zawierających wiązania podwójne i potrójne polega na zamianie wiązania
wielokrotnego na dwa lub trzy wiązania pojedyncze. Np. grupę >C=O traktuje się jako
C
(O)
O
(C)
, gdzie atomy w
nawiasach są powtórzeniem atomów związanych podwójnie.
Konfiguracja absolutna (konfiguracja RS):
C
H
3
C
2
H
5
H
Cl
C
2
H
5
C
H
3
H
Cl
1
2
3
4
1
2
3
4
(R)-2-chlorobutan
(S)-2-chlorobutan

Materiały z chemii organicznej
kg
- 5 -
Alkeny i alkiny
C C
C
H
miejsca reaktywne w alkenach
1. Reakcje A
E
do wiązania C=C
+ E +
E
+
Nu
-
:
dowolny nukleofil
obecny w roztworze
Trwałość karbokationów
CH
3
+
CH
3
-CH
2
+
CH
2
+
C
H
2
C
H
CH
2
+
C
CH
3
C
H
3
CH
3
+
C
H
3
CH
CH
3
+
<
<
<
<
<
stabilizacja karbokationu poprzez
efekt indukcyjny i hiperkonjugacje
stabilizacja karbokationu poprzez
efekt mezomeryczny
C
H
3
C
H
CH
2
+ E +
C
H
3
CH CH
2
E
+
C
H
3
C
H
CH
2
E
+
Nu
:
-
C
H
3
C
H
CH
2
E
Nu
Addycja przebiega poprzez
trwalszy karbokation i jest
addycją trans.
C
H
3
C
H
CH
2
+ X
2
C
H
3
C
H
CH
2
X
X
X= Br, Cl
Reakcje alkenów z bromem
i chlorem
C
H
3
C
H
CH
2
+ HX
C
H
3
C
H
CH
2
H
X
X= Cl, Br, I, OH, SH, HSO
4
, OR, SR
Reakcje alkenów z cząsteczkami niesymetrycznymi.
Addycja przebiega zgodnie z regułą Markownikowa
(Powstaje trwalszy karbokation). Mówimy, że reakcja
addycji do wiązań C=C jest regiospecyficzna.
C
H
3
C
H
CH
2
+ H-O-X
C
H
3
C
H
CH
2
X
OH
..
H-O-X + H
+
O
H
H
X
+
H
2
O + X
+
C
H
3
C
H
CH
2
+ X
+
C
H
3
CH CH
2
X
+
OH
2
..
..
C
H
3
C
H
CH
2
X
OH
2
+
-H
+
C
H
3
C
H
CH
2
X
OH
X= Br, Cl, H
Mechanizm addycji kwasu
chlorowego(I) (podchlora-
wy) (bromowego(I)) lub
wody
C
H
3
C
H
CH
2
+ X-Y
C
H
3
C
H
CH
2
X
Y
Y bardziej elektroujemne od X
ON-Cl, I-Cl
C
H
3
C
H
CH
2
+ HBr
nadtlenki
C
H
3
C
H
2
CH
2
Br
HBr
nadtlenki
∆
∆
Br
.
C
H
3
C
H
CH
2
+ Br
C
H
3
CH CH
2
Br
.
C
H
3
CH CH
2
Br
.
+ HBr
C
H
3
C
H
2
CH
2
Br
C
H
3
C
H
CH
2
Br
mniej trwały
+ Br
.
.
.
W wyniku reakcji rodnika
z alkenem powstaje rodnik
o większej rzędowości
(trwalszy)

Materiały z chemii organicznej
kg
- 6 -
2. Utlenianie alkenów
C
H
3
C
H
CH
2
+ MnO
4
-
C
H
3
C
H
2
C
H
O
Mn
O
O
O -
H
2
O
C
H
3
C
H
2
C
H
O
H
OH
C
H
3
C
H
CH
2
+ MnO
3
-
+ MnO
3
-
C
H
3
C
H
2
C
H
O
Mn
O
O -
H
2
O
C
H
3
C
H
2
C
H
O
H
OH
+ MnO
2
W łagodnych warunkach
(środowisko obojętne) w
wyniku utleniania alkenów
nadmanganianem potasu
powstają cis-diole
C
H
3
C
H
CH
2
H
2
O
C
H
3
C
H
2
C
H
O
H
OH
+ OsO
2
+ OsO
4
C
H
3
C
H
2
C
H
O
Os
O
O
O
Mechanizm reakcji
podobny do opisanego
wyżej. Produktem reakcji
są również cis-diole.
C
H
3
C
H
CH
2
+ H
2
O
2
HCOOH
C
H
3
C
H
CH
2
O
C
H
3
C
H
CH
2
O
H
+
H
+
OH
2
..
..
-H
+
C
H
3
C
H
C
H
2
OH
O
H
Reakcja może przebiegać poprzez
karbokation. Atak cząsteczki wody
następuje od strony przeciwnej do jonu
oksoniowego. Produktem reakcji są
trans-diole.
R
1
R
2
H
R
3
+ MnO
4
-
H
+
O
R
1
R
2
+ R
3
-COOH
W warunkach bardziej drastycznych, w
zależności od ilości grup R, powstają
ketony i/lub kwasy karboksylowe.
H
R
3
R
1
R
2
O
O
O
..
.. ..
..
..
..
:
+
-
O
O
O
R
2
R
1
H
R
3
O O
O
R
3
H
R
1
R
2
H
2
O, Zn
O
R
2
R
1
O
R
3
H
+
molozonek
ozonek
aldehydy, ketony
W zależności od grup R
powstają aldehydy lub
ketony, lub aldehyd i
keton.
3. Redukcja
C
H
3
C
H
CH
2
+ H
2
Pt
C
H
3
C
H
CH
2
H
H
C
H
3
C
H
CH
2
+ HN=NH
C
H
3
C
H
CH
2
H
H
+ N
2
W wyniku redukcji na
katalizatorze lub diimidem
powstaje produkt cis
addycji.
4. Reakcje w pozycji allilowej
C
H
3
C
H
CH
2
+ X
2
∆ lub hν
C
H
2
C
H
CH
2
X
+ HX
N-X
O
O
∆
C
H
3
C
H
CH
2
+
C
H
2
C
H
CH
2
X
N-H
O
O
+
Reakcja ma charakter rodnikowy i zachodzi
wg podanego schematu w przypadku bardzo
małego stężeni X
2
. Małe stężenie halogenku
można uzyskać z rozkładu N-halogenoimidu
kwasu bursztynowego.
Przegrupowanie karbokationów:
C
H
C
H
C
H
3
CH
3
CH
2
+ HBr
C CH
C
H
3
CH
3
CH
3
H
+
C CH
C
H
3
CH
3
CH
3
H
+
Br
_
_
|
|
C CH
C
H
3
CH
3
CH
3
H
Br
C C
H
C
H
3
CH
3
CH
2
C
2
H
5
+ HBr
C CH
C
H
3
CH
3
CH
3
C
2
H
5
+
C CH
C
H
3
CH
3
C
2
H
5
CH
3
C C
H
C
H
3
CH
3
CH
3
C
2
H
5
+
+
C CH
C
H
3
CH
3
C
2
H
5
CH
3
Br
C C
H
C
H
3
CH
3
CH
3
C
2
H
5
Br
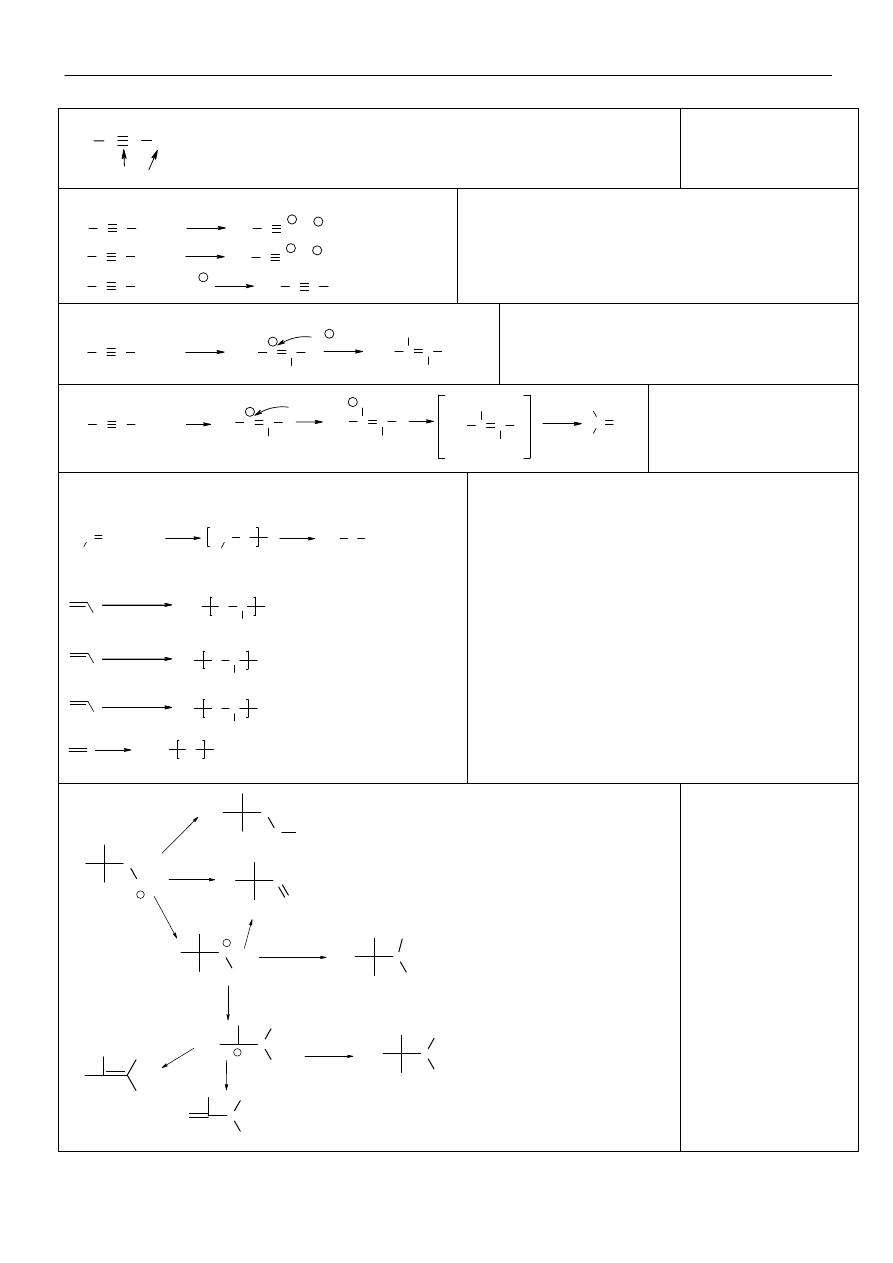
Materiały z chemii organicznej
kg
- 7 -
Alkiny
C C H
C
H
3
reaktywne centra
1. Reakcje wykorzystujące C-H kwasowość alkinów:
C C H
C
H
3
+ Na
NH
3
C C
C
H
3
Na
+
-
:
+ 1/2H
2
C C H
C
H
3
+ NaH
C C
C
H
3
Na
+
-
:
+ H
2
C C H
C
H
3
+ Ag(NH
3
)
2
C C
C
H
3
Ag
+
Z metalami alkalicznymi i ziem alkalicznych alkiny tworzą sole
w których wiązanie C-Me jest w znacznej mierze jonowe. Sole
te łatwo rozkładają się pod wpływem wody. Z innymi metalami
wiązania C-Me mają większy charakter kowalencyjny, nie
rozkładają się tak łatwo pod wpływem wody
2. Addycja do potrójnego wiązania
C C H
C
H
3
+ X-Y
C C H
C
H
3
X
+
Y
: -
C C H
C
H
3
X
Y
W połączeniu X-Y, atom (grupa) Y jest bardziej
elektroujemny. Powstały produkt może ulegać dalszej
reakcji z X-Y.
C C H
C
H
3
+ H
2
O
H
+
C C H
C
H
3
H
+
OH
2
:
C C H
C
H
3
H
OH
2
..
+
..
-H
+
C C H
C
H
3
H
OH
C O
C
H
3
C
H
3
enol
W środowisku kwaśnym do
alkinów ulega addycji woda.
Powstały enol jest nietrwały i
przekształca się do ketonu.
Inne reakcje:
Reakcje z borowodorem:
C
H
CH
2
C
H
3
3
+ BH
3
C
H
2
C
H
2
C
H
3
B
3
OH
-
C
H
3
C
H
2
CH
2
OH
Reakcje Polimeryzacji:
A
inicjator
wolnorodnikowy
C
H
2
C
H
*
*
A
n
A
inicjator
kationowy
C
H
2
C
H
*
A
*
n
A
inicjator
anionowy
C
H
2
C
H
*
A
*
n
AlEt
3
TiCl
4
*
C
H
2
*
n
Addycja borowodoru przebiega niezgodnie z regułą
Markownikowa
A=H, alkil, aryl, OOCCH
3
, Cl, CN
A= alkil, aryl
A =CN, Ph
Polimeryzacja Zieglera-Natty
C
H
3
CH
3
CH
3
C
H
2
CH
2
+
C
H
3
CH
3
CH
3
C
H
2
C
H
2
Nu
Nu
-
C
H
3
CH
3
CH
3
C
H
CH
2
C
H
3
CH
3
CH
3
CH
CH
3
+
C
H
3
CH
3
CH
3
CH
CH
3
Nu
C
H
3
CH
3
CH
CH
3
CH
3
+
C
H
3
CH
3
CH
3
CH
3
C
H
2
CH
3
CH
CH
3
CH
3
C
H
3
CH
3
CH
CH
3
CH
3
Nu
Nu
-
Nu
-
Reakcje, którym może
ulegać karbokation.
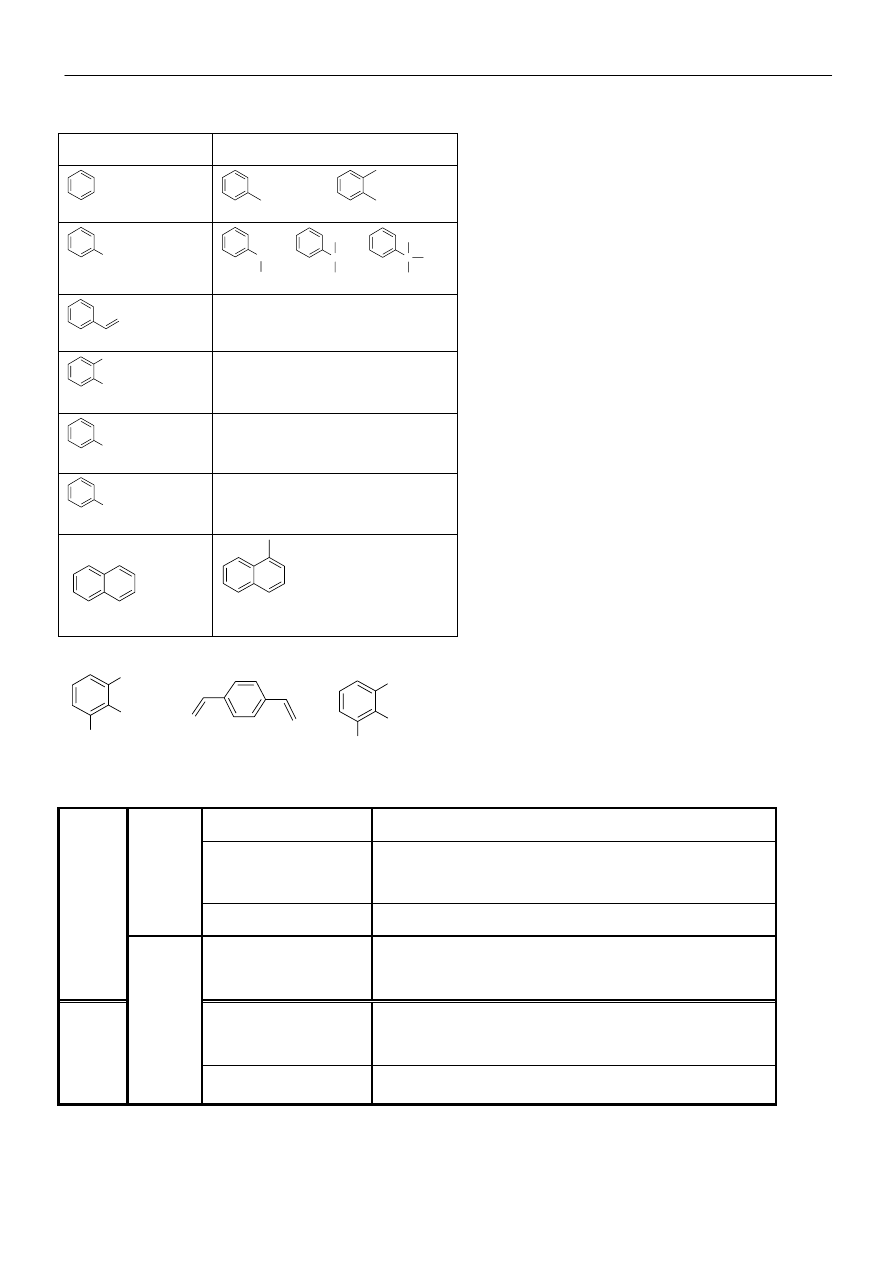
Materiały z chemii organicznej
kg
- 8 -
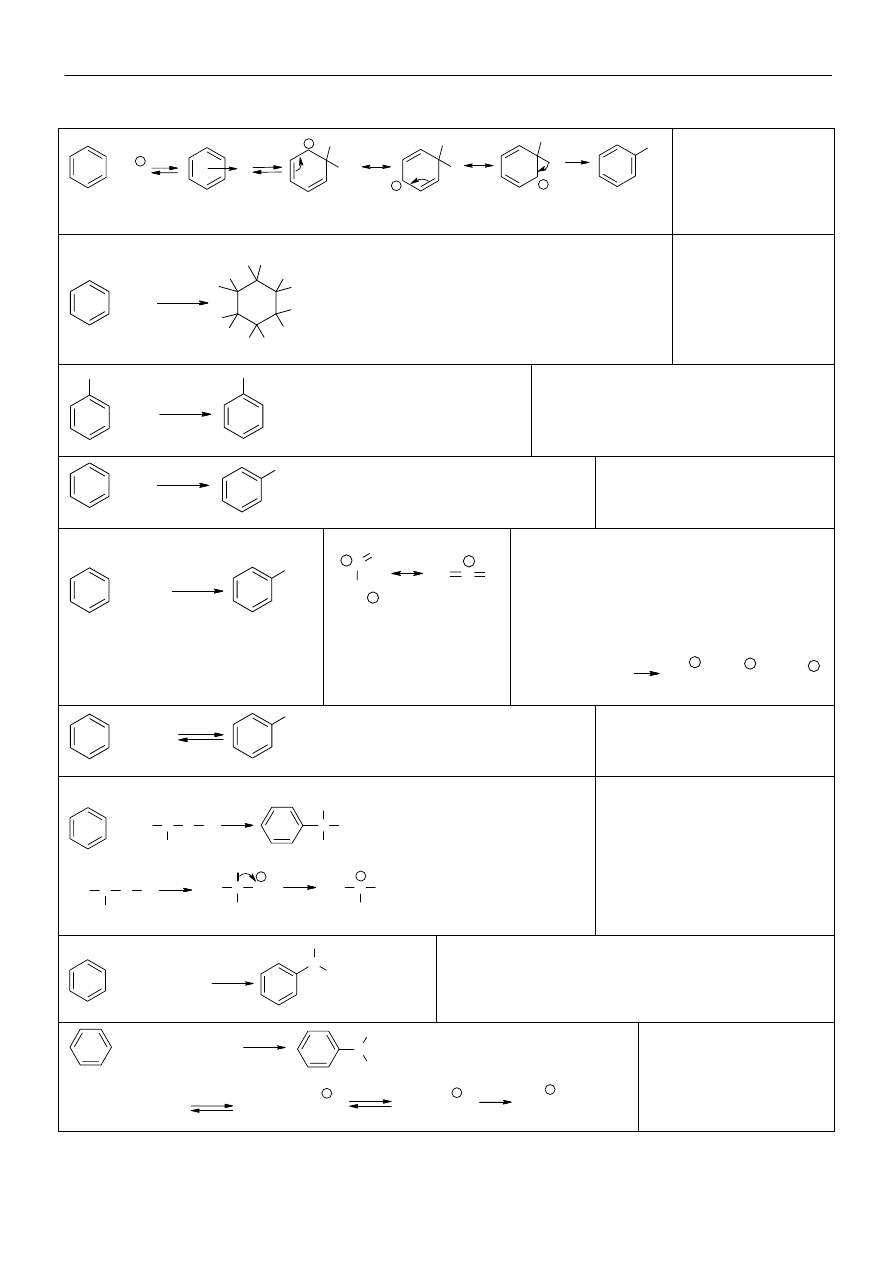
Powszechnie przyjęte nazwy zwyczajowe, oraz systematyczne dla węglowodorów aromatycznych (Ar):
Węglowodór
Grupa
benzen
fenyl (Ph) o-fenylen
CH
3
toluen
CH
2
C
CH
benzyl benzyliden benzylidyn
styren
CH
3
CH
3
o-ksylen
OH
fenol
NH
2
anilina
1
2
3
4
5
6
7
8
α
β
naftalen
1-naftyl,
α-naftyl
CH
3
CH
3
CH
3
CH
3
CH
3
Cl
3-chloro-o-ksylen 1,4-diwinylobenzen 1,2,3-trimetylobenzen
Wpływ podstawników na kierunek podstawienia elektrofilowego w benzenie
+I, +M
-O
-
+M > -I
-NR
2
, -NHR, -NH
2
, -OR, -OH, -NH-COR, -OCOR,
-SR, -CH=CH
2
, -C
6
H
5
,
aktywuj
ące
pi
er
ście
ń
+I, hiperkonjugacja -CR
3
, -CH
3
,
ki
er
uj
ące
w po
ło
żen
ie
-o i -p
-I > +M
-F, -Cl, -Br, -I, -CH
2
Cl, -CH=CH-COOH,
-CH=CH-NO
2
,
-M, -I
-CO-NH
2
, -COOR, -COOH, -COR, -CHO, -SO
3
H,
-CN, -NO
2
,
ki
er
uj
ące
w
po
ło
żenie -
m
dezaktywuj
ące pier
ście
ń
-I
-CCl
3
, -CF
3
, -NH
3
+
, -NR
3
+
,
Inne monocykliczne podstawione
węglowodory aromatyczne nazywane są
jako pochodne benzenu, albo też jako
pochodne jednego z pośród związków
wymienionych. Jeżeli podstawnik
wprowadzony do takiego związku jest
identyczny z jednym już obecnym, to
podstawiony związek nazywa się tak jak
pochodną benzenu.
Materiały z chemii organicznej
kg
- 9 -
Związki Aromatyczne
+ E +
E
E
H
+
E
H
+
E
H
+
E
-H
+
kompleks
π
kompleks
σ
Mechanizm reakcji
cząsteczki elektrofila ze
związkiem aromaty-
cznym. Kompleks
σ
stabilizowany jest przez
mezomerię.
Halogenowanie:
+ Cl
2
h
ν
H Cl
H
Cl
H
Cl
H
Cl
H
Cl
H
Cl
Areny w ciemności nie
reagują z fluorowczmi.
Jednak na świetle, lub w
obecności nadtlenków
następuje wolnorodni-
kowa addycja do wiązań
C=C.
CH
2
-CH
3
+ X
2
h
ν
CHX-CH
3
+HX
Jeżeli w pierścieniu obecny jest podstawnik
alkilowy, to na świetle lub w obecności
nadtlenków następuje wolnorodnikowa substy-
tycja fluorowca w pozycji benzylowej. X = Cl,
Br
+ X
2
Fe
X
+HX
W obecności kwasów Lewisa
fluorowce ulegają reakcji substytucji.
X = Cl, Br
Nitrowanie:
+ HNO
3
H
2
SO
4
NO
2
N
O
O
+
-
..
..
..
: :
2
N O
O
+
Areny ulegają łatwo nitrowaniu, w zależności od
reaktywności związku aromatycznego używa się
kwasu azotowego (V) (reaktywne), lub mieszaniny
nitrującej (mało reaktywne areny).
Czynnikiem nitrującym jest jon nitroniowy:
HNO
3
+ 2H
2
SO
4
NO
2
+ H
3
O + 2HSO
4
+
+
-
+ H
2
SO
4
SO
3
H
Sulfonowanie związków aroma-
tycznych jest reakcją odwracalną.
Alkilowanie:
C
H
3
C
H
CH
3
C
H
2
Cl
+
AlCl
3
C
CH
3
CH
3
CH
3
C
H
3
C
H
CH
3
C
H
2
Cl
AlCl
3
C
H
3
C
CH
3
CH
2
H
+
C
H
3
C
CH
3
CH
3
+
Alkilowanie metodą Friedla-Craftsa.
Reakcji alkilowania ulegają jedynie
aktywne zawiązki aromatyczne.
Reakcji tej towarzyszy prawie zawsze
przegrupowanie łańcucha.
+ CH
3
-CH=CH
2
H
3
PO
4
C
H CH
3
CH
3
Zamiast chlorowcopochodnych, do alkilowania pierścienia
związku aromatycznego można użyć alkenów.
W środowisku kwaśnym przyłączają one jon wodorowy z
wytworzeniem karbokationu, który atakuje pierścień arenu.
+ CH
3
-CH
2
-CH
2
-OH
H
3
PO
4
CH
CH
3
CH
3
CH
3
-CH
2
-CH
2
-OH
H
3
PO
4
CH
3
-CH
2
-CH
2
-OH
2
+
..
CH
3
-CH
2
-CH
2
+
CH
3
-CH-CH
3
+
-H
2
O
Karbokation można również
wytworzyć z alkoholu.
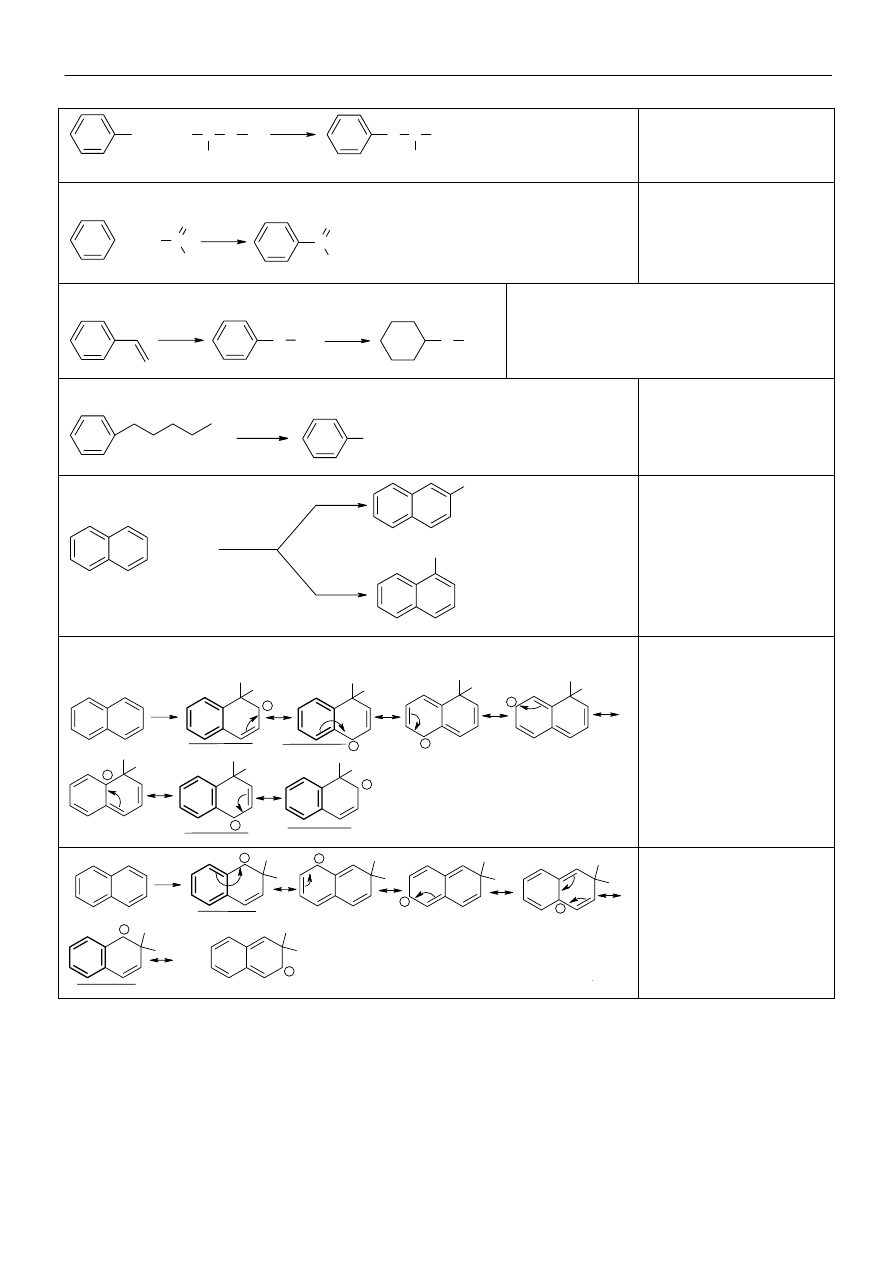
Materiały z chemii organicznej
kg
- 10 -
X +
C
H
3
C
H
CH
3
C
H
2
X
Na
C
H
2
C
H
CH
3
CH
3
Reakcja Wurtza Fittiga
syntezy alkiloarenów.
Acylowanie:
+ R C
X
O
C
R
O
AlCl
3
Reaktywne związki
aromoatyczne można łatwo
acylować metodą Friedla-
Craftsa.
X = Cl, Br, OR, R-COO
Redukcja:
H
2
/ Ni
20
o
C
C
H
2
CH
3
C
H
2
CH
3
H
2
/ Ni
100
o
C
Pierścień aromatyczny ulega trudniej redukcji od
wiązań C=C w alkenach. Jednak w ostrzejszych
warunkach możliwa jest również jego redukcja.
Utlenianie:
KMnO
4
/ H
+
COOH
+ CH
3
-CH
2
-CH
2
-COOH
Silne utleniacze utleniają
alkiloareny do kwasów.
Rozerwaniu ulega zawsze
wiązanie C-C w pozycji
C1-C2.
+ H
2
SO
4
SO
3
H
SO
3
H
100
o
C
160
o
C
Sulfonwanie związków
aromatycznych, w przeci-
wieństwie do nitrowania czy
chlorowania jest reakcją
odwracalną. W niższych
temperaturach powstaje
produkt kinetyczny, a w
wyższych termodynamiczny.
E
+
E
H
+
E
H
+
E
H
+
E
H
+
E
H
+
E
H
+
E
H
+
Kompleks
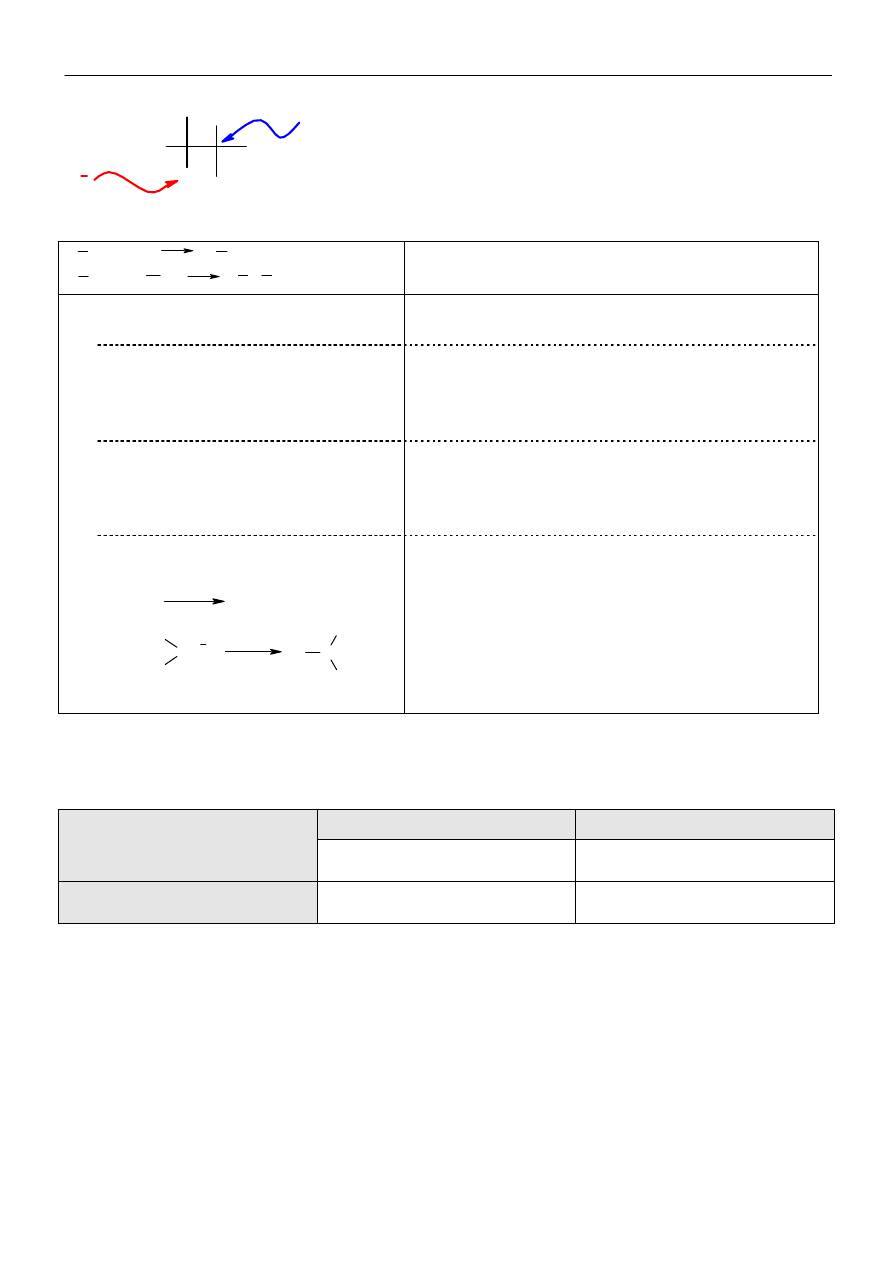
σ powstały w
wyniku ataku cząsteczki
elektrofila na pozycję
α w
naftalenie jest trwalszy od
kompleksu
σ powstałego w
wyniku ataku elektrofola na
pozycję
β.
E
+
H
E
+
H
E
+
H
E
+
H
E
+
H
E
+
H
E
+
Materiały z chemii organicznej
kg
- 11 -
R
H
X
:Nu
B
Centra reaktywne w halogenoalkanach
Podstawienie nukleofilowe przy nasyconym atomie węgla:
R OH + HX
R X + H
2
O
R OH +R'
OH
H
+
R O R' + H
2
O
estryfikacja alkoholi za pomocą kwasów chlorowcopochodnych i
innych kwasów nieorganicznych. Synteza eterów.
+ OH
-
→ R-OH + X
-
+
-
OR’
→ R-OR’ + X
-
hydroliza alkaliczna
synteza eterów Williamsona
+ R’COO
-
→ RO-COR’ + X
-
+
-
SH
→ R-SH + X
-
+
-
SR’
→ R-SR’ + X
-
+ SR’
2
→ R-S
+
R’
2
+ X
-
synteza estrów kwasów karboksylowych
synteza merkaptanów
synteza tioeterów
tworzenie związków sulfoniowych
+ R’NH
2
→ RR’NH + HX
+ NR’
3
→ RN
+
R’
3
+ X
-
+ NO
2
-
→ R-NO
2
+ X
-
+ X’
-
→ R-X’ + X
-
alkilowanie amin
czwartorzędowanie amin
synteza nitroalkanów (oraz azotynów R-O-NO)
reakcja Finkelsteina
R-X
+ CN
-
→ R-CN + X
-
+
-
C
≡
CH
→ R-C
≡
CH + X
-
+ C
6
H
6
AlCl
3
C
6
H
5
-R + H
-
CH
COR
COR
+
R
CH
COR
COR
synteza nitrylów Kolbego (oraz izonitrylów R-NC)
synteza alkinów
alkilowanie Fridela i Craftsa
alkilowanie związków
β-dwukarbonylowych
X = -Cl, -Br, -I, siarczan dwu i monoalkilowy, tosylan
Przykłady twardych i miękkich kwasów i zasad:
ZASADY
KWASY
MIĘKKIE
R
2
S, RSH, RS
-
, I
-
, SCN
-
, Br
-
, R
3
P, CN
-
,
alkeny, areny, H
-
, R
-
Ag
+
, Hg
2+
, RS
+
, I
+
, Br
+
, I
2
, Br
2
TWARDE
H
2
O, OH
-
, F
-
, RCOO
-
, SO
4
-2
, CO
3
-2
,
ROH, RO
-
, R
2
O, NH
3
, RNH
2
H
+
, Al
3+
, BF
3
, AlCl
3
, AlR
3
, SO
3
, RCO
+
,
CO
2
Zasady określa się jako miękkie wtedy, gdy atom, który jest donorem pary elektronów, wykazuje dużą
polaryzowalność, łatwo się utlenia i odznacza się małą elektroujemnością.
Kwasy określa się jako miękkie wówczas, gdy atom, który jest akceptorem pary elektronów, ma stosunkowo duże
rozmiary, niewielki ładunek dodatni (może też nie mieć ładunku) i dysponuje elektronami na zewnętrznej orbicie.
Korzystając z pojęć twardości i miękkości można sformułować ogólną regułę mającą zastosowanie we wszystkich
przypadkach kwasów i zasad: w układach w których istnieje możliwość wyboru, twarde kwasy reagują najchętniej z twardymi
zasadami, a miękkie kwasy z miękkimi zasadami. Związki twardych kwasów z twardymi zasadami i miękkich z miękkimi są
trwalsze i łatwiej się tworzą od związków twardych kwasów z miękkimi zasadami lub miękkich kwasów z twardymi zasadami.
Reguła ta jest pomocna w przewidywaniu kierunku reakcji, w której uczestniczą kwasy i zasady.
Materiały z chemii organicznej
kg
- 12 -
Typ
reakcji
Stereochemia reakcji
Warunki reakcji
S
N
2
R-CH
2
-X
R-CHX-R’
Przebiega z inwwersją konfiguracji, atak
cząsteczki nukleofila z przeciwnej strony do
grupy odchodzącej
Rozpuszczalniki aprotyczne o dużej stałej
ε.
Cząsteczki o wysokiej nukleofilowości.
E2
R-CH
2
-X
R-CHX-R’
Podstawniki ulegające odszczepieniu muszą
znajdować się w położeniu trans (reguła
Ingolda). Gdy X jest grupą obojętną
elektrycznie produktem głównym jest alken
bardziej podstawiony (produkt Zajcewa). W
przypadku gdy X jest grupą obdarzoną
ładunkiem dodatnim, produktem głównym jest
alken powstający wg reguły Hoffmana.
Silne zasady o małej nukleofilowości.
Rozpuszczalniki aprotyczne o małej stałej
ε.
Zasady o dużych cząsteczkach sprzyjają
powstawaniu alkenu mniej podstawionemu
(Hoffmana)
S
N
1
R
3
C-X
R-CHX-R’
Następuje racemizacja
Rozpuszczalniki protyczne o dużej
ε (woda,
alkohole, kwasy), niska temperatura reakcji.
Utworzenie karbokationów stabilizowanych
przez mezomerię (allilowe, benzylowe).
E1
R
3
C-X
R-CHX-R’
Rozpuszczalniki protyczne o dużej
ε (woda,
alkohole, kwasy), ogrzewanie mieszaniny
reakcyjnej. Utworzenie karbokationów stabili-
zowanych przez mezomerię (allilowe,
benzylowe).
X =
F, Cl, Br, I, OH, SO
2
Ph (tosylany)
+
NR
3
,
+
SR
2
X
D
E
A
δ-
δ-
Nu:
-
D
E
A
Nu
S
N
2 - inwersja konfiguracji
X
D
E
A
D
E
A
Nu:
-
Nu
D
E
A
D
E
A
Nu
S
N
1 - racemizacja
X
H
A
D
D
A
B
-
D
A
A
D
=
D
A
A
D
E2
X
Nu
D E
A
+
inwersja konfiguracji
retencja konfiguracji

Materiały z chemii organicznej
kg
- 13 -
Związki metaloorganiczne R-Me
Wiązanie C-Me
jonowe
kowalencyjne silnie spolaryzowane
kowalencyjne
R-Na alkilosód
R-K alkilopotas
R-Li alkilolit
R-MgX halogenek alkilomagnezowy
R
3
Al trialkiloglin
R
4
Sn tetralkilocyna
R
3
SnH wodorek trialkilocyny
R
3
SnX halogenek trialkilocyny
R
4
Pb tetralkiloołów
RHgX halogenek alkilortęciowy
R
2
Hg dialkilortęć
Metody syntezy związków metaloorganicznych:
• halogenek alkilu z metalem (wymiana chlorowca na metal)
R-X
+ Na
+ K
+ Li
+ Mg
+ Zn
+ Hg
→ R-Na + NaX
→ R-K + KX
→ R-Li + LiX
→ R-Mg-X
→ R-Zn-X
→ R-Hg-X R= allil, benzyl, X=I,Br
• związek metaloorganiczny z metalem (wymiana metalu na inny metal)
3R
2
Hg + Al
→ 2R
3
Al + 3Hg
R
2
Hg + 2Me
→ 2R-Me + Hg Me = K, Na, Li
• reaktywny związek metaloorganiczny z halogenkiem metalu
2R-Mg-X + HgX
2
→ R
2
Hg + 2MhX
2
4R-MgX + SnX
4
→ R
4
Sn + 4MgX
2
2R-MgX + ZnX
2
→ R
2
Zn + 2MgX
2
• metalacja węglowodorów
CH
3
-CH
2
-CH
2
-CH
2
-Na + C
6
H
6
→ C
4
H
10
+ C
6
H
5
Na
Zastosowanie związków metaloorganicznych:
• związki sodo- , potaso- i litoorganiczne wykorzystywane są najczęściej jako silne zasady w celu utworzenia C-nukleofila
(metalacji), który następnie może reagować np. z halogenkiem alkilu
R-Me + R’-H
→ R’-Me
R’-Me + R”-X
→ R’-R”
• związki magnezoorganiczne
synteza alkoholi: R-MgX + R’-CO-R”
→ R’-CRR”-OMgX
synteza kwasów: R-MgX + CO
2
→ RCOOMgX
synteza innych związków metaloorganicznych
• związki glinoorganiczne – jako katalizatory podczas polimeryzacji
• związki rtęcioorganiczne – synteza alkoholi
C CH
2
C
H
3
C
2
H
5
+ (CH
3
COO)
2
Hg
C CH
2
CH
3
C
2
H
5
Hg-OOCCH
3
OH
NaBH
4
/OH-
C CH
3
CH
3
C
2
H
5
OH
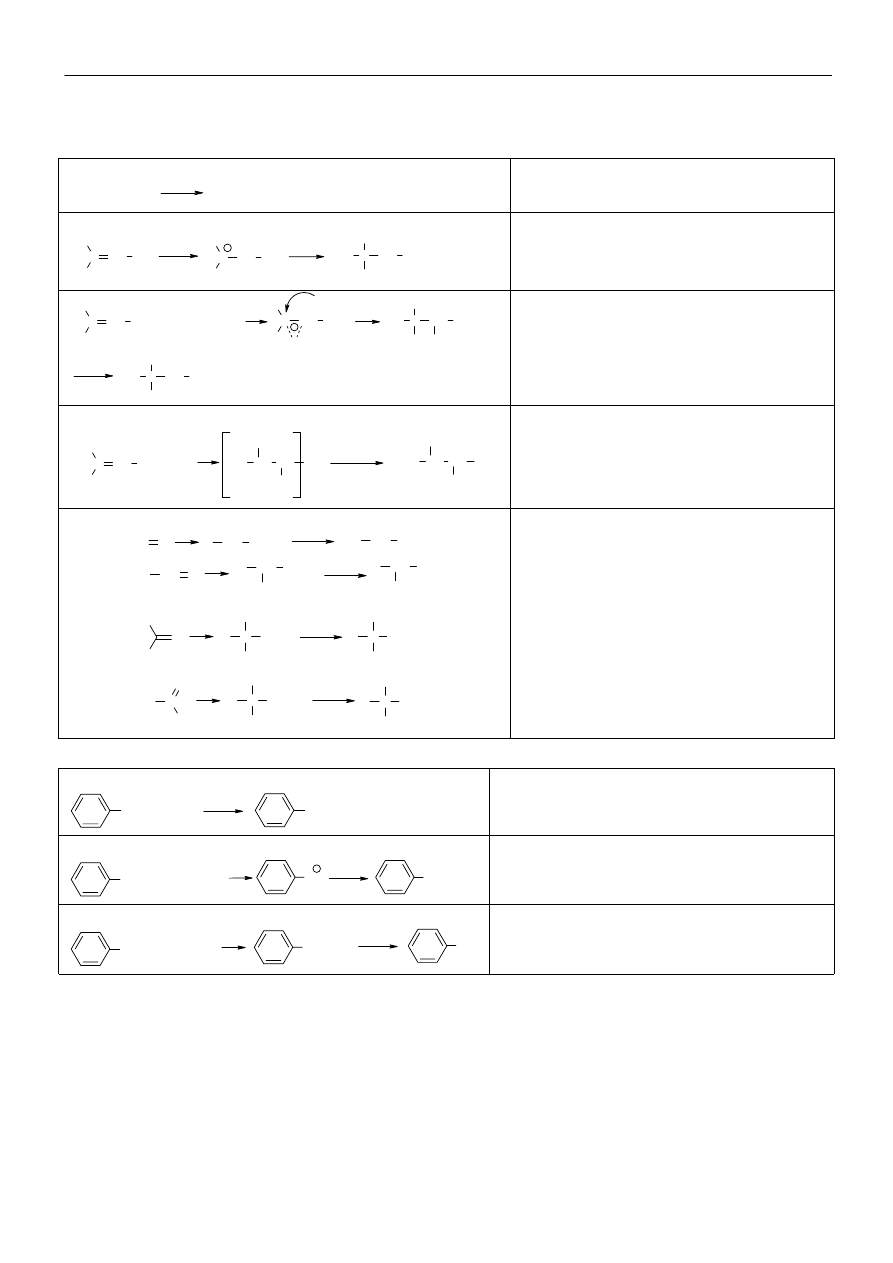
Materiały z chemii organicznej
kg
- 14 -
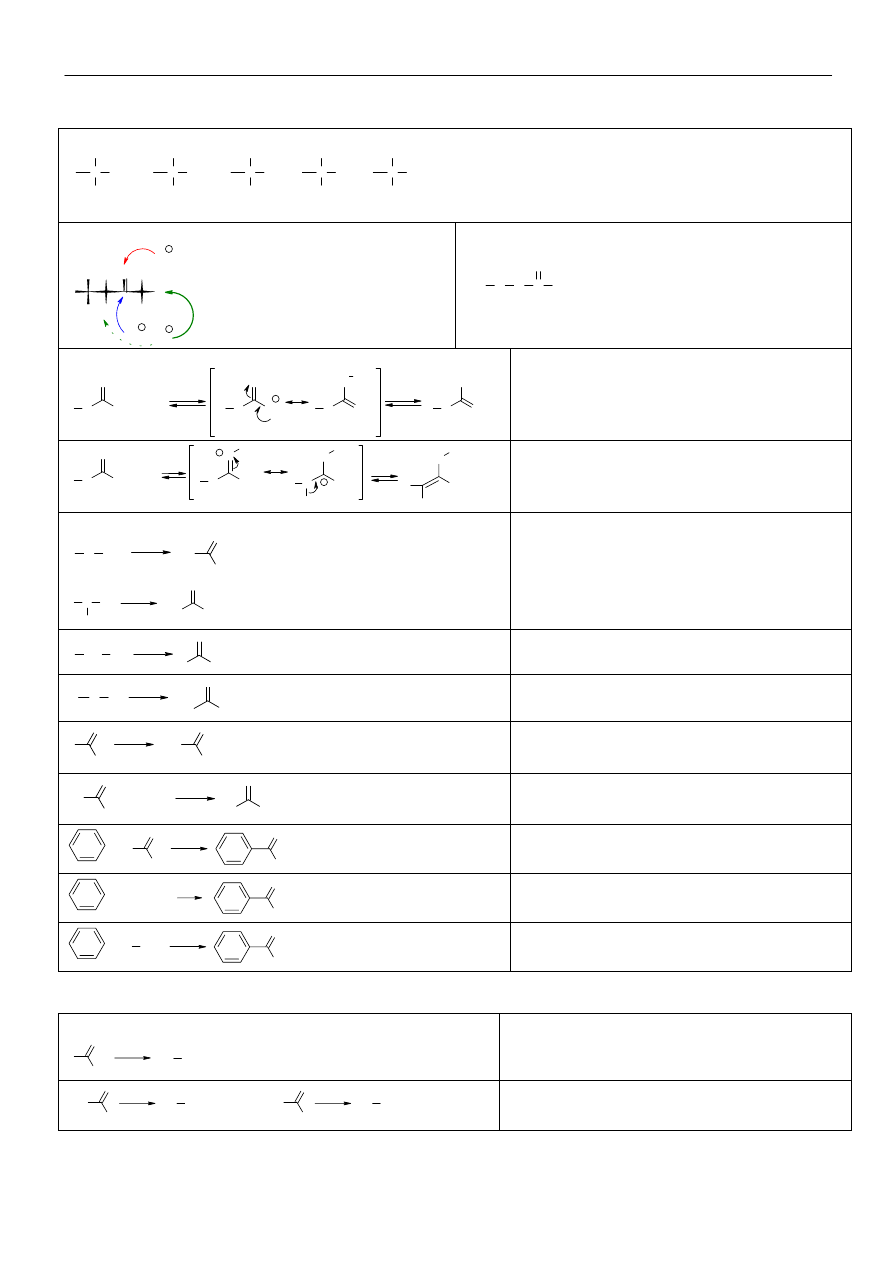
Alkohole i fenole.
Metody syntezy alkoholi:
hydroliza fluorowcopochodnych:
CH
3
-CH
2
-CH
2
-X
H
2
O
OH
-
CH
3
-CH
2
-CH
2
-OH
dla fluorowcopochodnych I
o
reakcja hydrolizy
przebiega wg mechanizmu S
N
2, dla III
o
wg
mechanizmu S
N
1.
hydratacja alkenów:
C CH
C
H
3
C
H
3
CH
3
H
2
SO
4
C CH
2
C
H
3
C
H
3
CH
3
+
H
2
O
-H
+
C CH
2
CH
3
C
H
3
CH
3
OH
addycja wody zgodnie z regułą Markownikowa.
Przejściowo tworzy się karbokation, który może
ulec izomeryzacji.
+
C CH
C
H
3
C
H
3
CH
3
C CH
CH
3
C
H
3
CH
3
OH Hg-OOCCH
3
+ (CH
3
COO)
2
Hg
C CH
C
H
3
C
H
3
CH
3
Hg-OOCCH
3
OH
2
NaBH
4
OH
-
C CH
2
CH
3
C
H
3
CH
3
OH
-H
+
dobre wydajności, addycja wody zgodnie z regułą
Markownikowa, bez izomeryzacji łańcuch
węglowego ponieważ w stanie przejściowym
tworzy się kompleks
σ.
borowodorowanie alkenów:
C CH
C
H
3
C
H
3
CH
3
+ BH
3
3
CH CH
CH
3
C
H
3
CH
3
B
3
CH CH
CH
3
C
H
3
CH
3
OH
H
2
O
2
/ OH-
3
addycja wody przebiega niezgodnie z regułą
Markownikowa (atak cząsteczki borowodoru na
wiązanie
π, a następnie przeniesienie jonu
wodorkowego).
addycja związków magnezoorganicznych do związków karbonylowych:
C
H
2
O
C
H
3
CH O
O
C
H
3
C
H
3
R-Mg-X +
R-Mg-X +
R-Mg-X +
R-Mg-X +
CH
2
OMgX
R
CH OMgX
R
CH
3
C OMgX
R
CH
3
CH
3
H
2
O
H
2
O
H
2
O
H
2
O
CH
2
OH
R
CH OH
R
CH
3
C OH
R
CH
3
CH
3
C
H
3
C
O
OMe
C OMgX
R
CH
3
R
C OH
R
CH
3
R
I
o
alkohole w reakcji związku magnezo-
organicznego z formaldehydem
II
o
alkohole w reakcji związku magnezo-
organicznego z aldehydami
III
o
alkohole w reakcji związku magnezo-
organicznego z ketonami i estrami.
metody syntezy fenoli:
hydroliza kwasów sulfonowych:
SO
3
H
+ NaOH
300
o
C
OH
zagotowanie soli diazoniowych:
NH
2
+ NaNO
2
+ H
+
N
2
+
H
2
O
OH
∆
użycie związków boroorganicznych:
MgX + B(OCH
3
)
3
B(OCH
3
)
2
H
2
O
2
/H
+
OH

Materiały z chemii organicznej
kg
- 15 -
Alkohole i fenole.
Właściwości chemiczne alkoholi i fenoli:
R=alkil,
alkohol
R=aryl,
fenol
1. Reakcje z zachowaniem wiązania C-O
R OH + Me
R O
-
Me
+
-H
2
R'X
R-O-R'
-MeX
Wykorzystywanie kwasowych
właściwości alkoholi i fenol
Me=Na, K, Mg, Al; R’=alkil
+
+
OH
Ar
+ NaOH
Ar O
-
Me
+
-H
2
O
R'X
Ar-O-R'
-MeX
Synteza eterów Williamsona
-
+
R
OH
O
R
1
OH +
H
+
R
OH
O
+
H
R
1
OH
R
OH
O
O
+
R
1
H
H
-H
2
O
R
OR
1
O
+
H
R
OR
1
O
-H
+
Estryfikacja alkoholi
+
-
R'-OH + R
X
O
NEt
3
R
OR'
O
Reakcje alkoholi i fenoli z
chlorkami kwasowymi i
bezwodnikami. X=Cl, R’-COO
+
+
2. Wymiana grupy OH na X
R-OH + HX
R O
H
H
+
X
-
X-R + H
2
O
HX= HCl, HBr, HI
I
o
alkohole reagują wg
mechanizmu S
N
2, II i III
o
wg
mechanizmu S
N
1.
+
-
H
C
H
3
C
2
H
5
OH + SOCl
2
-HCl
H
C
H
3
H
5
C
2
O
S
Cl
O
H
C
H
3
C
2
H
5
Cl
Reakcja przebiega wg
mechanizmu S
N
i – z retencją
konfiguracji
+
-
H
C
H
3
C
2
H
5
OH + SOCl
2
pirydyna
H
C
H
3
H
5
C
2
O
S
Cl
O
Cl
C
H
3
C
2
H
5
H
Cl
-
Reakcja przebiega wg
mechanizmu S
N
2 – z inwersją
konfiguracji
+
-
R-OH + PX
3
→ R-X + H
3
PO
3
Synteza halogenków
+
-
R-OH + PPh
3
Br
2
→ R-X + POPh
3
+ HBr
+
+
Eliminacja H
2
O
CH CH
2
C
H
3
C
H
3
CH
2
OH
H
+
-H
2
O
CH CH
2
C
H
3
C
H
3
CH
2
+
CH CH
C
H
3
C
H
3
CH
2
CH CH
C
H
3
C
H
3
CH
3
+
C
CH
C
H
3
C
H
3
CH
3
C
CH
2
C
H
3
C
H
3
CH
3
+
Eliminacji wody z alkoholi
towarzyszą często
przegrupowania szkieletu
węglowego (Wagnera -
Meerweina)
+
-
CH CH
2
C
H
3
C
H
3
CH
2
OH
CH CH
C
H
3
C
H
3
CH
2
+ CS
2
KOH
CH CH
2
C
H
3
C
H
3
CH
2
O
C
S
S
-
K
+
CH
3
I
CH C
C
H
3
C
H
3
CH
2
O
C
S
SCH
3
H
H
+ CH
3
SH + COS
Reakcja eliminacji Czugajewa
(eliminacja cis)
+
-
Reakcje utleniania alkoholi i fenoli
R CH
2
OH
[O]
R COOH
R CH
2
OH
[O]
R CHO
R CH OH
R'
[O]
O
R'
R
OH
[O]
O
O
[O]=KMnO
4
/H
+
; CrO
3
/H
+
[O]=MnO
2
; CrO
3
/pirydyna
dowolny utleniacz
[O]=MnO
2
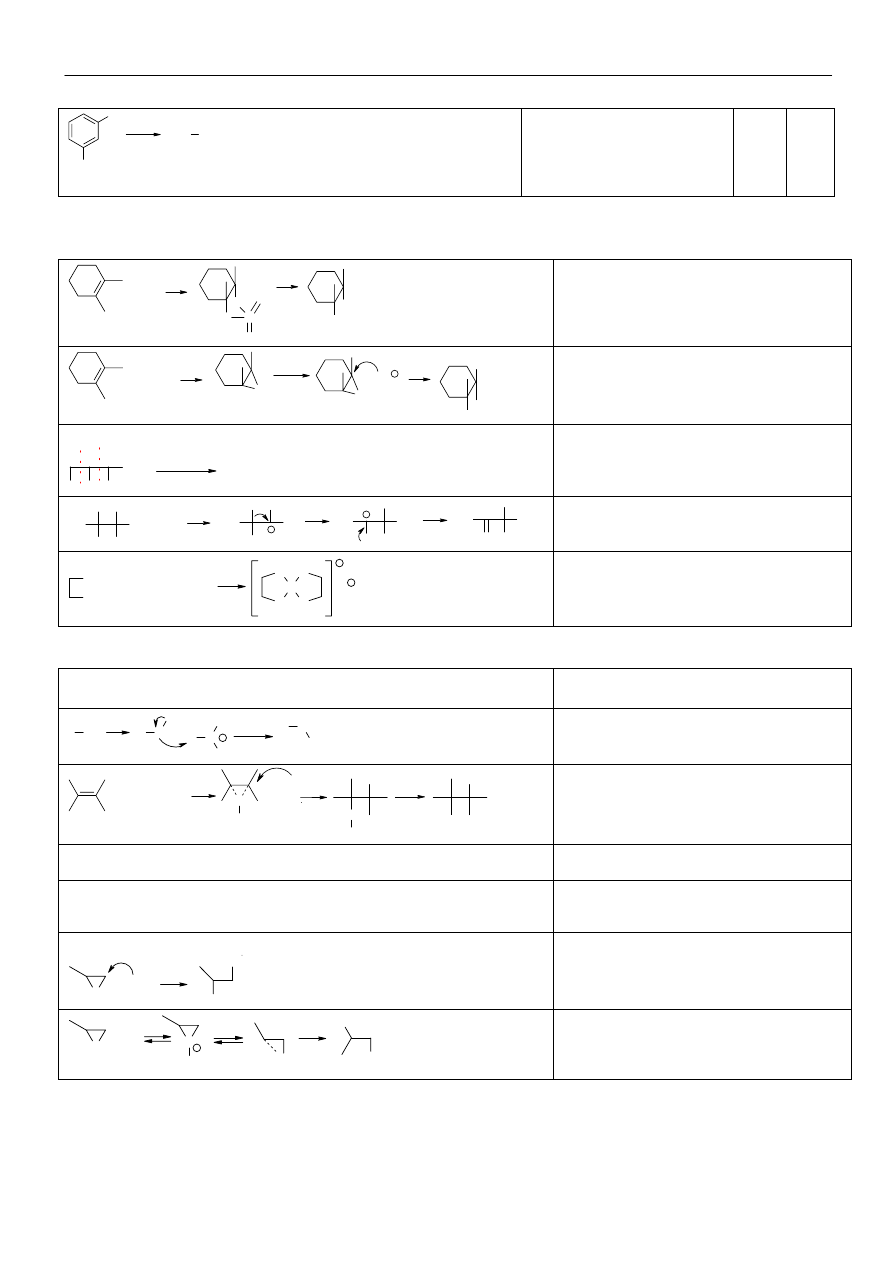
Materiały z chemii organicznej
kg
- 16 -
OH
CH
3
[O]
C
H
3
COOH
[O]=KMnO
4
/H
+
; CrO
3
/H
+
Alkohole wielowodorotlenowe
Otrzymywanie
+ KMnO
4
O
O
Mn
O
O
OH
OH
Synteza cis dioli
+ RCOOOH
O
H
2
O/OH
-
O
OH
-
OH
OH
Synteza trans dioli, reakcja otwarcia pierścienia
przebiega wg mechanizmu S
N
2
Właściwości chemiczne
CH
3
OH OH OH
HIO
4
(CH
3
COO)
4
Pb
CH
2
O + CHOOH + CH
3
-CHO
reakcji ulegają jedynie vic-polialkohole
C
H
3
OH
CH
3
CH
3
OH
H
+ H
+
-H
2
O
C
H
3
OH
CH
3
CH
3
H
+
C
H
3
OH
CH
3
CH
3
H
+
-H
+
C
H
3
O
CH
3
CH
3
H
Przegrupowanie pinakolinowe (pinakolonowe),
ulegają tylko vic-diole. Łatwość migracji
grupy: H>aryl>alkil
OH
OH
+ H
3
BO
3
(Na
2
B
4
O
7
)
O
B
O
O
O
-
H
+
Reakcji ulegają jedynie 1,2 lub 1,3-diole, oraz
cis-diole (cykloalkanodiole)
Etery
Metody syntezy
R-O-Me + R’-X
→ R-O-R’ + MeX
Metoda Williamsona, dobre wydajności tylko
dla halogenków I
o
R OH
H
+
-H
2
O
-H
+
R O
H
H
R O
H
R O
R
+
dehydratacja alkoholi, dobre wydajności
jedynie dla alkoholi I
o
, I i III
o
lub I i II
o
+ (CF
3
COO)
2
Hg
R-OH
Hg
OOCCF
3
HO-R
Hg
OR
OOCCF
3
-H
+
OR
NaBH
4
OH
-
alkoksyrtęciowanie alkenów
R-OH + CH
2
N
2
→ R-O-CH
3
+ N
2
Reakcja z diazometanem, pozwala tylko na
syntezę eterów metylowych
Właściwości chemiczne eterów
R-O-R’ HI
→ R-OH + R’-I (w przypadku nadmiaru HI, R-I + R’I)
Rozpad kwasowy eterów, gdy R=Ar tworzy się
wyłącznie fenol, z dwóch możliwych alkoholi
powstaje ten o niższej rzędowości
Właściwości chemiczne oksiranów
O
Nu-H
OH
Nu
W środowisku zasadowym lub obojętnym
cząsteczka nukleofila przyłącza się do węgla
mniej osłoniętego, reakcja wg mechanizmu
S
N
2.
O
+ H
+
O
H
+
O
Nu-H
-H
+
OH
Nu
δ+
W środowisku kwaśnym cząsteczka nukleofila
przyłącza się do węgla bardziej osłoniętego,
reakcja wg mechanizmu S
N
1, bez wytworzenia
typowego karbokationu.
Materiały z chemii organicznej
kg
- 17 -
Związki karbonylowe
Nietrwałe układy geminalne:
R
1
C
OH
OH
R
2
R
1
C
OH
NHR
R
2
R
1
C
OH
X
R
2
R
1
C
NHR
NHR
R
2
R
1
C
NHR
X
R
2
R
1
= H, alkil, aryl; R
2
= H, alkil, aryl; R=H, alkil, aryl
Miejsca reaktywne w związkach karbonylowych:
O
H
H
H
H
R
H
H
H
-
OH
-
Nu
+
E
Oznaczanie atomów węgla i wodoru w zależności od odległości
od grupy funkcyjnej:
C
H
3
C
H
2
C
H
2
C
O
R
α
β
γ
Równowaga keto-enolowa katalizowana zasadą:
+ OH
-
-H
2
O
O
C
H
2
CH
3
R
O
C
H
2
CH
2
R
..
-
H
2
O
OH
C
H
2
CH
2
R
O
C
H
2
CH
2
R
Najwolniejszym etapem reakcji jest reakcja oderwania
protonu od związku karbonylowego.
+ H
+
O
C
H
2
CH
3
R
O
C
H
2
CH
3
R
H
+
O
C
H
CH
3
R
H
H
+
-H
+
O
CH
3
R
H
H
Metody syntezy:
R C
H
2
OH
MnO
2
R
H
O
R C
H
OH
R'
K
2
Cr
2
O
7
/H
+
O
R
R'
Utlenianie alkoholi I
o
prowadzi do aldehydów
Utlenianie alkoholi II
o
prowadzi do ketonów
R CX
2
R'
H
2
O
O
R
R'
Hydroliza gem-dihalogenopochodnych.
Ar
C
H
2
R'
CrO
3
AcOAc/H
+
O
Ar
R'
Utlenianie pochodnych alkilobenzenu
R
Cl
O
H
2
Pd, BaSO
4
R
H
O
Katalityczna redukcja chlorków kwasowych na
zdezaktywowanym (zatrutym) katalizatorze (BaSO
4
)
reakcja Rosenmunda
R
Cl
O
+ R'
2
Cd
O
R
R'
2
2
Reakcja chlorków kwasowych ze związkami
kadmoorganicznymi
R
X
O
+
AlCl
3
R
O
-HCl
Reakcja Fridela-Craftsa (benzen i inne uaktywnione
pochodne aromatyczne)
+ CO + HCl
H
O
Reakcja Gattermanna-Kocha
R CN
+
1. HCl
R
O
2. H
2
O
Reakcje związków karbonylowych:
Reakcje utlenienia i redukcji:
R
H
O
[O]
R COOH
Utlenianie aldehydów zachodzi bardzo łatwo.
[O]= K
2
Cr
2
O
7
/H+; KMnO
4
/H
+
lub OH
-
; Br
2
a)
R
H
O
Ag(NH
3
)
2
+
R COO
-
+ Ag
b)
R
H
O
Cu(OH)
2
R COO
-
+Cu
2
O
Reakcje: a)Tollensa oraz b)Fehlinga
służą do odróżnienia aldehydów od ketonów
Materiały z chemii organicznej
kg
- 18 -
R
CH
3
O
SeO
2
R
O
H
O
Reakcja z dwutlenkiem selenu, ulegają jej związki
karbonylowe posiadające wodory w położeniu
α. Służy
do syntezy związków
α−β dikarbonylowych.
O
H
+
OH
KMnO
4
O
COOH
Utlenianie ketonów zachodzi tylko wobec energicznych
środków utleniających. W środowisku kwaśnym
powstaje enol trwalszy termodynamicznie.
O
H
2
/Pt
OH
Grupa karbonylowa, wodorem na platynie, ulega
trudniej redukcji od wiązania podwójnego C=C.
O
LiAlH
4
/eter
lub NaBH
4
/H
2
O
OH
Redukcje grupy karbonylowej bez naruszenia wiązań
C=C można przeprowadzić glinowodorkiem litu (LAH)
lub borowodorkiem sodu.
O
CH
3
NaBH
4
/H
2
O
CH
3
OH
H
CH
3
OH
=
Podczas redukcji borowodorkiem lub LAH atak jonu
wodorkowego następuje od strony mniej zatłoczonej –
reakcja stereoselektywna.
O
CH
3
Me/C
2
H
5
OH
CH
3
OH
H
OH
CH
3
=
Redukcja metalem, Me = Na, Mg/Hg; Zn/Hg. W reakcji
powstaje przejściowo termodynamicznie trwalszy
karboanion. Reakcja przebiega w obecności
rozpuszczalników protycznych.
O
Mg/Hg
benzen
2
O
O
.
.
Mg
+2
O O
Mg
+2
H
2
O
OH OH
W przypadku gdy reakcje prowadzi się w
rozpuszczalniku aprotycznym, karboanion nie może
powstać, następuje dimeryzacja anionorodników i
tworzą się pinakole.
H
O
CH
3
C
H
3
CH
3
+ OH
-
H
O
CH
3
C
H
3
CH
3
OH
H
O
CH
3
C
H
3
CH
3
OH
O
CH
3
C
H
3
CH
3
H
O
CH
3
C
H
3
CH
3
H
O
O
CH
3
C
H
3
CH
3
H
OH
CH
3
C
H
3
CH
3
H
Reakcja dysproporcjonowania aldehydów – reakcja
Cannizzaro. Reakcji tej ulegają tylko aldehydy nie
posiadające wodorów w pozycji
α.
Reakcje nukleofilowej addycji do grupy karbonylowej:
+ :Nu
-
O
R'
R"
R'
R"
O
Nu
R'
R"
O
Nu
+
+
R'
R"
OH
Nu
R'
R"
OH
Nu
H
2
O
-OH
-
Reakcje w środowisku alkalicznym
+ :Nu-H
O
+
R'
R"
H
R'
R"
OH
Nu
H
R'
R"
OH
Nu
H
+
+
R'
R"
OH
Nu
R'
R"
OH
Nu
-H
+
+
+
Reakcje w środowisku kwaśnym
Odczynnik nukleofilowy może atakować węgiel
karbonylowy z obydwu z jednakowym
prawdopodobieństwem.
O
R'
R"
+ R-OH
H
+
lub OH
-
OH
R'
R"
OR
H
+
OR
R'
R"
OR
tworzenie hemiacetali (hemiketali) oraz acetali (ketali)
O
R'
R"
+ CN
-
OH
-
OH
R'
R"
CN
Tworzenie cyjanohydryn
O
R'
R"
+ NaHSO
3
OH
R'
R"
SO
3
Na
Tworzenie soli kwasów
α-hydroksysulfonowych.
H
O
Ph
O
H
Ph
CN
OH
Ph
CN
-
H
O
Ph
OH O
H
Ph
Ph
CN
O OH
H
Ph
Ph
CN
O OH
H
Ph
Ph
NaCN(kat.)
:
Kondensacja
benzoinowa
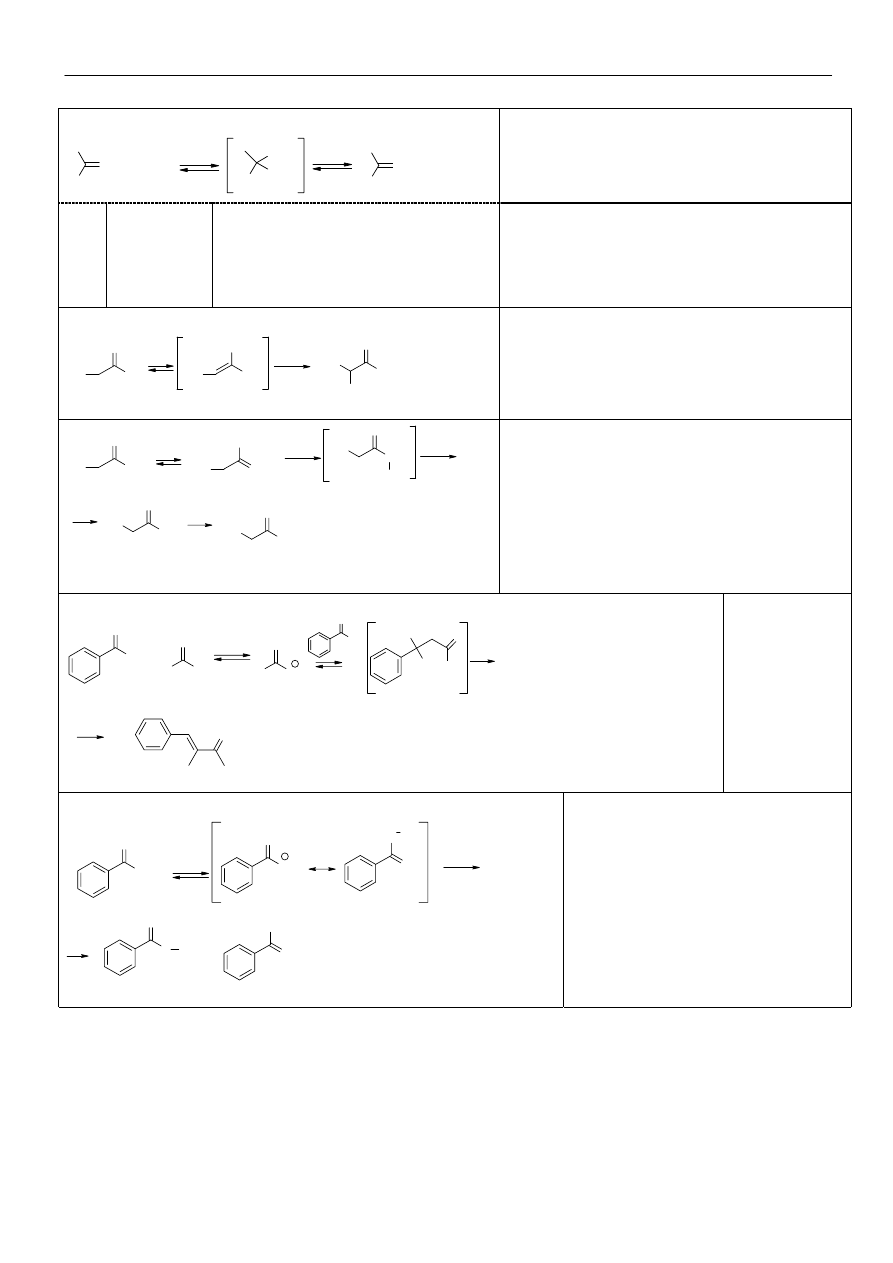
Materiały z chemii organicznej
kg
- 19 -
Reakcje z pochodnymi amin typu H
2
N-Y:
O
R'
R"
+ H
2
N-Y
H
+
R'
R"
OH
NH-Y
-H
2
O
N-Y
R'
R"
Y=:
OH
R
NH
2
NH-Ph
NH-CO-NH
2
hydroksyloamina
amina
hydrazyna
fenylohydrazyna
semikarbazyd
oksym
imina
hydrazon,
azyna (>C=N-N=C<)
fenylohydrazon
semikarbazon
Halogenowanie związków karbonylowych:
O
CH
3
C
H
3
H
+
OH
CH
3
C
H
3
X
2
O
CH
3
C
H
3
X
W
środowisku kwaśnym tworzy się enol
termodynamicznie trwalszy. Po wprowadzeniu jednego
atomu fluorowca, dalsza reakcja przebiega trudniej.
O
CH
3
C
H
3
OH
CH
2
C
H
3
OH
-
X
2
O
CH
2
C
H
3
X
X
2
O
CX
3
C
H
3
OH
-
O
O- + HCX
3
C
H
3
W środowisku alkalicznym reagują protony bardziej
kwaśne. Powstały a-halogenoketon enolizuje jeszcze
łatwiej niż wyjściowy keton. Reakcji nie można
zatrzymać na etapie monohalogenoketonu. Biegnie aż
do utworzenia trihalogenopochodnej, a następnie tworzy
się sól kwasu karboksylowego i haloform. Reakcja ta
nosi nazwę reakcji haloformowej. Ulegają jej tylko
metyloketony, acetaldehyd, etanol i
metyloalkilokarbinole (po utlenieniu halogenem do
metyloalkiloketonu).
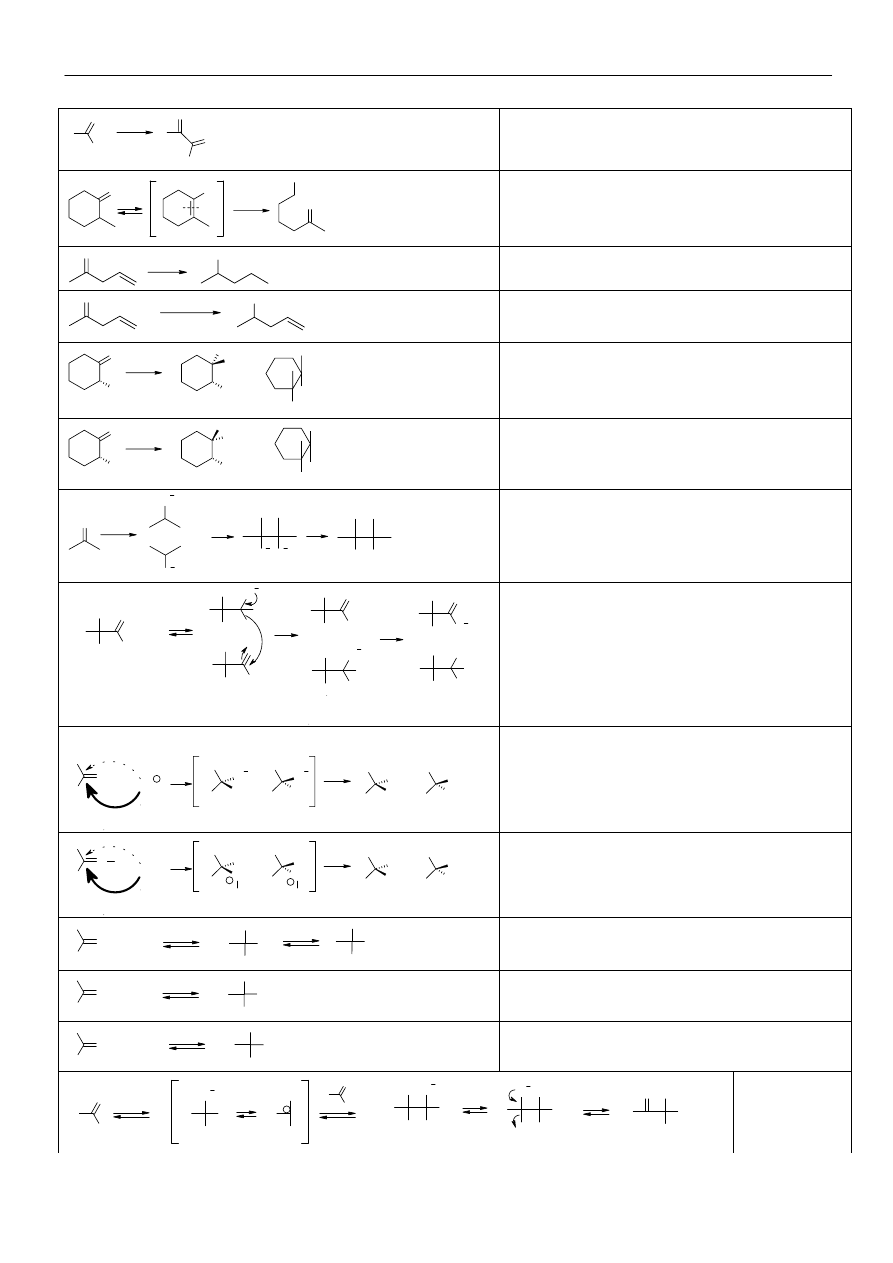
Kondensacja aldolowa:
O
CH
3
+
O
C
H
3
CH
3
OH
-
O
C
H
3
CH
2
-
O
CH
3
O
H
CH
3
O
CH
3
O
H
+
-H
2
O
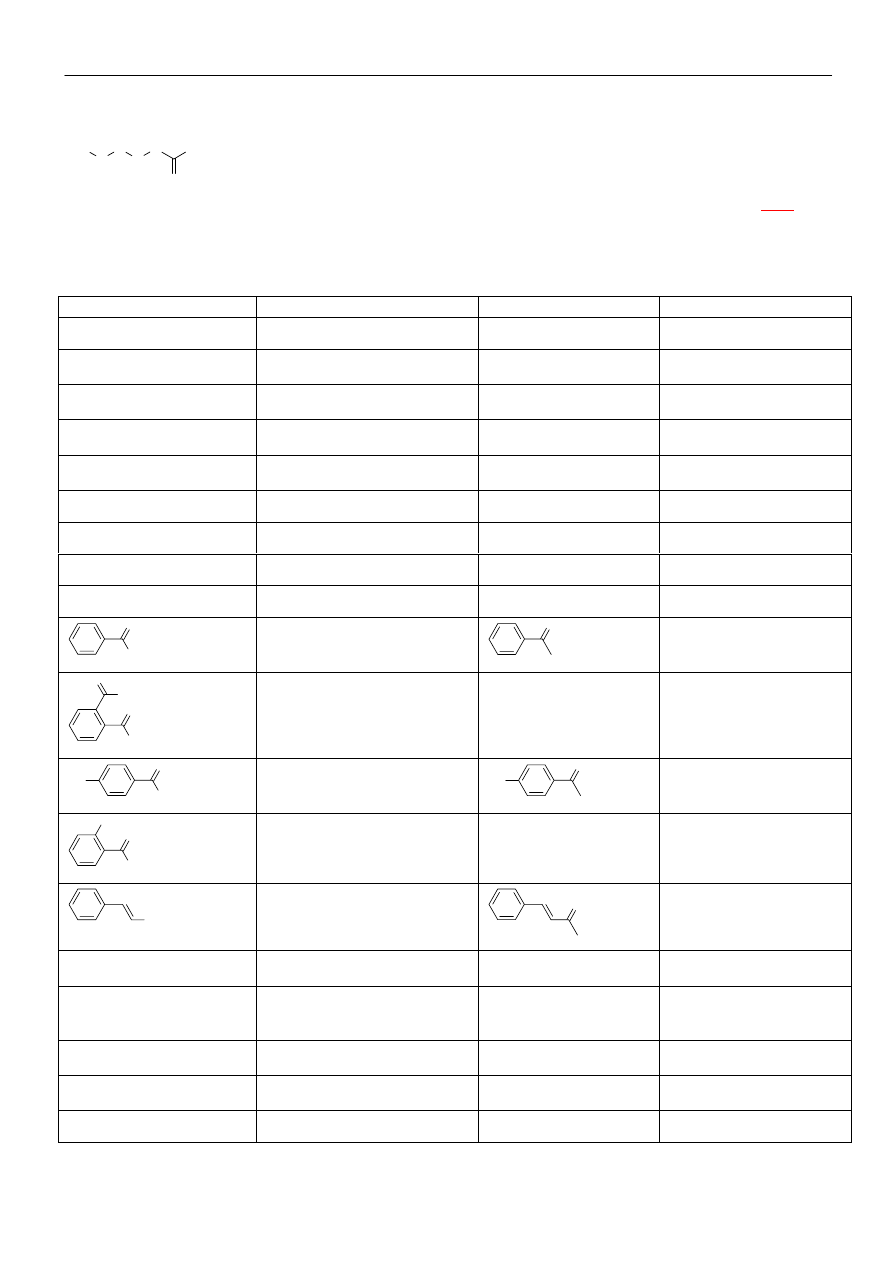
Alkilowanie związków karbonylowych
O
CH
3
OH
-
O
CH
2
O
CH
2
-
R-X
O
C
H
2
R
O-R
CH
2
+
W przypadku związków
monokarbonylowych, produktem głównym
jest produkt C-alkilowania (reakcja między
miękkim kwasem i miękką zasadą Pearsona)
Materiały z chemii organicznej
kg
- 20 -
Kwasy karboksylowe
C
H
3
C
H
2
C
H
2
C
H
2
C
H
2
O
OH
α
β
γ
δ
ω
Nazwę kwasu tworzymy:
• dodając do nazwy odpowiedniego węglowodoru końcówkę owy, a przed nazwą dodajemy słowo kwas: Alkan+
owy,
(atom
węgla w grupie karboksylowej jest wtedy atomem nr 1)
• traktując grupę jako podstawnik, do nazwy alkanu z końcówką karboksylowy dodajemy słowo kwas (pierwszym atomem
węgla jest atom połączony z grupą karboksylową)
Najczęściej spotykane kwasy karboksylowe:
Wzór kwasu
Nazwa kwasu
Wzór grupy acylowej
Nazwa grupy acylowej
H-COOH
metanowy (mrówkowy)
H-CO-
formyl
CH
3
-COOH
etanowy, metanokarboksylowy
(octowy)
CH
3
CO-
acetyl
C
2
H
5
COOH
propanowy, etanokarboksylowy
(propionowy)
CH
3
CH
2
CO-
propionyl
CH
3
CH
2
CH
2
COOH
butanowy, propanokarboksylowy,
(masłowy)
CH
3
CH
2
CH
2
CO-
butyryl
CH
3
CH
2
CH
2
CH
2
COOH
pentanowy, butanokarboksylowy,
(walerianowy)
HOOC-COOH
etanodiowy (szczawiowy)
-OC-CO-
oksalil
HOOC-CH
2
-COOH
propanodiowy (malonowy)
-OC-CH
2
CO-
malonyl
HOOC-CH
2
CH
2
-COOH
butanodiowy (bursztynowy)
CH
2
=CH-COOH
propenowy (akrylowy)
OH
O
benzenokarboksylowy
(benzoesowy)
O
benzoil
OH
O
OH
O
benzeno-1,2-dikarboksylowy
(ftalowy)
OH
O
C
H
3
4-metylobenzenokarboksylowy
(p-toluilowy)
O
C
H
3
p-toluil
OH
O
OH
2-hydroksybenzenokarboksylowy
(salicylowy)
COOH
trans-3-fenylopropenowy
(cynamonowy)
O
cynamoil
CH
3
-CH(OH)-COOH
2-hydroksypropanowy (mlekowy,
α-hydroksypropionowy)
CH
3
-CHCl-CH
2
-COOH
3-chloroprobutanowy,
2-chloropropanokarboksylowy
(
β-chloromasłowy)
HOOC-CH
2
-CH(OH)-COOH
2-hydroksybutanodiowy
(jabłkowy)
HOOC-CH(OH)-CH(OH)-COOH
2,3-dihydroksybutanodiowy
(winowy)
CH
3
-CO-COOH
2-oksopropanowy (pirogronowy)

Materiały z chemii organicznej
kg
- 21 -
Metody syntezy:
Utlenianie I
o
alkoholi lub aldehydów:
R CH
2
OH
[O]
R
OH
O
R
OH
O
[O]
R
OH
O
;
Utlenianie prowadzi się najczęściej dwuchromianem w
środowisku kwaśnym
Utlenianie alkiloarenów:
C
H
2
R
Na
2
Cr
2
O
7
/H
+
COOH
W przypadku gdy w pierscieniu znajduje się podstawnik
eltrodonorowy, wtedy pierścień ulega utlenieniu
Hydroliza nitryli:
R X
CN
-
R CN
H
+
, lub OH
-
R COOH
Jeżeli odpowiednie nitryle są łatwo dostępne
W reakcji związków metaloorganicznych z CO
2
:
R X
Mg
R MgX
CO
2
R COOMgX
H
2
O
R COOH
Wykorzystanie estrów malonowych:
C
H
2
O
O
R
O
O
R
R-O
-
CH
O
O
R
O
O
R
..
-
R'-X
CH
O
O
R
O
O
R
R'
C
H
2
OH
O
R'
H
+
Wodory a w estrze malonowym są kwaśne, łatwo je
oderwać, zwłaszcza silną zasadą
Reakcje kwasów karboksylowych:
Reakcje na grupie karboksylowej:
R COOH + H2N-R'
R COO
-
H
3
N-R'
+
R COOH + NaOH
R COO
-
Na + H
2
O
+
Kwasy karboksylowe z odczynnikami o charakterze
zasadowym reagują w klasyczny sposób.
R
O
O
-
+ R'-X
R
O
O
R'
Reszta kwasu karboksylowego wykazuje właściwości
nukleofilowe (jest to dość silny nukleofil)
Reakcje polegające na wymianie grupy –OH na inną
grupę
R
OH
O
R
OH
O H
+
H-O-R'
R
OH
OH
O R'
H
+
R
O
OH
2
O R'
H
+
R
O
O R'
H
R
O
O R'
+ H
+
- H
2
O
- H
+
Estryfikacja kwasów karboksylowych, kwaśna hydroliza
estrów przebiega wg identycznego mechanizmu. Każdy
etap reakcji jest odwracalny.
R
OH
O
2
R
O
O
O
R
-H
2
O
R
OH
O
PCl
3
lub SOCl
2
R
Cl
O
;
Synteza bezwodników i chlorków kwasowych
Reakcje dekarboksylacji:
R
O
R'
O
O
H
-CO
2
∆
R
O
R'
H
R
O
R'
R
O
O
O
O
H
H
R
O
O
H
H
-CO
2
∆
R
O
H
O
kwasy propanodiowe oraz 3-oksoalkanowe bardzo łatwo
ulegają dekarboksylacji
R-COOAg + Br
2
→ R-Br + CO
2
+ AgBr
Reakcja Hundsdieckera, podstawieniu ulegają tylko
wodory
α.
2R-COOH
→ R-R + CO
2
Elektroliza Kolbego kwasów karboksylowych lub ich soli
OH
O
C
H
2
R
Br
2
, P
OH
O
C
H
R
Br
Reakcja Helle, Volherda i Zielińskiego
OH
O
HNO
3
, H
2
SO
4
OH
O
O
2
N

Materiały z chemii organicznej
kg
- 22 -
Aminy
Aminy alifatyczne
Aminy aromatyczne
I
o
R NH
2
II
o
R N
H
R'
III
o
R N
R'
R"
I
o
Ar
NH
2
II
o
Ar
N
H
R'
III
o
Ar N
R'
R"
Metody syntezy:
R X +
R NH
3
X
+
-
OH
-
R NH
2
R X
R X
NH
3
R X
R
NH
R
R
R
+
X= halogen, siarczan, todylan
Ar NO
2
Fe /H
+
Ar NH
2
Redukcja aromatycznych nitrozwiązków
R X + N
3
-
-X
-
R N
3
LAH
R NH
2
redukcja azydków alkilowych
R X + CN
-
-X
-
R CN
LAH
R CH
2
-NH
2
redukcja nitryli (cyjanków)
O
R'
R
+ NH
2
-OH
R
R'
N
OH
R
R'
N
OH
H
2
/Ni
R CH
R'
NH
2
Redukcja oksymów. W oksymach grupa hydroksylowa może być
naprzeciw grupy R, lub naprzeciw grupy R’. Mamy do czynienia z
izomerią geometryczną E i Z. Najczęściej jednak ten typ izomerii
nazywa się izomerią syn i anti. Grupa hydroksylowa jest
odpowiednio syn w stosunku do R (anti w stosunku do R’) lub anti
w stosunku do R (syn do R’)
O
R'
R"
+ NH
2
-R
N
R'
R"
R
N
R'
R"
R
red.
C
H
N
H
R'
R"
R
Redukcja imin lub enamin, otrzymujemy aminy II
o
lub III
o
. Podobnie jak w przypadku oksymów, w
iminach występuje izomeria syn i anti.
O + HCOO
-
NH
4
+
C
H
NH
2
Aminy I
o
można otrzymać w jednoetapowej reakcji
związku karbonylowego z mrówczanem amonu.
Reakcja Leuckarta.
Właściwości chemiczne amin:
R X + H
2
N-R'
R N
H
R'
Alkilowanie amoniaku i amin. Reakcja może
przebiegać
aż do uzyskania soli
czwartorzędowych.
R"
X
O
+ R-NH-R'
R"
NRR'
O
X=Cl, OR, OOCR. Reakcję przeprowadza się
najczęściej w obecności aminy III
o
, celem
usunięcia powstałego chlorowodoru lub kwasu.
Reakcje z kwasem azotawym (azotowym III)
R N
2
+
R NH
2
+ HNO
2
R
+
+ N
2
Z I
o
aminami alifatycznymi powstają nietrwałe sole
diazoniowe, które natychmiast rozkładają się na
karbokation, który ulega wtórnym reakcjom. Z I
o
amin
aromatycznych powstają sole diazoniowe, które są
trwałe w niskiej temperaturze.
R N
H
R' + HNO
2
R N R'
NO
Z II
o
amin alifatycznych i aromatycznych powstają
związki N-nitrozowe.
N
R
R
+ HNO
2
N
R
R
ON
III
o
aminy alifatyczne w reakcji z kwasem azotawym dają
mieszaninę różnych produktów rozkładu, natomiast aminy
aromatyczne ulegają reakcj C-nitrozowaniu w pozycji para.
Jeżeli pozycja para jest zajęta to w pozycji orto.
Reakcje ze związkami karbonylowymi:
R C
H
2
O
R'
+ NH
2
-R"
R C
H
2
R'
N R"
R C
H
R'
NH R"
Powstała imina jest w równowadze tautomerycznej
z enaminą.
C
H
O
R'
+ R"NHR"
R'
NR'
2
Aminy II
o
tworzą enaminy
Grupa aminowa jako grupa odchodząca:
R N
+
CH
3
CH
3
CH
3
+ Nu
-
R Nu + N(CH
3
)
3
Grupa aminowa nie może być grupą odchodzącą w
postaci anionu NH
2
-
, podobnie jak grupa hydroksylowa
w postaci OH
-
. Stosunkowo łatwo może odejść
cząsteczka obojętna (woda, amina III
o
)
H
C
H
2
N
+
CH
3
CH
3
CH
3
OH
-
∆
CH
2
+ N(CH
3
)
3
Reakcja Hoffmana. W eliminacji powstaje zawsze alken
najmniej podstawiony. Oderawniu ulegają najbardziej
kwasne protony. Podobnie ulegają eliminacji N-tlenki
amin (reakcja Cope’a). Reakcja Hoffmana jest
eliminacją trans, natomiast eliminacja Cope’a jest
eliminacją cis.

Materiały z chemii organicznej
kg
- 23 -
Oksymy, iminy, nitryle, izonitryle, cyjaniany i izocyjaniany
Związek
Grupa (przyrostek)
Podstawnik (przedrostek)
Oksymy
R=N-OH
oksym + związek karbonylowy R
hydroksyimino + nazwa R
C
H
2
C
2
H
5
N OH
oksym 1-fenylobutan-2-onu
C
H
3
N
COOH
OH
a)
N
COOH
OMe
b)
a) kwas 3-hydroksyiminobutanowy
b) kwas 2-metoksyiminocykloheksano-
karboksylowy
Iminy
R=NH
Nazwa R + imina
Nazwa R= + amina
imino + nazwa R
C
H
3
C
H
2
NH
H
propanoimina
propylidenoamina
(imina propanalu)
C
H
3
C
H
2
N
COOCH
3
C
2
H
5
2-etyloiminopropionian metylu
2-(N-etyloimino)propionian metylu
Nitryle
R
≡Ν
R-CN
alkan + nitryl
alkan + karbonitryl
acyl - (-yl, -oil) + onitryl
cyjanek + alkil
cyjanek + nazwa R
C
H
3
C
H
2
N
propanonitryl
etanokarbonitryl
propionitryl
cyjanek etylu
C
H
3
C
H
N
COOH
kwas 2-cyjanopropanowy
Izocyjanki
R-NC
izocyjanek + alkil
izocyjano- + nazwa R
NC
izocyjanek cyklopentylu
COOH
NC
kwas m-izocyjanobenzoesowy
Cyjaniany
R-OCN
cyjanian + alkil
cyjaniano + nazwa R
OCN
cyjanian benzylu
O
NCO
p-cyjanianobenzaldehyd
Izocyjaniany
R-NCO
izocyjanian alkilu
izocyjaniano + nazwa R
NCO
izocyjanian allilu
NCO
HOOC
kwas cis-4-izocyjanianobut-2-enowy

Materiały z chemii organicznej
kg
- 24 -
Synteza oksymów
O
C
H
3
C
2
H
5
H
+
O
C
H
3
C
2
H
5
H
+
..
N
H
2
OH
..
C
H
3
C
2
H
5
OH
2
NH-OH
+
..
..
-H
2
O
N
C
H
3
C
2
H
5
OH
H
N
C
H
3
C
2
H
5
OH
H
+
+
N
C
H
3
C
2
H
5
OH
N
C
H
3
C
2
H
5
OH
C
H
3
C
2
H
5
OH
NH-OH
H
+
..
..
1
2
2
1
1
2
2
1
:
:
(E)-oksym butan-2-onu
(Z)-oksym butan-2-onu
Przegrupowanie Beckmana oksymów
N
C
H
3
C
2
H
5
OH
H
+
N
C
H
3
C
2
H
5
O H
H
+
..
-H
2
O
:
N
H
C
H
3
C
2
H
5
+
+H
2
O
N
C
H
3
C
2
H
5
O
H
H
+
N
C
H
3
C
2
H
5
:
+
..
N
C
H
3
H
O
C
2
H
5
:
..
..
-H
+
N-etyloacetamid
..
N
C
H
3
O
C
2
H
5
H
:
..
forma tautomeryczna
amidu
N
C
H
3
C
2
H
5
OH
H
+
N
C
H
3
C
2
H
5
O
H
H
+
C
2
H
5
N
O
H
CH
3
N-metylopropanoamid
Przegrupowanie Beckmana jest reakcją stereospecyficzną. W reakcji stereospecyficznej dany izomer ulega przemianie
prowadzącej do jednego produktu, a inny stereoizomer – do innego produktu.
Reakcja w której ze zbioru możliwych stereoizomerów tworzy się wyłącznie lub w przeważającej ilości tylko jeden z nich,
nazywa się reakcją stereoselektywną.
R
O
NH
2
+ Br
2
OH
-
R
O
N
H
Br
OH
-
:
:
N
R
C O
..
R
O
N
:
..
izocyjanian
H
2
O
R N
O
O
H
H
..
..
+
-
R N
O
O
H
H
kwas karbaminowy
..
R N H
H
..
-CO
2
Degradacja amidów Hoffmana
nitryle, izonitryle, cyjaniany i izocyjaniany
H C N
:
C N
:
+ H
+
:
twarde centrum
zasadowe
miękkie centrum
zasadowe
O C N
H
kwas cyjanowy
kwas izocyjanowy
:
O C N
H
:
..
..
O C N
..
..
:
:
+ H
+
O C N
..
..
:
..
+ H
+
cyjanian
izocyjanian
-
-
..
..
S C N
S C N
..
..
:
:
tiocyjanian
izotiocyjanian
-
-
..
..
Nitryle
R C
H
H
C N
E
+
OH
:..
..
-
Nu
:
-
:
Centra reaktywne w nitrylach
Reakcje rozpoczynające się atakiem odczynnika o charakterze elektrofilowym
R C N: + H
+
R C N H
+
OH
2
:
..
R C N H
O
H
H
+
..
-H
+
R C N H
O
H
R C N H
O H
..
H
+
/
H
2
O
R COOH + NH
4
+
forma tautomeryczna amidu
kwasowa hydroliza nitryli

Materiały z chemii organicznej
kg
- 25 -
R C N: + H
+
R C N H
R C N H
+
+
Y
Y
R
NH
H
+
/
H
2
O
Y
R
O
Reakcje acylowania związków aromaty-
cznych.
R=alkil, aryl – metoda Hoescha
R=H metoda Gattermana
Y = -OH, -OMe, -NH-Ac, alkil
OH
H
+
OH
2
: ..
R
N
+
+
R
N
O
H
H
+
..
N
R
:
-H
+
R
O
N
H
R
O
N
H
H
+
/
H
2
O
NH
3
+
+ RCOOH
Reakcja Rittera – pozwala na syntezę
amin z grupą aminową przy III
o
atomie
węgla. Ulegają jej jedynie związki z
których można otrzymać „trwały”
karbokation (alkohole III
o
, alkeny,
karbokationy stabilizowane przez
mezomerię).
Reakcje nitryli z nukleofilami
R
N
OH
-
:
..
..
:
R
N
O H
:
..
-
H
2
O
R
N
O H
H
..
R
N
O
H
H
OH
-
/H
2
O
R
O
O
+ NH
3
Zasadowa hydroliza nitryli
R
N:
CH
3
-Mg-X
R
N
CH
3
MgX
..
H
2
O
R
N
CH
3
H
..
H
+
/H
2
O
-MgOHX
R
O
CH
3
Reakcje ze związkami metaloorgani-
cznymi.
Reakcje rozpoczynające się atakiem zasady na kwaśny atom wodoru
R C
H
H
N
-BH
B
:
-
R C
H
N
-
CH
3
-X
R C
H
N
R
-X
-
Alkilowanie nitryli
R C
H
2
N
B
-
BH
R CH
N
-
..
R'
N
:
R CH
N
R'
CN
:
..
BH
-B
-
R CH
NH
R'
CN
..
R
NH
2
R'
CN
..
H
2
O
R CH
O
R'
CN
Reakcje kondensacji
Redukcja nitryli
R
N
LAH
R C
H
2
NH
2
R
N
LAH
-50
o
C
R
H
NH
H
2
O
R
H
O
R
N
1. SnCl
2
/HCl
2. H
2
O
R
H
O
Synteza nitryli
C
H
3
C
H
2
C
H
2
Br
+ NaCN
miękkie centrum kwasowe
C
H
3
C
H
2
C
H
2
CN
Reakcja halogenków alkilowych z
cyjankiem sodu
C
H
3
C
H
CH
3
Br + AgCN
C
H
3
CH
CH
3
+
twarde centrum kwasowe
C
H
3
C
H
CH
3
NC
W przypadku stosowania cyjanku srebra
produktem głównym jest izonitryl
R
O
NH
2
P
2
O
5
R
N
N
2
+
Cu
2
(CN)
2
CN + N
2
Reakcja Sandmeyera
Materiały z chemii organicznej
kg
- 26 -
Centra reaktywne w izonitrylach
R N C
:
-
+
centrum nukleofilowe
R N C
:
-
+
H
+
R N C H
+
H
2
O
R NH
2
+ HCOOH
Izonitryle nie reagują z odczynnikami o
charakterze nukleofilowym, jednak
bardzo łatwo reagują z odczynnikami o
charakterze elektrofilowym
Centra reaktywne izocyjanianów i izotiocyjanianów
R N C Y
centrum elektrofilowe
Y=O
Nu= -OH kwas karbaminowy
Nu= -OR karbaminian
Nu= _NHR pochodne mocznika
R N C Y + Nu-H
..
..
R N C Y
Nu
H
..
..
+
-
R N
H
Y
Nu
Y=S
Nu= -OR tiokarbaminian
Nu= _NHR pochodne tiomocznika
Związki nitrowe
R
C
H
H
N
O
O
+
-
OH
..
..
:
-
Miejsca reaktywne w związkach nitrowych.
Metody syntezy:
C
H
3
C
H
2
CH
3
+ HNO
3
C
H
3
C
H
2
C
H
2
NO
2
C
H
3
C
H
CH
3
NO
2
C
H
3
C
H
2
NO
2
CH
3
O
2
N
+
+
+
Nitrowanie alkanów zachodzi tylko w fazie
gazowej. Jest to reakcja o mechanizmie
rodnikowym. Rzadko się ją stosuje do
syntezy nitroalkanów.
O N
O
..
..:
:
-
miękkie centrum zasadowe
twarde centrum zasadowe
anion azotynowy (azotowy III), podobnie jak
jon cyjankowy, czy wodorosiarczynowy, jest
jonem ambidentnym (dwuzębnym).
R C
H
2
X + NO
2
-
R C
H
2
NO
2
R C
H
C
H
2
X
R' + NO
2
-
R C
H
C
H
2
NO
2
R'
R
C
H
2
X
R'
R"
+ NO
2
-
R
C
H
2
NO
2
R'
R"
R
C
H
2
O-NO
R'
R"
C
H
R'
R
R"
+
+
I
o
halogenki reagując z azotynem sodu dają
nitropochodne. W miejsce NaNO
2
można
użyć AgNO
2
. Jony srebrowe nie wspomagają
w tworzeniu się karbokationów, ale
przyśpieszają i ułatwiają reakcję poprzez
polaryzacje wiązania C-X. II i III
o
halogenki
dobre wydajności nitrozwiązków dają tylko z
NaNO
2
(miękkie centra reaktywne). Z III
o
halogenków, obok nitrozwiązków tworzą się
jednak azotyny i alkeny (reakcja poprzez
karbokation).
A
+ HNO
3
A
O
2
N
A
NO
2
+
Nitrowanie związków aromatycznych jest
najczęstszą metodą syntezy aromatycznych
związków nitrowych. Podstawniki
aktywujące pierścień kierują kolejny
podstawnik w położenie orto i para.
D
+ HNO
3
NO
2
D
H
2
SO
4
Nitrowanie arenów z podstawnikiem
dezaktywującym wymaga bardziej
drastycznych warunków (mieszanina
nitrująca, wyższa temp. reakcji)
+ HNO
3
NO
2
Naftalen ulega nitrowaniu w pozycji 1 (
α)

Materiały z chemii organicznej
kg
- 27 -
A
+ HNO
3
A
NO
2
A
NO
2
+
Podstawnik aktywujący kieruje grupę nitrową
w położenie 4.
+
+
A
+
A
:
+
A
:
A
:
+
A
:
+
A
+
A
:
Miejsce podstawienia się kolejnego
podstawnika można przewidzieć rozpatrujące
struktury graniczne kompleksu
σ.
A
:
+
A
:
+
A
+
A
:
+
A
:
+
A
:
+
A
:
+
Podstawnik aktywujący A, bardziej aktywuje ten
pierścień, w którym się znajduje. Należy
rozpatrzyć więc sytuacje gdy kolejny podstawnik
podstawi się w pozycje 2, 3 i 4. Rozpatrując
struktury graniczne widać, że kompleks
σ , w
pozycji 4 jest trwalszy, więcej struktur
granicznych w których został zachowany
aromatyczny charakter jednego pierścienia.
OH + HNO
2
OH
ON
HNO
3
HNO
3
- HNO
2
OH
O
2
N
Podczas nitrowania fenolu nawet rozcieńczonym kwasem
azotowym, obok o- i p-nitropochodnych tworzy się dużo
produktów utlenienia fenolu. nitrowanie kwasem azotowym
z małą domieszką kwasu azotawego powoduje, że z dobrą
wydajnością powstaje p-nitrofenol.
N
H
COCH
3
HNO
3
N
H
O
2
N
COCH
3
1. H
+
2. HNO
2
N
2
+
O
2
N
NO
2
-
O
2
N
NO
2
Nitrozwiązki (nieosiągalne innymi metodami)
można otrzymać w wyniku reakcji soli
diazoniowych ze stężonym roztworem
azotynu sodu.
Właściwości chemiczne:
R C
H
2
N
O
O -
+
OH
..
..
:
-
R C N
O
O
H
-
+
-
..
R C N
O
O
H
-
+
-
R C N
OH
O
H
-
+
H
2
O
R C
H
2
N
O
O -
+
anion
aci nitrozwiązku
aci nitrozwiązek
Nitralkany, posiadające w położeniu
α atomy
wodoru wykazują C-H kwasowość
(rozpuszczają się w wodnych roztworach
zasad). Występuje u nich tautomeria nitro-
aci nitro.
R C
H
2
NO
2
B
-
BH
R
H
NO
2
-
..
R'-X
R
N
O
O
R'
-
+
Nitroalkanów nie można C-alkilować,
otrzymuje się raczej O-alkilowe pochodne.
R C
H
2
NO
2
B
-
BH
R
H
NO
2
-
..
R'
O
R
R
O
R'
R
O
2
N
H
-
R
OH
R'
R
O
2
N
H
BH
B
-
R
OH
R'
R
O
2
N
-
..
R
O
2
N
R'
R
Nitroalkany ulegają natomiast łatwo
reakcji kondensacji ze związkami
karbonylowymi. (Tworzący się produkt
ataku atomem tlenu na karbonylowy atom
węgla jest nietrwały i rozpada się na
substraty).
C
H
3
NO
2
Cl
2
/OH
-
Cl
3
C NO
2
Nitroalkany posiadające atomy wodorów w położeniu
α ulegają łatwo halogenowaniu. Reakcji
w środowisku alkalicznym nie można zatrzymać na etapie monohalogenowania. Z nitrometanu
w wyniku chlorowania otrzymuje się chloropikrynę (trichloronitrometan), związek o b. silnych
właściwościach łzawiących (lakrymator), stosowany przez wojsko i policję.
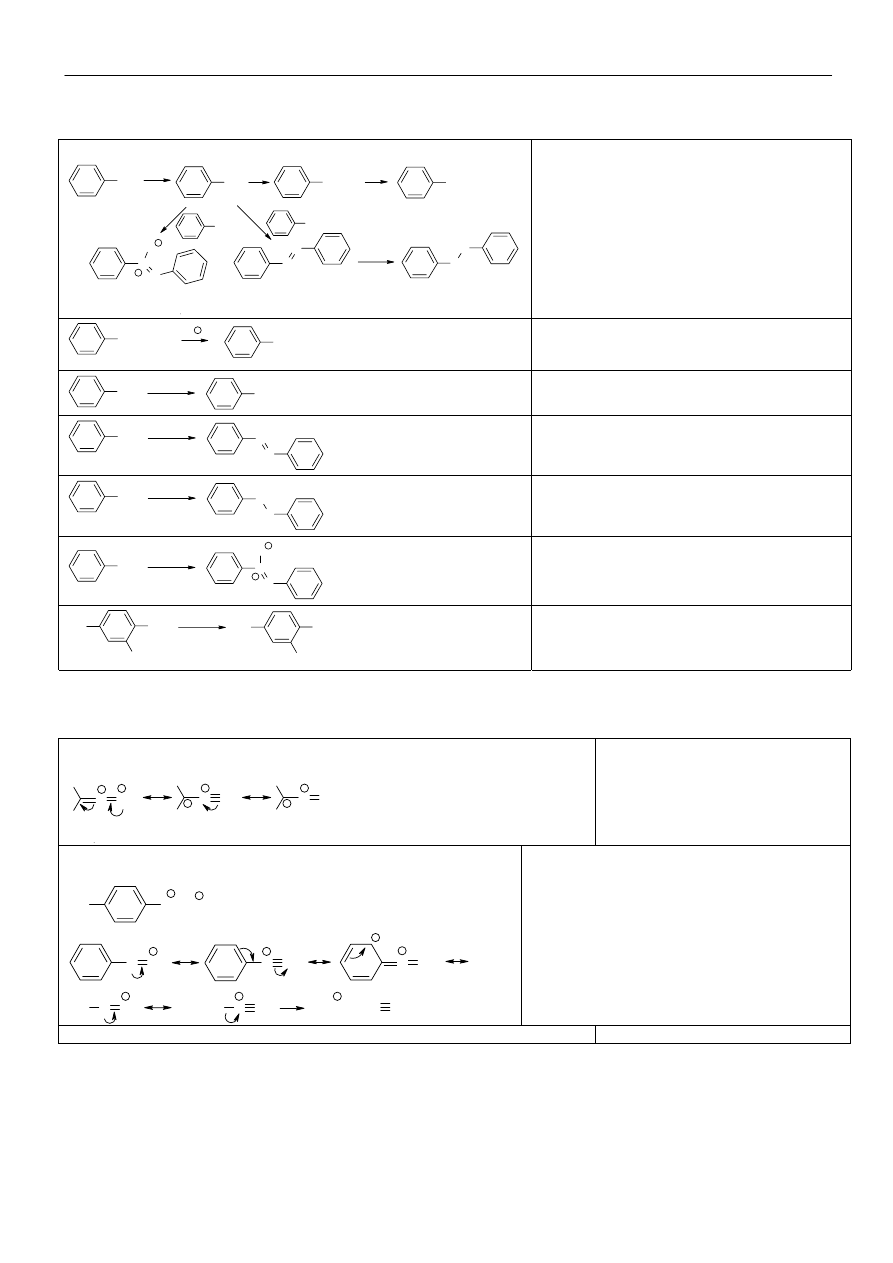
Materiały z chemii organicznej
kg
- 28 -
Redukcja związków nitrowych:
NO
2
NO
NH-OH
NH
2
NH-OH
N
N
O -
+
NH
2
N
N
N
H
N
H
Redukcja związków nitrowych może doprowadzić
do szeregu związków w zależności od warunków
reakcji:
nitrozobenzen, fenylohydroksyloamina, anilina,
azoksybenzen, azobenzen (izomer trans),
hydrazobenzen
NO
2
H
+
Fe
+ Zn
SnCl
2
NH
2
W środowisku kwaśnym, grupa nitrowa redukuje
się do grupy aminowej, bez możliwości
wydzielenia produktów pośrednich.
NO
2
Zn/NH
4
Cl
NH-OH
Zmniejszenie kwasowości środowiska reakcji
umożliwia wydzielenie fenylohydroksyloaminy
NO
2
Zn/OH
-
,alk.
N
N
W środowisku alkalicznym zachodzą reakcje
kondensacji
NO
2
Mg/alk.
N
H
N
H
Magnez, bardziej aktywny od cynku, powstający
azobenzen redukuje do hydrazobenzenu
NO
2
As
2
O
3
N
N
O
-
+
Arszenik w srodowisku zasadowym (arsenin sodu)
umożliwia wydzielenie azoksybenzenu.
NO
2
O
2
N
CH
3
Na
2
S
x
H
2
N
CH
3
NO
2
Wielosiarczek sodowy (amonowy) selektywnie
redukuje jedną grupę nitrową.
Związki diazowe i azowe
R=N
2
diazoalkan, np.:
CH
2
N
2
diazometan; CH
3
-CO-CH=N
2
diazoaceton; N
2
=CH-COOC
2
H
5
diazooctan etylu
N
H
H
N
..
-
:
+
N
H
H
N
..
-
:
+
N
H
H
N
..
-
:
+
..
R-N
2
+
X
-
sól alkilodiazoniowa np.: chlorek, bromek....
p-toluilodiazoniowy
C
H
3
N
2
+
X
-
N N
..
:
+
N N
:
+
N N
:
+
..
+
C
H
3
N N
..
:
+
C
H
3
N N
:
+
N N
:
+
:
CH
3
+
X = Cl, I, Br, SO
4
-2
, BF
4
,
aromatyczne sole diazoniowe są trwałe
(stabilizowane poprzez rezonans)
alifatyczne sole diazoniowe rozkładają się nawet w
niskiej temperaturze na karbokationy,które ulegają
wtórnej reakcji i azot
Ar-N=N-Ar, R-N=N-R związki azowe, azoalkan np.:
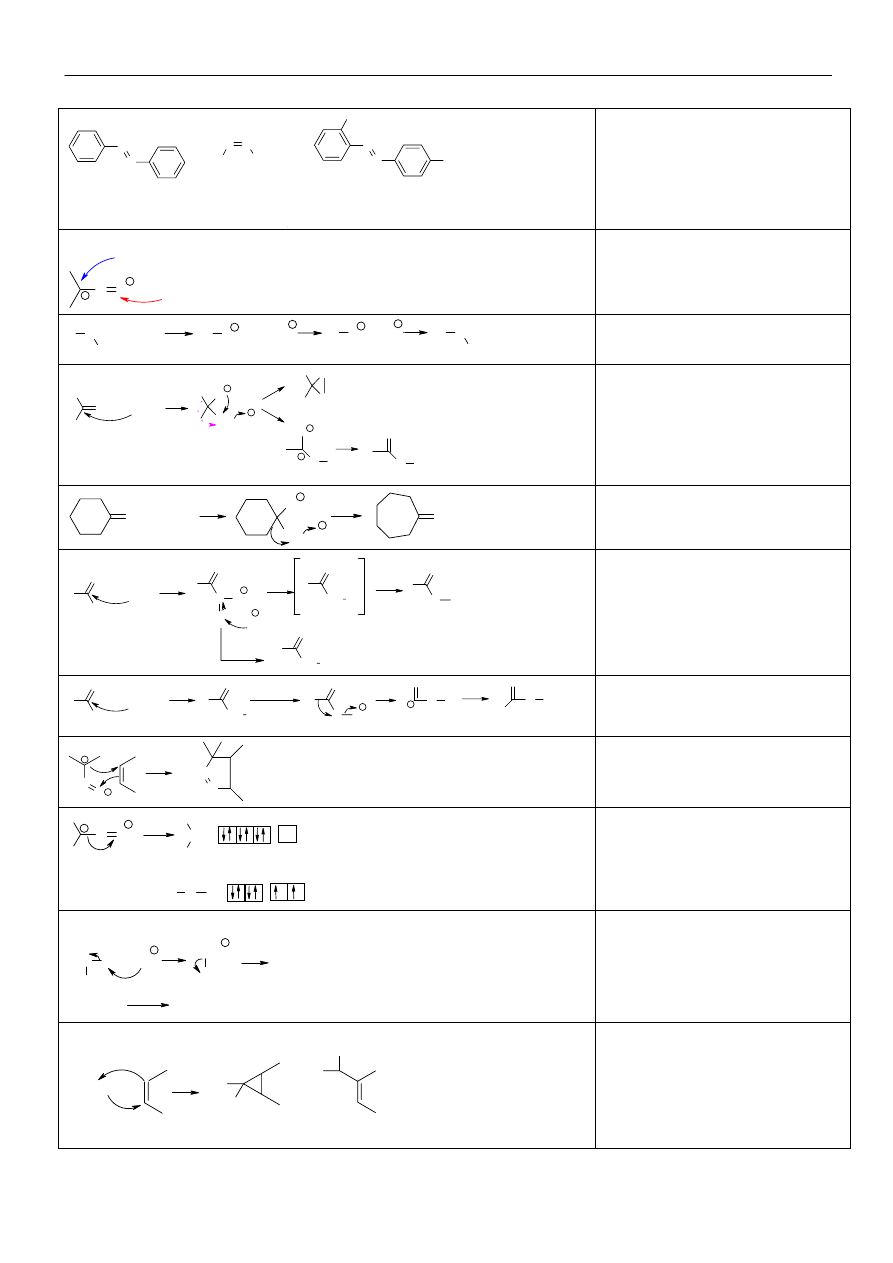
Materiały z chemii organicznej
kg
- 29 -
N
N
C
H
3
N N
CH
3
N
N
CH
3
NH
2
azobenzen
azometan
2-amino-4'-metyloazobenzen
Właściwości chemiczne diazozwiązków:
N N
..
-
+
..
:
centrum nukleofilowe
centrum elektrofilowe
R O
H
+ CH
2
N
2
R O
+ CH
3
N
2
-
+
R O + CH
3
-
+
-N
2
R O
CH
3
R= alkil, aryl, acetyl
Synteza eterów i estrów
O
R
R'
+ CH
2
N
2
R
R'
O
CH
2
-N
2
-
+
-N
2
R
R'
O
C
H
2
R'
O
C
H
2
R
-
+
R'
O
C
H
2
R
Reakcja przedłużenia łańcucha ketonów
O + CH
2
N
2
O
CH
2
-N
2
-
+
O
-N
2
R
Cl
O
+ CH
2
N
2
R
C
H
O
H
N
2
+
Cl
-
-HCl
R
CH
O
N
2
+HCl
R
C
H
2
O
Cl
+ CH
2
N
2
R
CH
O
N
2
+ N
2
+ CH
3
Cl
Metoda syntezy chlorometyloketonów,
lub diazoketonow w przypadku
nadmiaru diazometanu
R
Cl
O
+ 2 CH
2
N
2
R
CH
O
N
2
-CH
3
Cl
- N
2
Ag
+
/H
2
O
R
C
H
2
O
N
2
+
O
C
H
2
R
+
H
2
O
O
C
H
2
R
O
H
Przedłużenie
łańcucha węglowego
kwasów metodą Arndta Eisterta
N
N
-
:
:
..
+
N
N
Reakcje addycji 1,3-dipolarnej. Służą do
syntezy układu pirazoliny
N
H
H
N
..
-
:
..
+
h
ν
C
H
H
:
-N
2
karben
C H
H
.
.
hybrydyzacja sp
hybrydyzacja sp
2
S=0
M=2S+1, M=1 (stan singletowy)
S=1
M=2S+1, M=3 (stan trypletowy)
Podczas naświetlania diazometanu
(fotolizy), rozkłada się on na karben
(singletowy) i azot.
Metody syntezy karbenów:
Cl
2
C H
Cl
+ OH
-
Cl
2
C
Cl
:
-Cl
-
Cl
2
C:
Ph-CHCl
2
OH
-
-HCl
Ph-CCl:
-
karbeny otrzymuje się w wyniku
α-eliminacji. Mają one harakter
elektrofilowy.
Reakcje karbenów
Ph-CCl: +
Cl
Ph
+
Ph
Cl
produkt insercji
do wiązania C-H
produkt addycji
do wiązania C=C
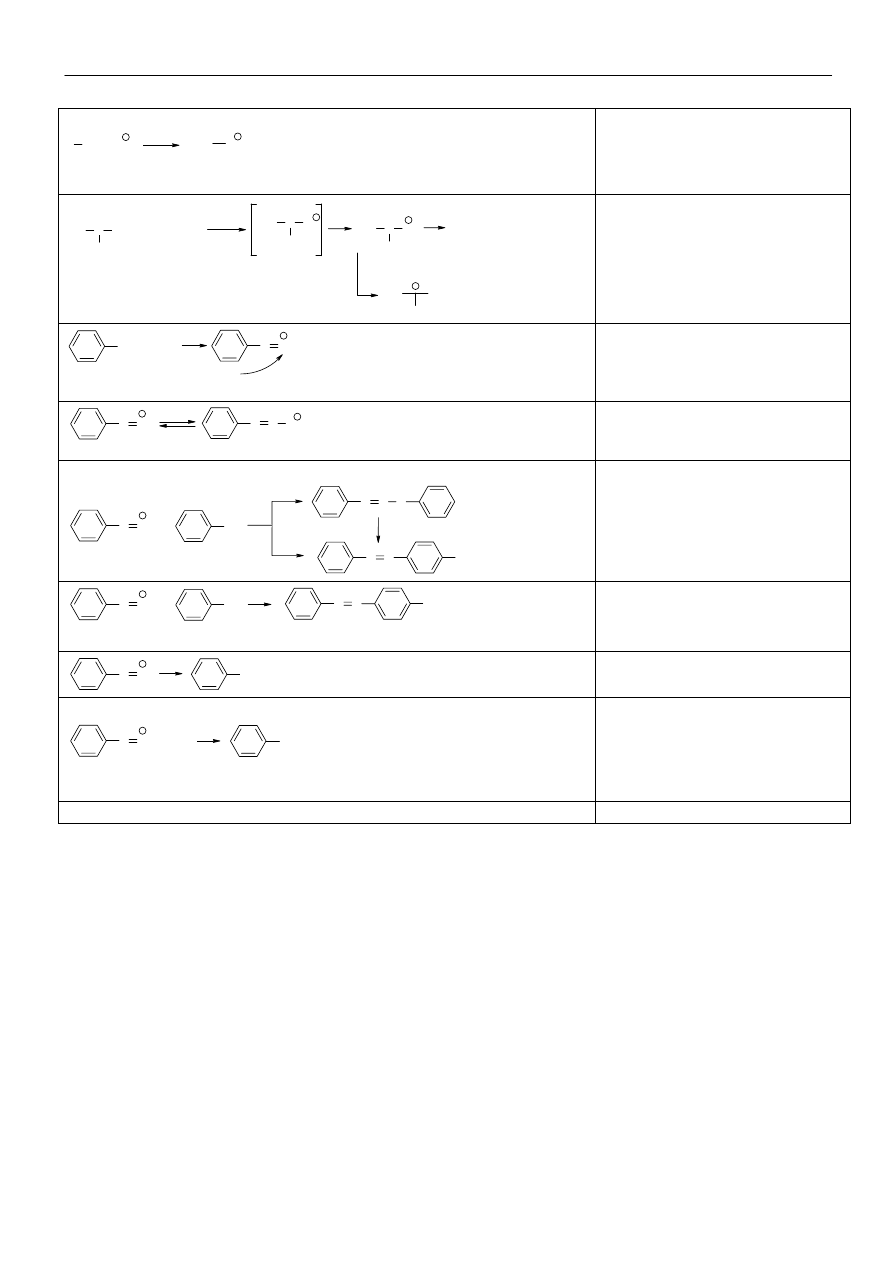
Materiały z chemii organicznej
kg
- 30 -
Sole diazoniowe:
R NH
2
+ NO
+
:
-H
2
O
R
N
2
+
Gdy R=aryl, sole są trwałe w niskich
temperaturach, gdy R=alkil, rozkładają
się z wydzieleniem azotu.
HNO
2
+2HCl= NO
+
+ 2Cl
-
+ H
3
O
+
R-ONO + HCl = ROH + NO
+
+ Cl
-
C
H
3
C
H
CH
2
-NH
2
CH
3
+ NO
+
Cl
-
-H
2
O
C
H
3
C
H
CH
3
N
2
+
-N
2
C
H
3
C
H
CH
3
CH
2
+
alkohol + chlorek + alken
C
H
3
CH
3
CH
3
+
alkohol + chlorek + alken
NH
2
+ NO
+
N N
+
:
..
słabe centrum
elektrofilowe
Sole diazoniowe istnieją tylko w
środowisku kwaśnym i obojętnym. W
roztworach alkalicznych przekształcają
się w pochodne kwasów diazowych.
N N
+
:
..
pH>7
pH<7
N N O
.. ..
-
fenylodiazan
Po zalkalizowaniu roztworu powstaje
izomer cis-diazanu, który przekształca
się powoli do trwalszego trans-diazanu.
Reakcje soli diazoniowych przebiegające bez wydzielania azotu:
N N
+
:
..
NH
2
+
N N N
H
.. .. ..
N N
NH
2
pH
≈ 7
pH<7
pH<7
W środowisku obojętnym lub słabo
alkalicznym powstaje diazoamino-
benzen, który w środowisku kwaśnym
ulega przegrupowaniu do p-aminoazo-
benzenu. Aminy aromatyczne w
środowisku umiarkowanie kwaśnym
ulegają sprzęganiu (w pozycji para).
N N
+
:
..
OH
+
N N
OH
pH
≈ 7
Podobnej reakcji ulegają fenole, jednak
największa szybkość sprzęgania
zachodzi w środowisku słabo
alkalicznym.
N N
+
:
..
NH-NH
2
red.
Reakcje przebiegające z wydzieleniem azotu:
N N
+
:
..
+ X
X
X = I
-
; NO
2
-
; H
2
O
+ N
2
Wymiana grupy diazoniowej na jod,
grupę nitrową lub hydroksylową.
(Wymiana grupy diazoniowej na
nitrową zachodzi prawdopodobnie wg
mechanizmu S
N
1, pozostałe
mechanizmy są rodnikowe)
Wyszukiwarka
Podobne podstrony:
Materiały do wykładu 4 (27 10 2011)
MATERIALY DO WYKLADU CZ IV id Nieznany
MATERIALY DO WYKLADU CZ VIII i Nieznany
MATERIALY DO WYKLADU CZ V id 2 Nieznany
Materiały do wykładu z Rachunkowości
Materiały do wykładu 4 (28 10 2011)
Podstawy budownictwa materialy do wykladu PRAWO wydr
15.02.06-Anemia-materiały do wykładu, studia, 4 rok, farmakologia, materiały, C21W15-niedokrwistosci
Logika materiały do wykładów
Materiały do wykładu nr 1
Materialy do wykladu nr 5 id 28 Nieznany
Rezerwa z tytułu odrocznego podatku - materiały do wykładu 2014, UE KATOWICE ROND, I stopień, VI sem
Rezerwy na świadczenia pracownicze - materiały do wykladu 2014, UE KATOWICE ROND, I stopień, VI seme
Rachunkowośc obrotu towarowego - materiały do wykladu 2012, Uniwersytet Ekonomiczny w Katowicach, Fi
Materiały do wykładów z filozofii, AJD - PEDAGOGIKA, I rok, I semestr, Wstęp do filozofii
podatki w rachunkowości, Materialy do wykladu - VAT w rachunkowosci 2009 rok, Szkoła Główna Handlowa
więcej podobnych podstron