Priony
Tytuł

Historia
Termin "prion" został wprowadzony
do literatury w 1982 na łamach
Science przez Stanleya B.
Prusinera.
W tym samym roku wykryto białko
prionu (PrP) i stwierdzono korelację
między ilością białka a
infekcyjnością materiału
zakaźnego. Określenie sekwencji
białka PrP poprzedziło
zidentyfikowanie genu kodującego
PrP (u człowieka ma nazwę PRNP
i znajduje się na 20 chromosomie)

Czym są priony?
Jedna z hipotez zakłada, że są to czynniki chorobotwórcze
zbudowane wyłącznie z niewielkiego białka (27 – 30 kDa)
bez kwasów nukleinowych. Wymaga to przyjęcia całkowicie
innego sposobu ich powielania. Odkrycie genu PRNP u
człowieka i analogicznych genów (Prn-p) u większości
zwierząt wyższych przyniosło wniosek, że kodowane przez
ten gen białko jest niezbędne do funkcjonowania niektórych
procesów fizjologicznych.

Czym są priony?
Wielu badaczy uważa jednak, że prion może zawierać
niewielką ilość kwasu nukleinowego, dobrze chronionego przez
strukturę białkową, a jego mechanizm powielania jest podobny
jak u wirusów.
W odróżnieniu od wirusów, cząsteczka prionu nie traci
zdolności do namnażania się po działaniu czynników
uszkadzających kwasy nukleinowe (nukleazy, promieniowanie
UV)

Czym są priony?
Białko prionowe (PrP) występuje powszechnie w każdym
organizmie i jest niegroźne. Wchodzi w skład otoczek komórek
nerwowych oraz limfocytów. Dopiero w sytuacji, gdy zmienia
ono swoją naturalną konformację, staje się białkiem prionowym
infekcyjnym.

Białko prionowe a priony
Porównanie cząsteczek białek PrP obecnych w tkankach
w warunkach fizjologii i w patologiach, (np. scrapie)
dowiodło,
że
mają
one
identyczną
strukturę
pierwszorzędową (czyli sekwencję aminokwasów), ale
różnią
się
strukturą
drugorzędową
(konformacją
przestrzenną), co wiąże się z odmiennymi właściwościami
fizykochemicznymi.

Decydująca jest konformacja białka prionowego. wg jednej z
hipotez PrPsc oddziałują na formę białka endogennego,
przyczyniając się do zmiany jego struktury. Jak w „reakcji
łańcuchowej” dochodzi do przemiany konformacji utworzonej
głównie z
α-helisy do postaci nieprawidłowej z przewaga
struktury
β-helikalnej.
Powstałe cząsteczki białka wykazują odmienne właściwości, są
trudno rozpuszczalne, tworzą agregaty (przyłączają się do
glikozaminoglikanów), a ich nagromadzenie w komórce
prowadzi do jej zmian morfologicznych i śmierci.
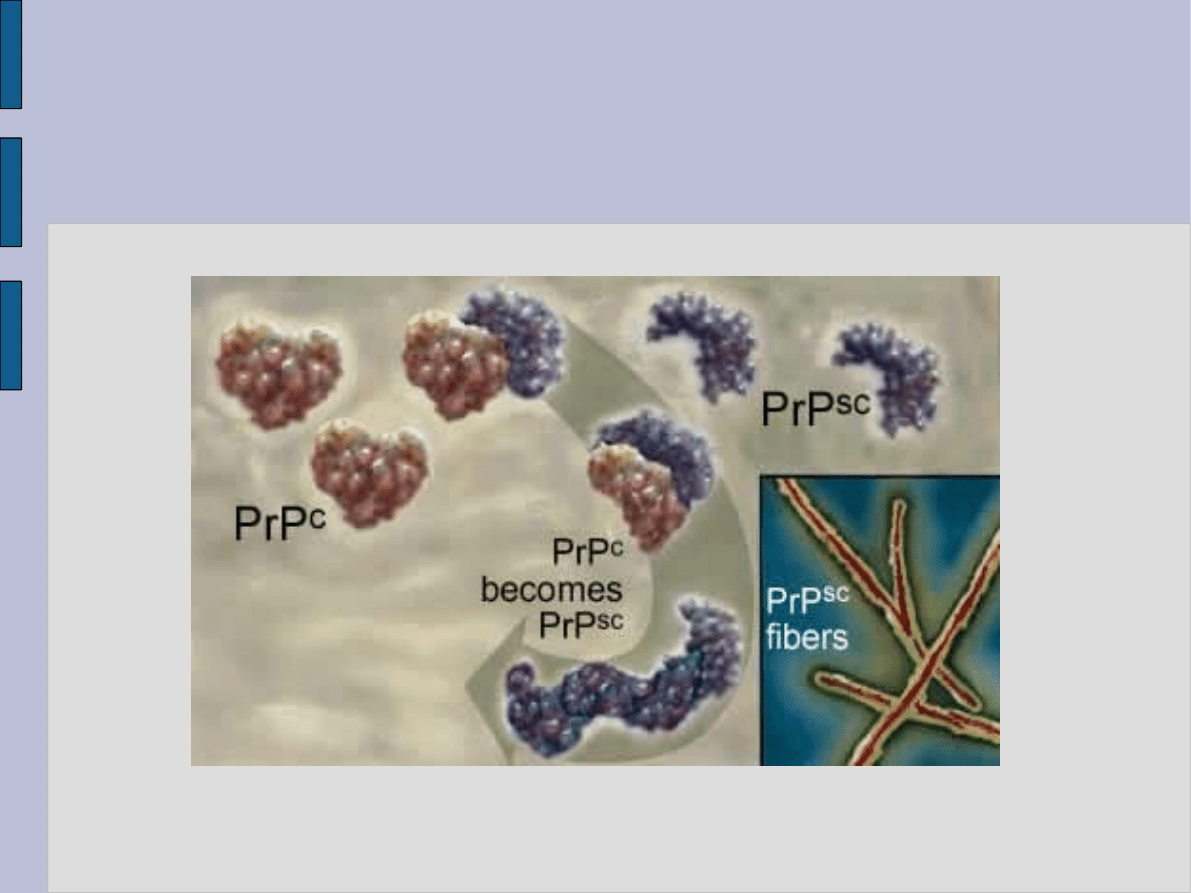

Białko PrP nie wywołujące
choroby (oznaczane PrPc, c z
ang. cellular - komórkowe)
posiada trzy
α-helisy i dwie tzw.
ß- harmonijki
Białko PrP o domniemanym
patogennym charakterze
(oznaczane PrPsc, od
scrapie) zawiera przewagę
struktury tzw. ß- harmonijki
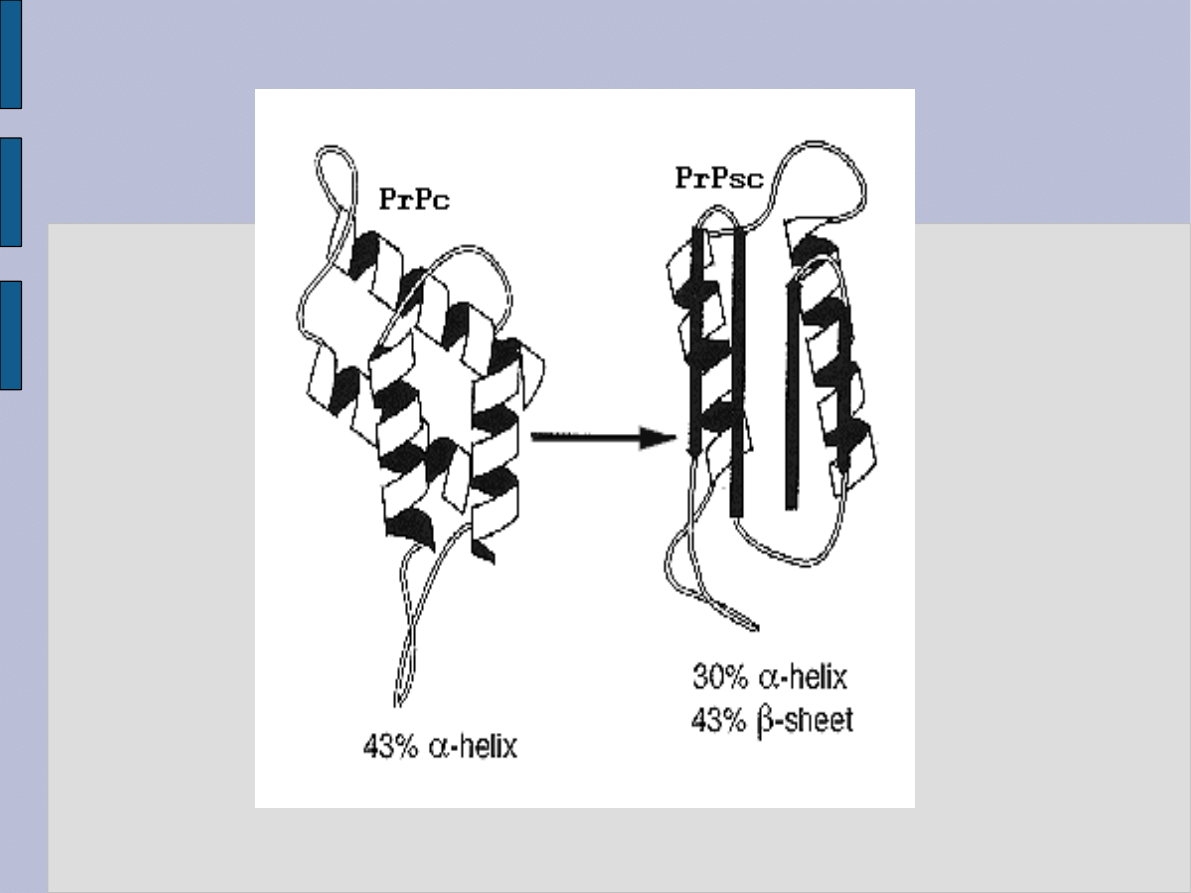

PrPc jest całkowicie rozpuszczalne w wodzie i denaturujących
detergentach,
PrPSc jest nierozpuszczalne w wodzie
(stąd jedynie hipotetyczna jest struktura tego białka -
nierozpuszczalność w wodzie uniemożliwia poddanie białka
badaniu z użyciem spektroskopii NMR)

Priony a drożdże
W 1997 roku naukowcy odkryli obecność prionów
towarzyszących DNA drożdży, którym jest ono niezbędne w
cyklu rozwojowym. Po odwirowaniu DNA z komórek
drożdży, na dnie probówek z DNA zostawały mętne resztki,
które później zidentyfikowano właśnie jako priony. Podczas
podziału komórki, DNA jest kopiowane, zaś priony ulegają
podzieleniu na dwa identyczne białka, które następnie
dobudowują sobie drugą część przez przemianę innej
proteiny. Badacze doszli do wniosku, że priony są
niezbędne do rozmnażania komórek drożdży.

Choroby prionowe
prawidłowa struktura białka prionowego ludzi jest konieczna
do właściwego funkcjonowania układu nerwowego
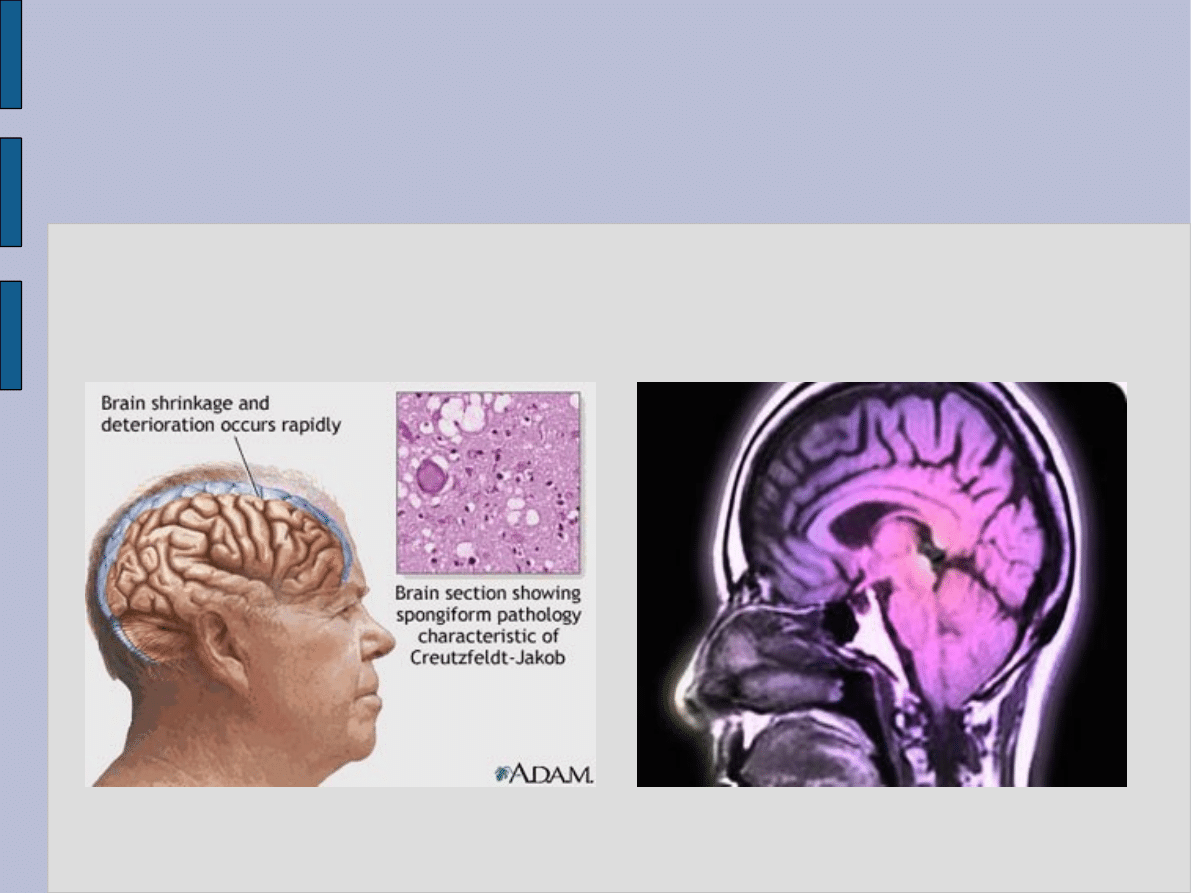
Wszystkie choroby prionowe charakteryzują się zmianami
zwyrodnieniowymi układu nerwowego (TSE- Transmissible
Spongiform Encephalopathies) i długim okresem inkubacji
(od kilku miesięcy do kilkudziesięciu lat)
mutacje białka PrP występujące u cierpiących na choroby
prionowe( tj. CDJ, FFI(śmiertelna dziedziczna bezsenność)
czy Kuru) są przyczyną zaburzeń przejawiających się
ataksją, otępieniem lub zanikami pamięci.
Choroby prionowe są chorobami najczęściej osobników
starych, ponieważ z biegiem czasu wzrasta ilość białka PrP-Sc

• Białka takie mogą powstawać na skutek somatycznej (więc
niedziedzicznej) mutacji genu je kodującego, a także bez
występowania mutacji wskutek powolnego, samorzutnego
przekształcania się białek prawidłowych w nieprawidłowe lub
też na skutek odziedziczenia wadliwego genu. Ponadto w
pewnych warunkach wszczepienie zdrowemu człowiekowi
lub zwierzęciu białka prionowego (od chorego zwierzęcia lub
człowieka) powoduje narastanie ilości zmienionego białka i w
rezultacie pojawienie się objawów encefalopatii gąbczastej.
Pozwala to podzielić choroby prionowe na sporadyczne,
rodzinne i przepasażowane
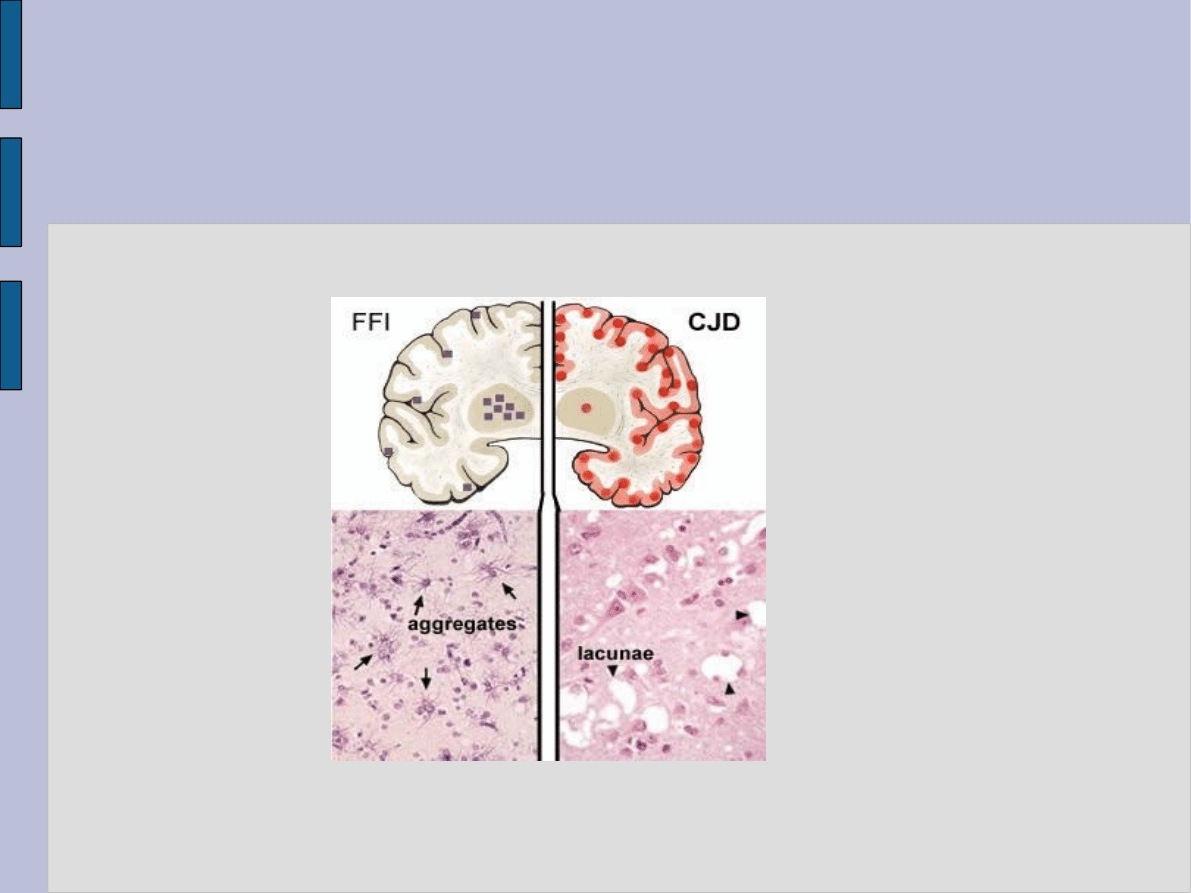

Choroby prionowe
Postacie przepasażowane u ludzi występują bardzo rzadko.
Obecnie związane mogą być albo z działalnością medyczną
(głównie: leczenie hormonem wzrostu lub gonadotropiną
uzyskaną z ludzkich przysadek, implantacje opony twardej,
rogówki), albo z nowym wariantem CJD gdzie dochodzi do
przeniesienia zakażenia ze zwierząt (bydła z BSE) na ludzi.
Najprawdopodobniej historycznym przykładem jest kuru,
choroba występująca u nowogwinejskiego plemienia
praktykującego rytualny kanibalizm.

•SPORADYCZNE:Choroba
Creutzfeldta-Jakoba (sCJD) 85 – 90%
przypadków,
Śmiertelna bezsenność (sFI)
•RODZINNE:Śmiertelna rodzinna
bezsenność (FFI)
•PRZEPASAŻOWANE:jatrogenna
(iCJD) i nowy wariant (vCJD), Kuru
(nie występuje)
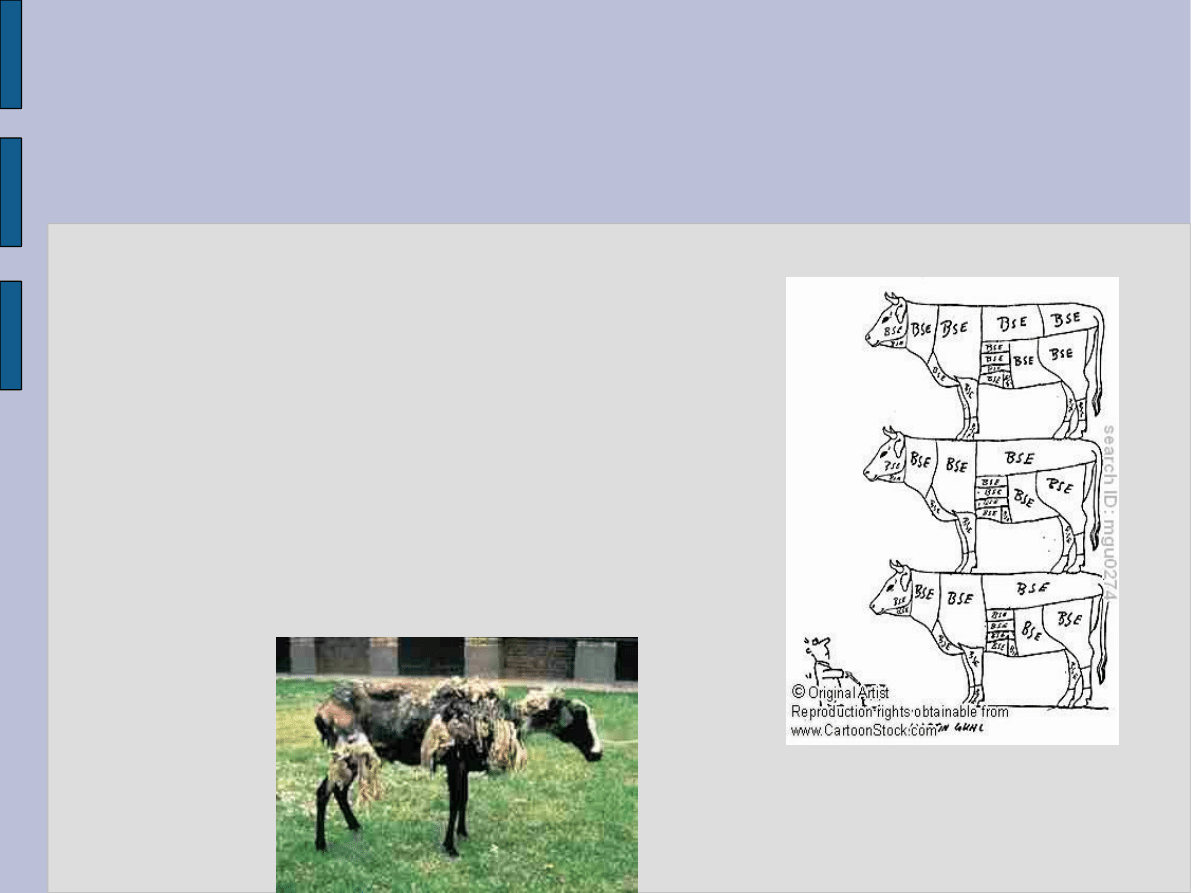
Choroby prionowe u zwierząt
Do najbardziej znanych prionowych chorób zwierząt
należy tzw. „scrapie” występująca wśród owiec oraz
pochodząca od niej choroba szalonych krów – BSE.
Naukowcy uważają, że BSE u bydła rozwinęła się w
wyniku podawania im mączek mięsno – kostnych
wyprodukowanych z ciał owiec padłych na scrapie. Jako
czynnik przenoszący chorobę uznane zostało właśnie
białko prionowe.

ELISA test
(Enzyme Linked ImmunoSorbent Assay) jest
powszechnie używany do wykrycia prionów w
badanym materiale z użyciem przeciwciał
monoklonalnych (wykazują jednakową swoistość
względem danego antygenu ewentualnie takie
samo lub podobne powinowactwo ) lub
poliklonalnych połączonych z odpowiednim
enzymem.

Źródła
http://pl.wikipedia.org/wiki/Priony
http://www.biotechnolog.pl/news-1021.htm
http://www.biotechnolog.pl/slownik-14.htm

Dziękuję za uwagę!
Document Outline
- Slajd 1
- Slajd 2
- Slajd 3
- Slajd 4
- Slajd 5
- Slajd 6
- Slajd 7
- Slajd 8
- Slajd 9
- Slajd 10
- Slajd 11
- Slajd 12
- Slajd 13
- Slajd 14
- Slajd 15
- Slajd 16
- Slajd 17
- Slajd 18
- Slajd 19
- Slajd 20
- Slajd 21
- Slajd 22
Wyszukiwarka
Podobne podstrony:
grzyby wirusy priony
priony
mikrobiologia zdj 5, WIRUSY, WIROIDY, RIKETSJE I PRIONY
Priony, Apy 2 sem
Priony, Mikrobiologia
priony
priony-materiały, biologia mol różne
R01 Wstęp priony
priony i wirusy TR44IXXNDFG3OWZQT5LT5AUPO43XTC4UCWBIBSY
Creutzfeldta Jakoba Priony
prezentacja priony
onkogeny priony id 335421 Nieznany
priony
wirusy priony
mikro priony
więcej podobnych podstron