
NIEDOBORY
ODPORNOŚCI

Niedobory odpornościowe
powstają
w wyniku
braku
lub
niewłaściwej
funkcji
jednego lub więcej
elementów układu
odpornościowego
PRZYCZYNY
NIEDOBORÓW
ODPORNOŚCI:
PODZIAŁ NIEDOBORÓW
ODPORNOŚCI
Swoiste
Nieswoiste
odchylenia funkcji limfocytów
T
lub B
zaburzenia dotyczące
składników dopełniacza lub
fagocytozy, które zapewniają
odporność nieswoistą
Pierwotne
Wtórne
zależne od wewnętrznych
defektów
komórek układu
odpornościowego
i są w większości uwarunkowane
genetycznie
spowodowane czynnikami
zewnętrznymi, takimi jak:
leki, promieniowanie,
niedożywienie,
czy zakażenie.
AIDS jest wtórnym
niedoborem odporności,
wynikającym z zakażenia
wirusowego
NIEDOBOR
Y

PODZIAŁ PIERWOTNYCH
NIEDOBORÓW ODPORNOŚCI,
OPARTY NA TYPIE KOMÓREK
DOTKNIĘTYCH WADĄ
Niedobory zależne od limfocytów B
Niedobory zależne od limfocytów T
Niedobory zależne od komórek żernych
Niedobory zależne od składników
dopełniacza
Złożone niedobory odporności

Zakres niedoborów limfocytów B
waha się od opóźnionego dojrzewania Ig, poprzez
niedobory pojedynczych izotypów, do
agammaglobulinemii sprzężonej z chromosomem X,
gdy dotknięci schorzeniem chłopcy nie mają ani
limfocytów B
ani Ig w surowicy
Chorzy z defektami funkcji limfocytów B
mają nawracające zakażenia ropne, takie jak:
zapalenie płuc, zapalenie ucha środkowego oraz
zatok. Bez leczenia, mogą oni rozwinąć poważną
obstrukcyjną chorobę płuc (rozstrzenie), gdyż
nawracające zapalenia płuc niszczą elastyczność
dróg oddechowych
PIERWOTNE NIEDOBORY
LIMFOCYTÓW B

PIERWOTNE NIEDOBORY
LIMFOCYTÓW B
Agammaglobulinemia sprzężona z
chromosomem X
Agammaglobulinemia dziedziczona
autosomalnie recesywnie
Pospolity zmienny niedobór odporności
Izolowany niedobór IgA
Niedobór odporności z podwyższeniem IgM
Niedobór podklas IgG
Przejściowa hipogammaglobulinemia
niemowląt
Agammaglobuline
mia sprzężona z
chromosomem X
(X-LA)
µ
IgM
µ
IgM
IgM
IgD
IgM
IgG1,2,
3
IgM
IgA1 lub
2
Ig
M
IgE
IgM
µ
IgM
Ig
M
IgD
Ig
M
IgD
IgG1,2,
3
IgE
IgM
IgM
IgM
µ
IgM
IgG1,2,
3 lub 4
IgM
IgA1
lub 2
Ig
M
IgE
Niedobór IgA
Niedobór
odporności ze
wzrostem IgM
(HIGM)
Pospolity zmienny
niedobór odporności
Wydzielane
przeciwciał
o
Limfocyt
pre-B
Niedojrzały
limfocyt B
Limfocyt B
Dojrzały
limfocyt B
Komórka
plazmatyczna
IgM IgG1,2,3,4
IgE
IgM
IgM
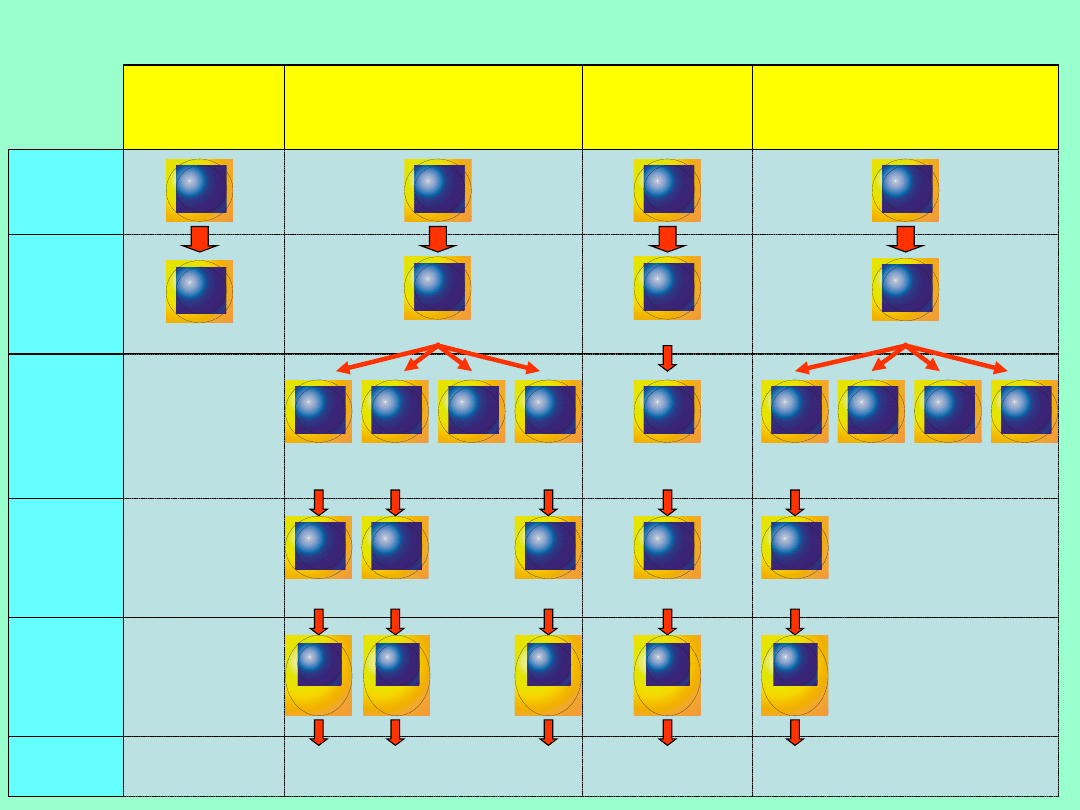
Dojrzewanie limfocytów B w wybranych niedoborach
odporności

Agammaglobulinemia sprzężona z
chromosomem X (agammaglobulinemia
Brutona)
Choroba dotyczy chłopców i charakteryzuje się
całkowitym brakiem przeciwciał oraz śladową obecnością
limfocytów B w krążeniu (<1%).
Dominują w niej nawracające zakażenia bakteryjne dróg
oddechowych.
Bez leczenia prowadzi do przewlekłego zapalenia zatok i
zmian rozstrzeniowych oskrzeli. Najczęstszą przyczyną
śmierci jest przewlekłe enterowirusowe zapalenie opon
mózgowych i mózgu.
Za chorobę odpowiada
mutacja w genie Btk
,
kodującym
kinazę tyrozynową należącą do rodziny
cytoplazmatycznych kinaz Tec. Aktywność kinazy Btk
wykryto w komórkach hematopoetycznych
z wyjątkiem limfocytów B. Kinaza ta jest potrzebna do
wzrostu prekursorów limfocytów B i ich dojrzewania.
Brak jest leczenia przyczynowego. Stosuje się terapię
zastępczą polegającą na podawaniu przeciwciał.
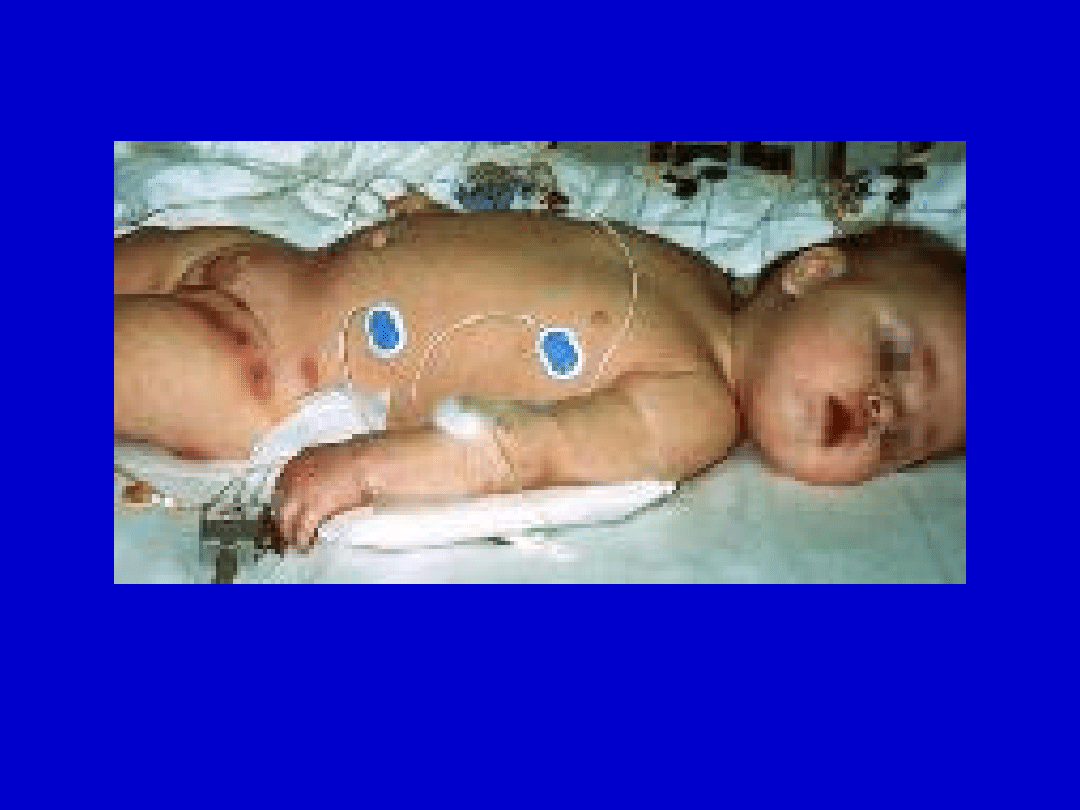
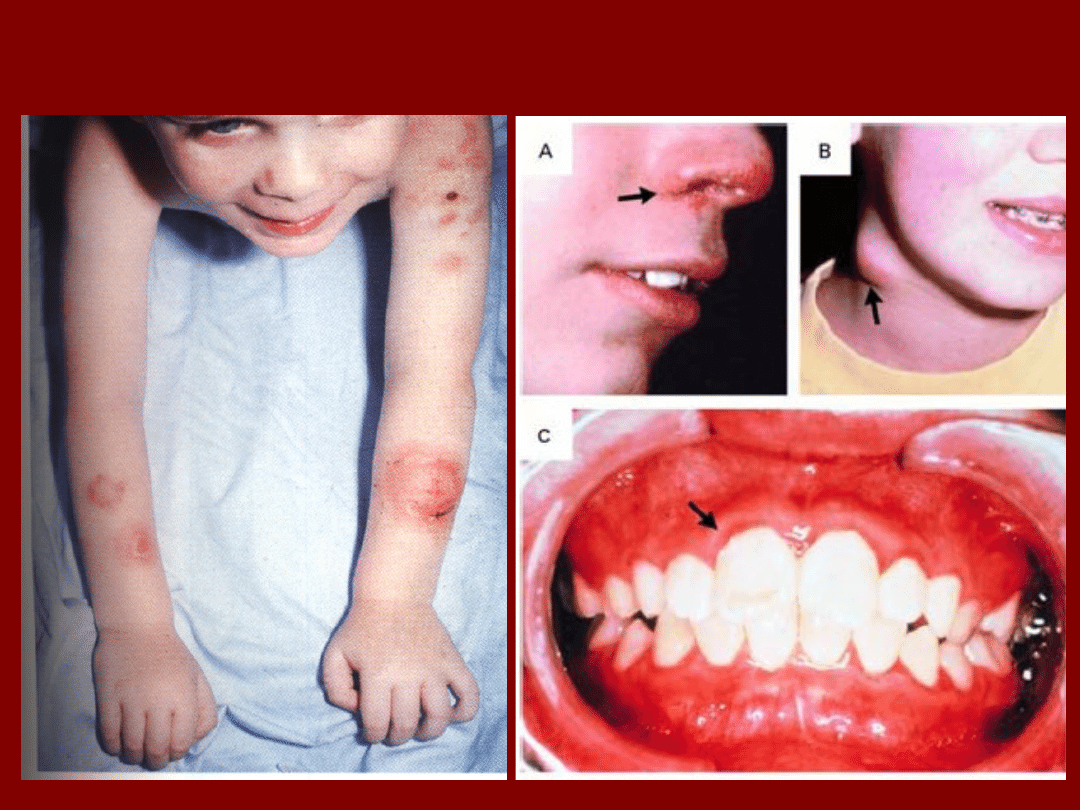
Nawracające zapalenie ucha oraz obszary zapalenia tkanki łącznej. U
dziecka wykryto infekcje Pseudomonas aeruginosa i Staphylococcus
aureus, anemię hemolityczną (choroba autoimmunizacyjna) i neutropenię.
Stwierdzono mutację w egzonie 15 genu Btk, polegającą na zamianie
asparaginy na walinę.
Agammaglobulinemia sprzężona z
chromosomem X

Agammaglobulinemia dziedziczona
autosomalnie recesywnie
Obraz kliniczny identyczny jak w przypadku
agammaglobulinemii Brutona
Dziedziczenie ma charakter autosomalny
U pacjentów wykryto różnego typu mutacje, np:
U pacjentów nie wykrywa się mutacji genu Btk
genu µ dla łańcucha ciężkiego przeciwciał, leżącego na
14.
chromosomie,
genu α dla łańcucha ciężkiego przeciwciał,
białka adaptorowego limfocytów B – Blnk (B-linker
adapter protein),
niezbędnego na etapie dojrzewania limfocytów z pro-B do
pre-B

Pospolity zmienny niedobór odporności
(
C
ommon
V
ariable
I
mmuno
D
eficiency –
CVID
)
Nazwą tą określa się grupę wrodzonych zaburzeń dojrzewania
limfocytów B.
Charakteryzuje ją brak komórek plazmatycznych w narządach
limfatycznych, hipo- lub agammaglobulinemia i nawracające
zakażenia układu oddechowego.
We krwi zwykle obserwuje się znaczne obniżenie poziomu
immunoglobulin wszystkich klas, aczkolwiek produkcja IgM
bywa najmniej upośledzona.
Węzły limfatyczne przejawiają charakterystyczną hiperplazję.
Od agammaglobulinemii sprzężonej z płcią, CVID odróżnia
autosomalny sposób dziedziczenia oraz ujawnianie się
pierwszych objawów choroby
w późniejszych latach życia.
U około 10% chorych na CVID występują choroby
autoimmunizacyjne.
W zespole tym wyższe jest też ryzyko wystąpienia chłoniaków
i nowotworów przewodu pokarmowego.
Brak jest leczenia przyczynowego, chorzy z CVID wymagają
podawania immunoglobulin.
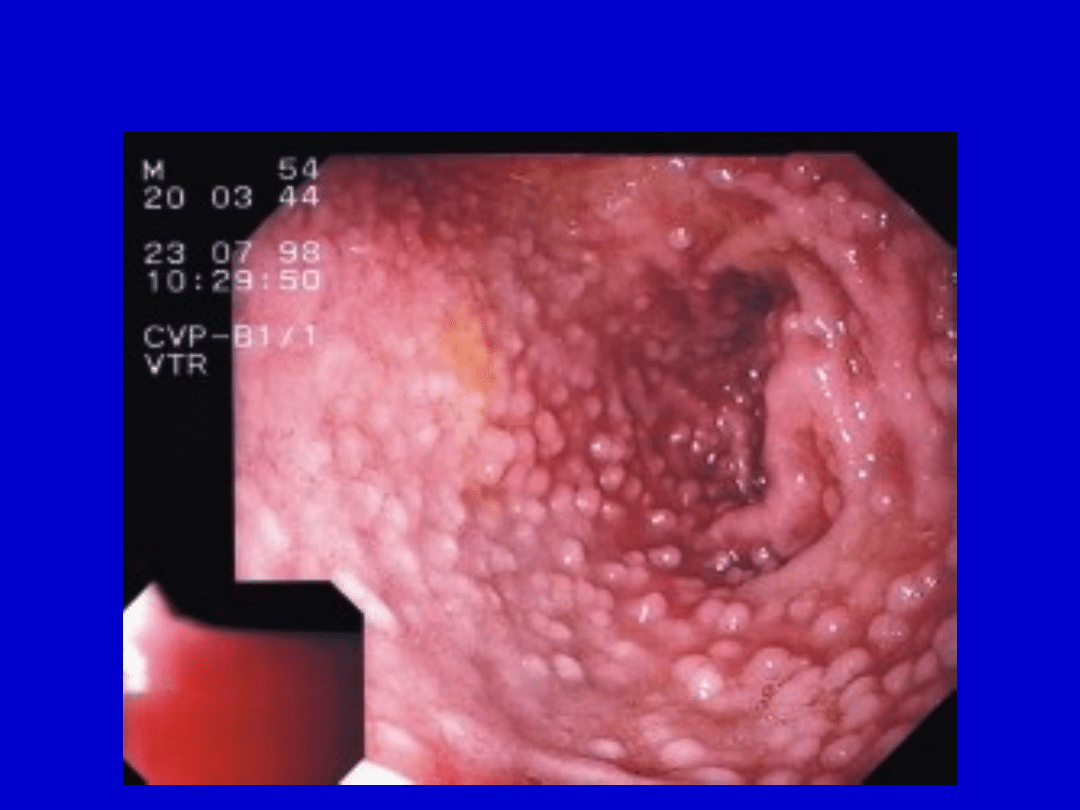
Hiperplazja węzłów limfatycznych w
dwunastnicy
u pacjenta chorego na CVID

Izolowany niedobór IgA
Najczęstszy wrodzony niedobór odporności, występuje
z częstością 1 na 600 urodzeń w Europie i USA.
Rozpoznaje się go wówczas, gdy stężenie IgA w
surowicy jest mniejsze niż 0,05 g/l
(norma – 2 g/l)
.
Stężenie przeciwciał innych klas pozostaje prawidłowe.
Objawami są nawracające bakteryjne zakażenia dróg
oddechowych.
Obecnie CVID i izolowany niedobór IgA uważa się za
dwie różne postacie tej samej choroby, o nie
wyjaśnionym dotychczas podłożu genetycznym.

Niedobór odporności związany z
chromosomem X ze zwiększonym stężeniem
IgM (HIGM)
Charakteryzuje się bardzo niskim poziomem IgA oraz
IgG,
przy zwiększonym lub normalnym poziomie IgM oraz
normalnym poziomie limfocytów B.
W zespole tym dominuje zapalenie płuc powodowane
przez Pneumocystis carinii, zakażenie Cryptosporidium
parvum.
Częste są ponadto choroby wątroby.
Za chorobę odpowiedzialna jest
mutacja genu
kodującego ligand
dla CD40 (CD154)
.
Cząsteczka ta jest obecna na
aktywowanych limfocytach T. Wiąże się ona z CD40 na
limfocytach B, przekazując sygnał niezbędny do zmiany
izotypu wytwarzanych przeciwciał z IgM na IgG, IgA lub
IgE. Brak tego receptora powoduje
zatrzymanie
limfocytów B na etapie syntezy IgM
. Brak również
ośrodków rozmnażania w węzłach chłonnych.
Leczenie polega na podawaniu przeciwciał.
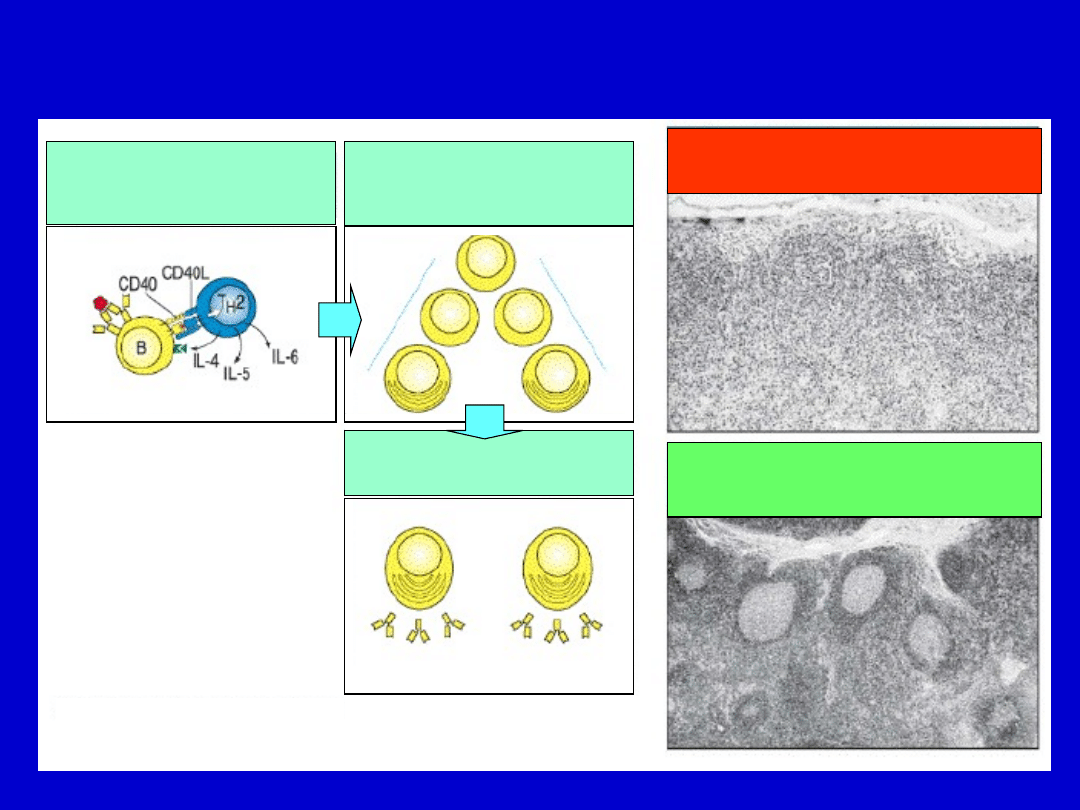
Limfocyty B pacjentów z niedoborem odporności ze
zwiększonym stężeniem IgM nie są zdolne do pełnej
aktywacji
Węzeł limfatyczny pacjenta z
podwyższonym stężeniem IgM
(brak ośrodków rozmnażania)
Normalny węzeł limfatyczny
(obecne ośrodki rozmnażania)
Rozpoznanie antygenu
indukuje ekspresję cząsteczek
efektorowych przez limfocyty T,
co z kolei aktywuje limfocyty B.
Proliferacja limfocytów B
Różnicowanie do komórek
plazmatycznych,
wydzielających przeciwciała

Niedobory podklas IgG
Najczęściej są to niedobory asymptomatyczne
U dzieci najczęściej występuje niedobór IgG2,
a u dorosłych IgG3. Izolowany niedobór IgG1
wykrywa się niezwykle rzadko.
Niedobór IgG2 jest związany ze słabszą
odpowiedzią
na antygeny polisacharydowe - składnik
otoczki wielu bakterii. U niektórych pacjentów
nie musi temu towarzyszyć zmniejszona
podatność na zakażenia bakteriami –
rolę IgG2
mogą przejąć IgG1.
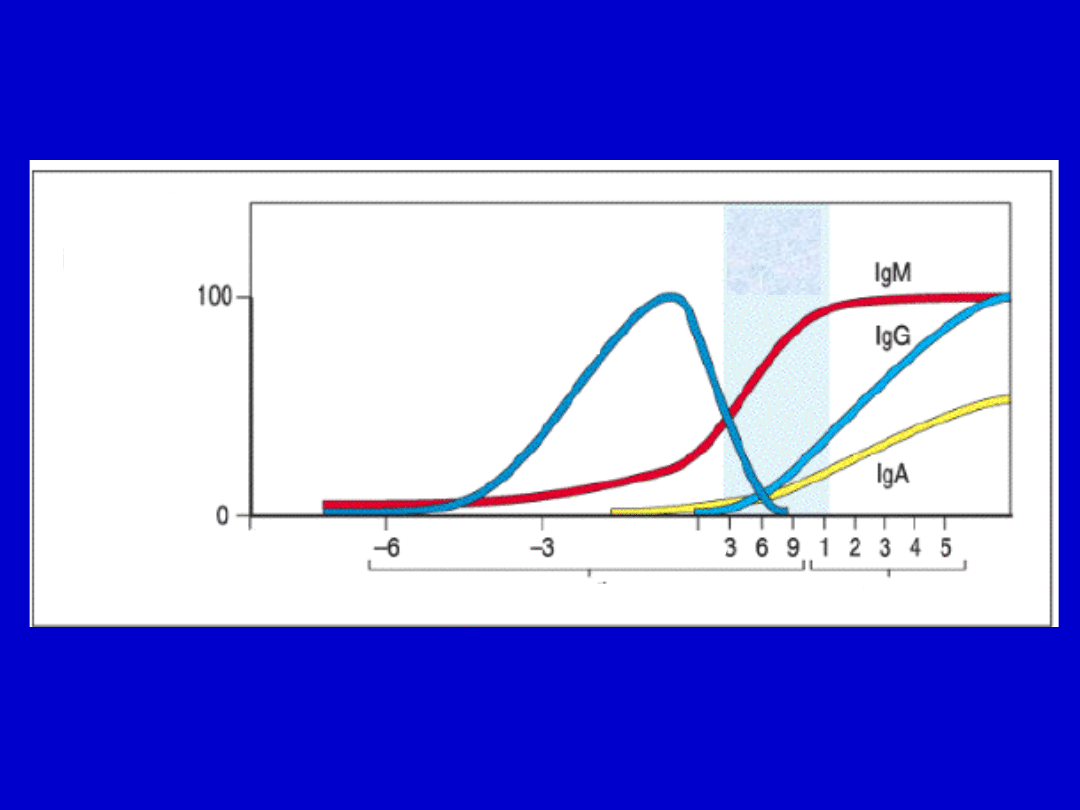
Przejściowa hipogammaglobulinemia
niemowląt
Przejściowa hipogammaglobuinemia niemowląt pojawia
się
od 6-12 miesiąca życia i może prowadzić do większej
podatności
na infekcje.
Poziom
immunoglobulin
poczęcie
Przekazane
biernie
matczyne IgG
narodzin
y
miesiące
lata
dorosłoś
ć
Przejściowy
niski
poziom IgG

PIERWOTNE NIEDOBORY
LIMFOCYTÓW T
Chorzy nie posiadający limfocytów T lub z
upośledzoną funkcją tych komórek są
szczególnie podatni
na zakażenia oportunistyczne.
Prawidłowa funkcja ludzkich limfocytów B jest
uzależniona od limfocytów T, toteż niedobory
tych ostatnich prowadzą również do zaburzeń
odporności humoralnej.
W wyniku niedoborów limfocytów T dochodzi do
złożonych zaburzeń odpowiedzi
zarówno komórkowej
jak i humoralnej.

Niedobory limfocytów T
Niedobór
limfocytów T
Choroba
Brak grasicy
Zespół DiGeorge`a; zaburzenia
w embriogenezie grasicy
Defekt komórki
macierzystej
SCID; 50% chorych ma
defekt
łańcucha γ
wchodzącego w skład wielu
receptorów dla cytokin, łącznie z IL-2R
Śmierć tymocytów
SCID; 25% chorych wykazuje
niedobory enzymu
dezaminazy
adenozynowej
lub niedobory
fosforylazy nukleozydów purynowych
;
toksyczność spowodowana
zwiększeniem metabolitów puryn,
hamujących syntezę DNA

PIERWOTNE NIEDOBORY
LIMFOCYTÓW T
Zespół DiGeorge’a
Zespół Wiskotta-Aldricha
Dziedziczna ataksja teleangiektazja
Ciężkie złożone niedobory immunologiczne
Niedobór fosforylazy nukleozydów
purynowych
Niedobór cząsteczek MHC klasy II

Zespół DiGeorge’a
Wrodzony defekt narządów pochodzących z 3 i 4
kieszonki gardłowej (grasicy i przytarczyc); 6 tydzień
życia płodowego.
Dzieci urodzone z zespołem DiGeorge’a mają
deformację części twarzowej czaszki, wady serca i
dużych naczyń
oraz brak lub niedorozwój grasicy i przytarczyc.
Przyczyną niedoboru są głównie delecje fragmentu
chromosomu 22. Jednym z genów odpowiedzialnych
za niedobór jest
gen TbX1
, kodujący czynnik
transkrypcyjny.
Tylko u 20% chorych występuje zmniejszona liczba
i/lub aktywność limfocytów T. Z czasem u chorych
pojawiają się limfocyty T i dochodzi do korekcji
niedoboru wskutek ektopowego rozwoju grasicy.

Zespół DiGeorge’a

Zespół Wiskott-Aldricha (WAS)
Zespół dziedziczy się z płcią.
Charakterystyczny objaw to skaza krwotoczna.
Pierwsze objawy - zazwyczaj krwawe biegunki i
wybroczyny skórne.
W późniejszych latach obserwuje się choroby
autoimmunizacyjne oraz nowotwory (głównie chłoniaki).
Ponadto ma miejsce niedobór płytek krwi.
Upośledzona
jest odporność komórkowa
(zmiany ropne i zakażenia
oportunistyczne).
U chorych występują defekty limfocytów T (
zaburzenia
w strukturze cytoszkieletu
) i nieprawidłowe stężenie Ig.
Molekularną przyczyną jest mutacja
białka WASP
(
W
iskott-
A
ldrich
s
yndrome
p
rotein), którego wybiórczą
ekspresję obserwuje się
w limfocytach i megakariocytach.
W makrofagach i komórkach dendrytycznych z mutacją w
WASP obserwuje się upośledzoną zdolność do polaryzacji
oraz upośledzoną ruchliwość. Makrofagi mają również
upośledzoną fagocytozę przez receptor FcγR.
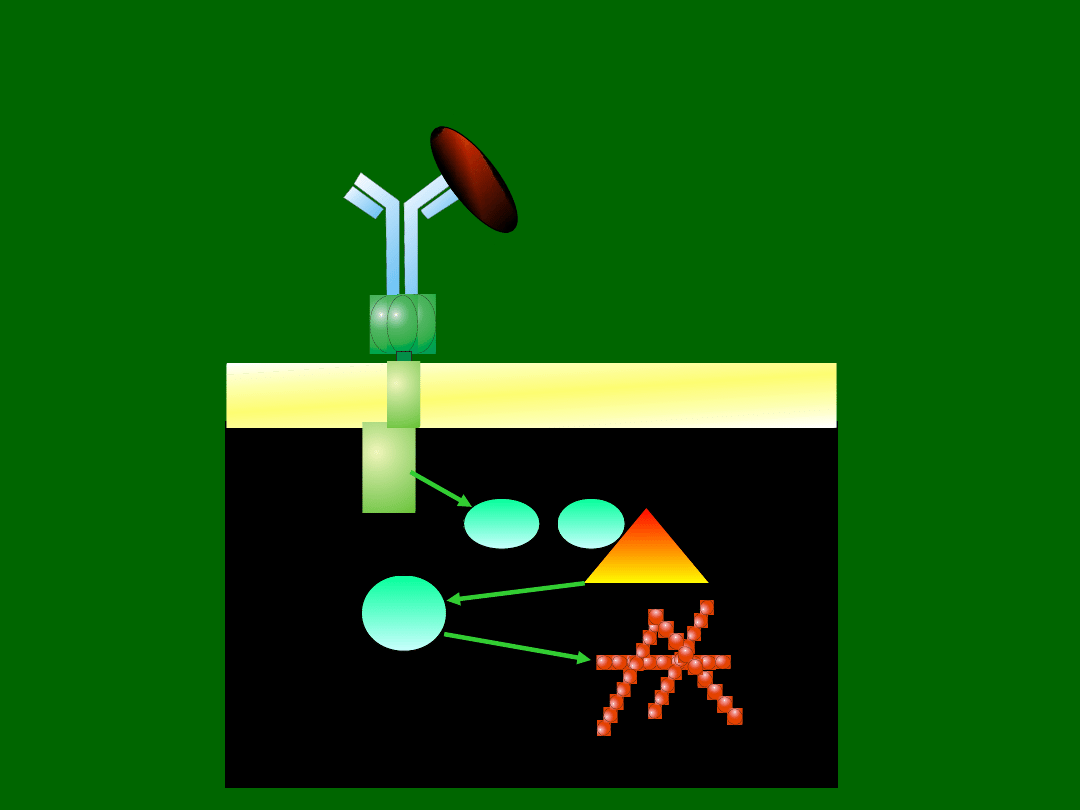
Rac 1 Cdc42
Arp 2/3
WASP
/
FcγR I
ba
kt
er
ia
cytoplazma
Filamenty aktynowe
Udział białka WASP w przekazywaniu sygnału z
receptorów błonowych do struktur cytoszkieletu
komórki
Zespół
Wiskott-Aldricha

Dziedziczna ataksja
teleangiektazja
Choroba dziedziczona autosomalnie recesywnie.
Charakterystyczne cechy to:
U chorych występuje niedobór limfocytów T i
zmniejszone
stężenie przeciwciał.
Jedną z przyczyn niedoboru jest
pęknięcie chromosomów
7 i 14
.
w obrębie genów dla TCR i łańcuchów ciężkich Ig.
Prowadzi
ono do defektu mechanizmów naprawy DNA
objawy neurologiczne (ataksja – chwiejny chód),
teleangiektazje – rozszerzone obwodowe naczynia
krwionośne
na skórze i gałkach ocznych,
hipogonadyzm,
zwiększona częstość występowania nowotworów.

Stanowi grupę uwarunkowanych
genetycznie schorzeń o odmiennej
patogenezie, ale o podobnym
obrazie klinicznym, który wynika z
braku czynności zarówno limfocytów
T, jak i B
Ciężki złożony niedobór odporności
(
S
evere
C
ombined
I
mmuno
D
eficiency –
SCID
)
Objawy SCID pojawiają się zwykle
w czasie pierwszych 6 miesięcy życia
Bez próby korekcji niedoboru, dzieci chore na SCID
umierają zazwyczaj w pierwszym roku życia
Często pierwszym objawem jest
przewlekłe zakażenie
błon śluzowych grzybami Candida
.
W dalszej
kolejności dochodzi
do uporczywych biegunek i zapaleń płuc

SCID: Agammaglobulinemia
typu „szwajcarskiego”
Klasyczna postać SCID
Dziedziczy się autosomalnie recesywnie
U podłoża niedoboru leży zaburzenie dojrzewania i
różnicowania komórek macierzystych limfopoezy.
Stwierdza się:
Istnieją dane, które wskazują na spowodowany mutacją
defekt enzymu –
rekombinazy DNA
– co wywołuje
zaburzenia rekombinacji genów dla receptora limfocytu T i
łańcua ciężkiego immunoglobulin. W trakcie tego procesu
rekombinaza DNA łączy ze sobą tak zwane sekwencje
sygnałowe, które graniczą z odpowiednimi sekwencjami
kodującymi genów
V
,
D
i
J
zanik centralnych (grasica) i obwodowych narządów
limfatycznych (węzły limfatyczne, grudki limfatyczne,
migdałki, śledziona)
limfopenię T i B oraz agammaglobulinemię we krwi
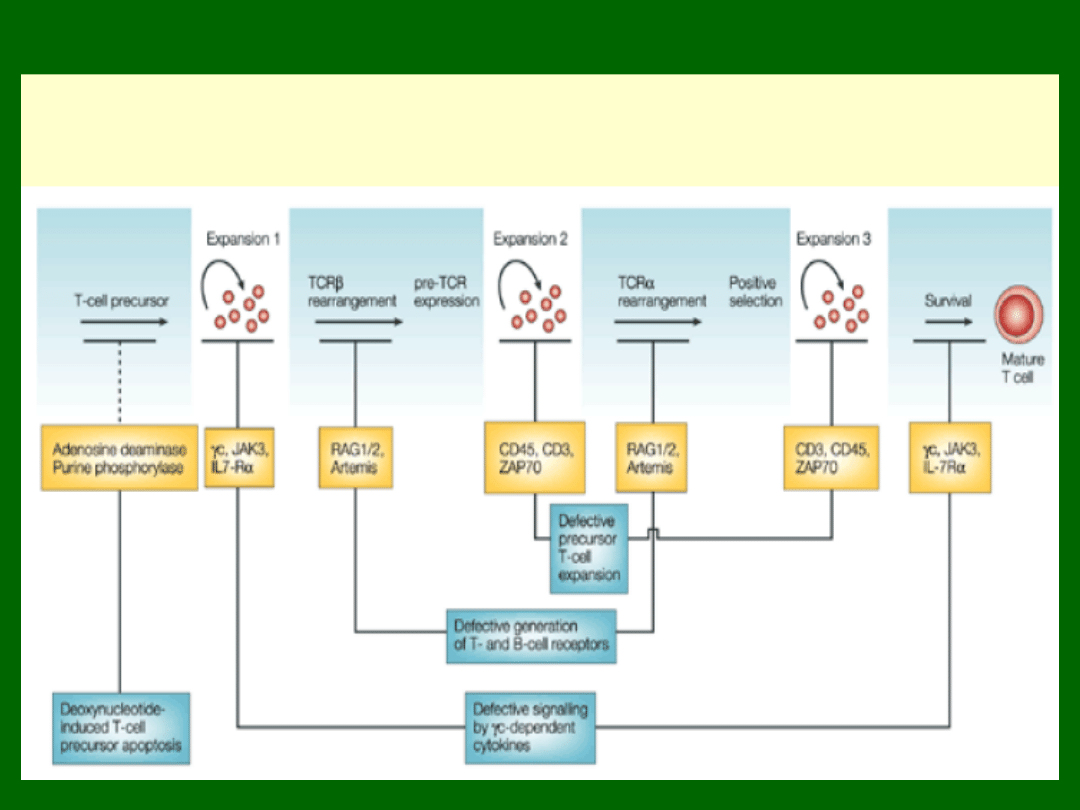
Defekty w rozwoju limfocytów T,
prowadzące do SCID

Postępująca ospa wietrzna u niemowlęcia
z ciężkim złożonym niedoborem odporności

Ciężki złożony niedobór odporności
związany z chromosomem X ze zmniejszoną liczbą
limfocytów T i prawidłowym poziomem limfocytów
B
Najczęściej spotykana postać SCID.
Obraz kliniczny nie różni się od klasycznego zespołu SCID.
Recesywny gen warunkujący wystąpienie objawów niedoboru
zlokalizowano na proksymalnej części długiego ramienia
chromosomu X
. W połowie przypadków zespół stanowi wynik
spontanicznej mutacji.
Przyczynę choroby stanowi mutacja w obrębie łańcucha
receptora
dla IL-2 (
IL-2Rγ
). Łańcuch γ wchodzi w skład receptora dla IL-2
o średnim
i wysokim powinowactwie do tej interleukiny oraz
prawdopodobnie znajduje się także w receptorach dla innych
cytokin.
Brak łańcucha γ, poprzez nieznany mechanizm, powoduje
eliminację szpikowych prekursorów limfocytów T w trakcie
dojrzewania
w grasicy. Brak limfocytów T pomocniczych oraz zaburzenia
aktywacji limfocytów B są przyczyną upośledzonej produkcji
przeciwciał. We krwi obwodowej przeważają limfocyty B
(czasami stanowią 90% wszystkich krążących limfocytów), lecz
wykrywa się jedynie niewielkie ilości IgA i IgG.

Ciężki złożony niedobór odporności
ze zmniejszoną liczbą limfocytów T, B oraz komórek
NK
Niedobór dziedziczony autosomalnie recesywnie
Związany jest on z niedoborem
deaminazy
adenozynowej (ADA)
. Niedobór spowodowany jest
mutacją genu dla ADA, leżącym na 20 chromosomie.
ADA jest enzymem uczestniczącym w metabolizmie
puryn
Niedobór ADA charakteryzuje się wcześniejszymi
objawami klinicznymi w porównaniu do pozostałych
postaci SCID
Defekt dotyczy limfocytów T, B i komórek NK.
Limfopenia jest głębsza w porównaniu do innych
postaci SCID. Oprócz klasycznych objawów SCID
(zakażenia i zatrzymanie wzrostu)
u połowy chorych występują zaburzenia w układzie
kostnoszkieletowym oraz zaburzenia neurologiczne
takie jak ślepota korowa czy dystonia

Niedobór fosforylazy nukleozydów
purynowych
Zespół dziedziczony jest autosomalnie recesywnie
Spowodowany jest defektem w genie kodującym
fosforylazę nukleozydów purynowych (
PNP
), leżącym
na chromosomie 14
Przy braku tego enzymu dochodzi do gromadzenia się
toksycznych metabolitów, deoksyguanozyny i
deoksyGTP
Limfocyty T są bardziej wrażliwe na akumulację tych
związków
w porównaniu do limfocytów B, dlatego niedobór ma
łagodniejszy charakter niż niedobór innego enzymu
uczestniczącego w metabolizmie puryn – deaminazy
adenozynowej
U chorych dominują nawracające zakażenia wirusowe
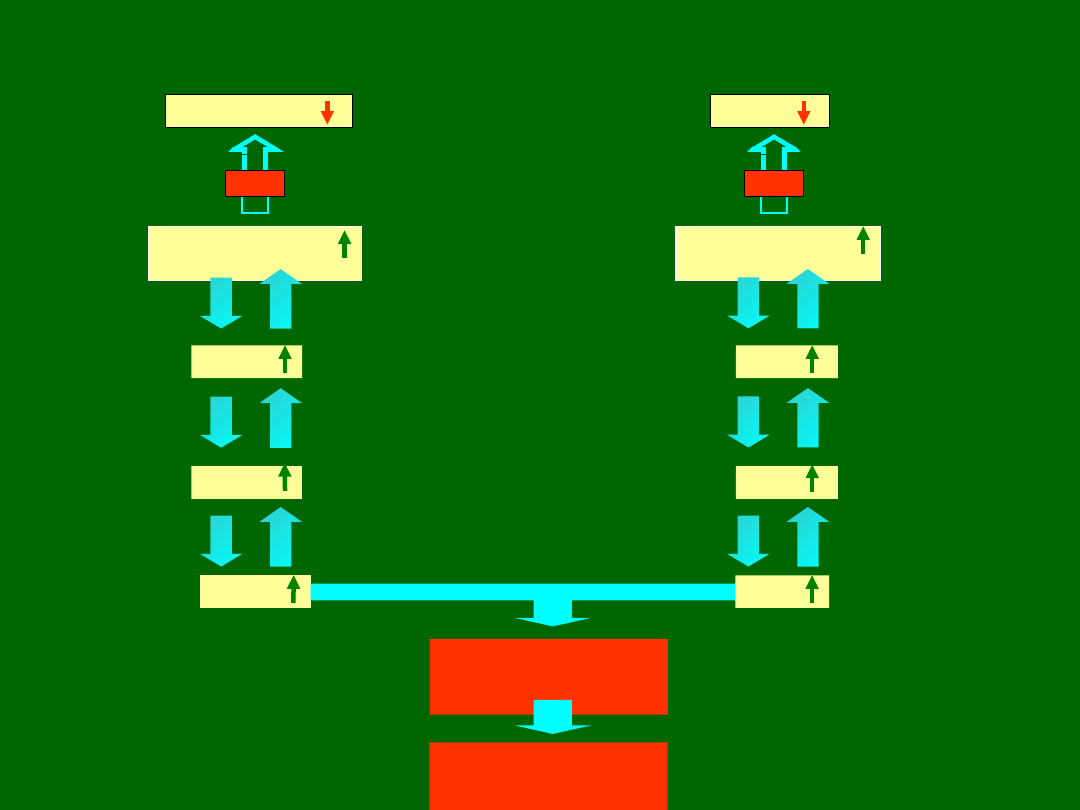
Rola niedoboru ADA i PNP w SCID
dezoksyinozyna
Niedobó
r
ADA
dezoksyadenozyn
a
dAMP
dADP
dATP
guanina
Niedobór
PNP
dezoksyguanozyn
a
dGMP
dGDP
dGTP
Hamowanie
reduktazy
rybonukleotydu
Hamowanie syntezy
DNA
i podziału komórki
TOKSYCZNE
METABOLITY

Ciężki złożony niedobór odporności
ze zmniejszoną liczbą limfocytów T, B
oraz prawidłową liczbą komórek NK
Przyczyną niedoboru jest
mutacja genów
RAG1
lub RAG2
, kodujących białka swoiste dla
limfocytów, odpowiedzialne za aktywacje
rearanżacji genów V(D)J, kodujących
fragmenty zmienne TCR lub BCR.
Charakterystyczne objawy kliniczne to
erytrodermia, hepatosplenomegalia,
eozynofilia i podwyższone stężenie IgE i brak
limfocytów B oraz ośrodków rozmnażania w
węzłach limfatycznych

IL2RG
T-
B+
NK-
JAK3
T-
B+
NK-
IL7R
T-
B+
NK+
CD45
T-
B+
NK+
RAG1
T-
B-
NK+
RAG2
T-
B-
NK+
ARTEMIS
T-
B-
NK+
ADA
T-
B-
NK-
Reticular Dysgenesis
T-
B+
NK+
SCID, multiple bowel atresias T-
B+/-
NK+
SCID, congenital abnormalities
T-
B+/-
NK+
Severe DiGeorge Syndrome
T- B+/- NK+
CD3 Deficiency
Tlow/+
B+
NK+
CD8 Deficiency
T+
B+
NK+
Severe Ataxia Telangiectasia Tlow B+/-
NK+
GENY ODPOWIEDZIALNE ZA SCID

Ciężki złożony niedobór odporności
związany z brakiem cząsteczek MHC
klasy II
Bardzo rzadka, dziedziczona autosomalnie
recesywnie postać SCID.
Charakteryzuje się brakiem lub niewielką liczbą
cząstek MHC klasy II na powierzchni
makrofagów, limfocytów B, komórek
dendrytycznych (stąd wcześniej używana
nazwa –
zespół „nagich” limfocytów
).
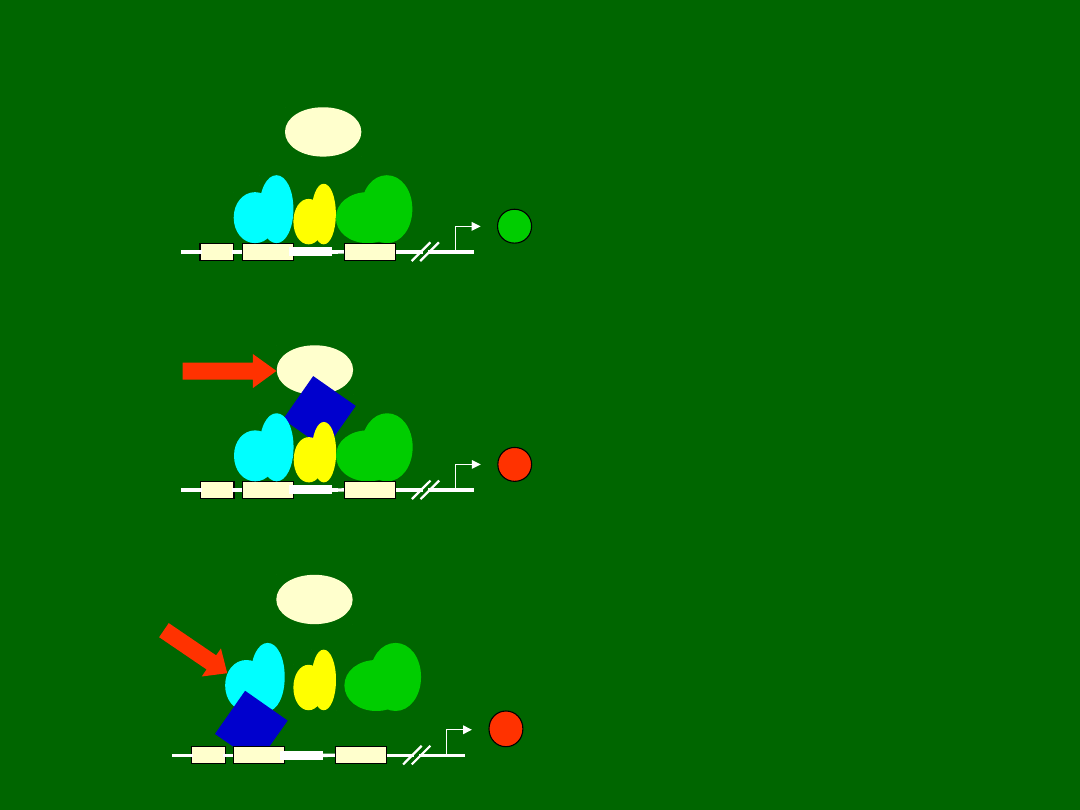
+
RFX
X2BP
NF-Y
W
X
X2
Y
CIITA
CIITA
-
RFX
X2BP
NF-Y
W
X
X2
Y
MUTACJA
RFX
X2BP
NF-Y
CIITA
-
W
X
X2
Y
Mutacja w obrębie genu dla CIITA
powoduje zahamowanie
transkrypcji, pomimo
prawidłowego połączenia się
czynników transkrypcyjnych
Mutacja genu dla RFX blokuje
wiązanie się innych białek
transkrypcyjnych z rejonem
promotora dla cząsteczek MHC
klasy II
Inicjacja transkrypcji genów dla MHC
klasy II wymaga przyłączenia do
promotora białek RFX, X2BP, NF-Y oraz
udziału molekuły CIITA, nie łączącej się
z sekwencją promotora
Ciężki złożony niedobór odporności związany z
brakiem cząsteczek MHC klasy II
MUTACJA
Niedobór odporności związany
z niedoborem cząsteczek MHC klasy I
Bardzo rzadki niedobór odporności
Typowe objawy to nawracające
zakażenia bakteryjne
i zmiany ziarninujące w skórze
ze skłonnością do martwicy.
Jego przyczyną jest
mutacja białek
transportujących TAP1 i TAP2
(
t
ransporters
a
ssociated with antigen
p
rocessing). Białka te
dostarczają peptydy antygenowe do siateczki
śródplazmatycznej, gdzie dochodzi do ich
wiązania w rowku łańcucha α cząsteczki MHC
klasy I. Przy braku peptydu cząsteczka MHC
ulega degradacji w cytplazmie.

Zaburzenia czynności komórek
żernych
Niedobór immunologiczny komórek żernych powstaje pod
wpływem:
zmniejszenia liczby komórek żernych (np. neutropenia),
upośledzenia czynności: chemotaksji, ruchliwości, fagocytozy i
zabijania
drobnoustrojów.
Funkcja komórek żernych zależy lub pozostaje pod kontrolą
innych czynników:
produktów aktywacji dopełniacza (chemotaksja, fagocytoza),
przeciwciał (fagocytoza),
cytokin (chemotaksja, cytotoksyczność).
Defekt obejmujący jeden z wymienionych układów powoduje
wtórne zaburzenia czynności komórek żernych i objawy
niedoboru immunologicznego.
Charakterystycznym objawem pierwotnych i wtórnych zaburzeń
czynności komórek żernych jest zwiększona podatność na
zakażenia bakteryjne i grzybicze.

Przewlekła choroba ziarniakowa (
CGD
)
Niedobory adhezji leukocytów (
LAD
)
Komórki fagocytujące (żerne) – leukocyty
wielojądrzaste
i komórki linii monocytów (makrofagów) są bardzo
ważne
w obronie gospodarza przeciw bakteriom
ropotwórczym
i innym drobnoustrojom wewnątrzkomórkowym.
Ciężki niedobór leukocytów wielojądrzastych
(neutropenia) powoduje uogólnione zakażenie
bakteryjne.
Z klinicznego punktu widzenia ważne są dwa
defekty dziedziczne fagocytów, powodujące
wrażliwość na poważne infekcje, często śmiertelne:
Zaburzenia czynności komórek
żernych

Przewlekła choroba ziarniakowa
(CGD)
Charakterystyczne cechy tej choroby to nawracające
zakażenia tkanek miękkich, płuc i innych organów
pomimo intensywnej terapii antybiotykowej.
Częstymi objawami są: trądzik, zapalenie naręczy,
zapalenie dziąseł, zapalenie płuc
i tworzenie się ziarniaków
Infekcje powodowane są przez mikroorganizmy,
takie jak: Staphylococcus aureus, Burkholderia
cepacia, Aspergillus species, Nocardia species i
Seratia marcescens
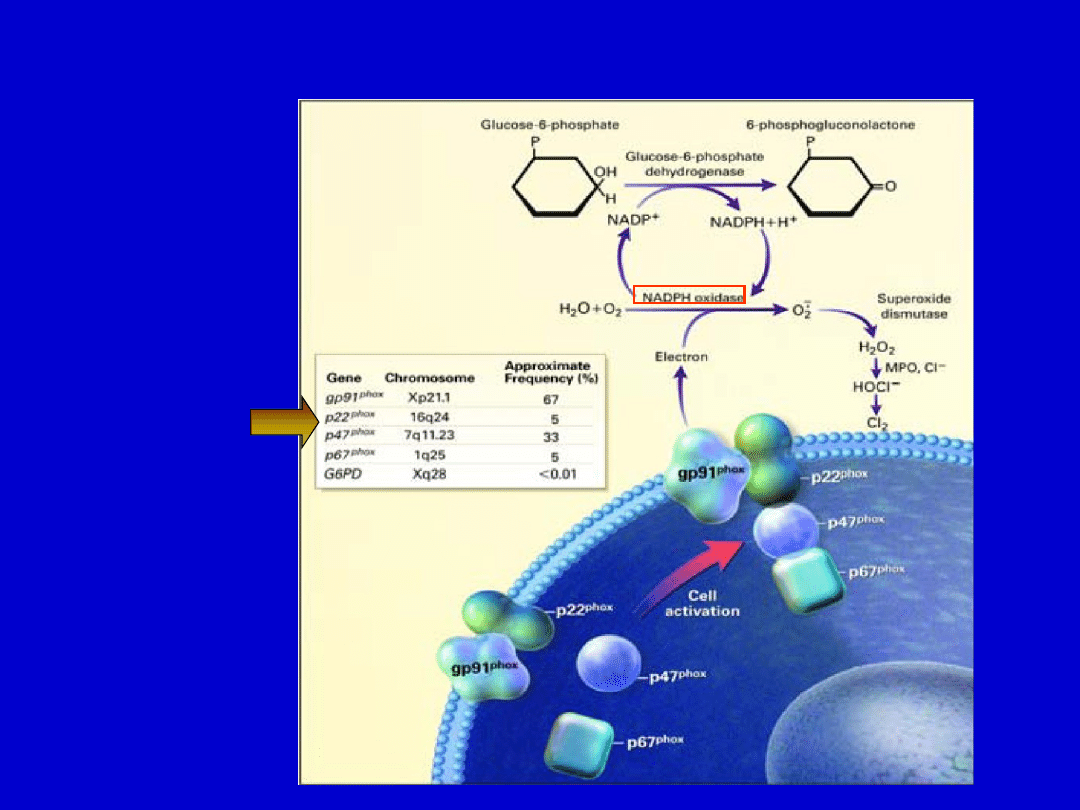
W chorobie tej zaburzony jest mechanizm zabijania
wewnątrzkomórkowego patogenów przez wolne
rodniki tlenowe: anion ponadtlenkowy, nadtlenek
wodoru i tlen singletowy. Powstają one w reakcji
katalizowanej przez
oksydazę NADPH
(kompleks
enzymatyczny), której defekt prowadzi do niedoboru
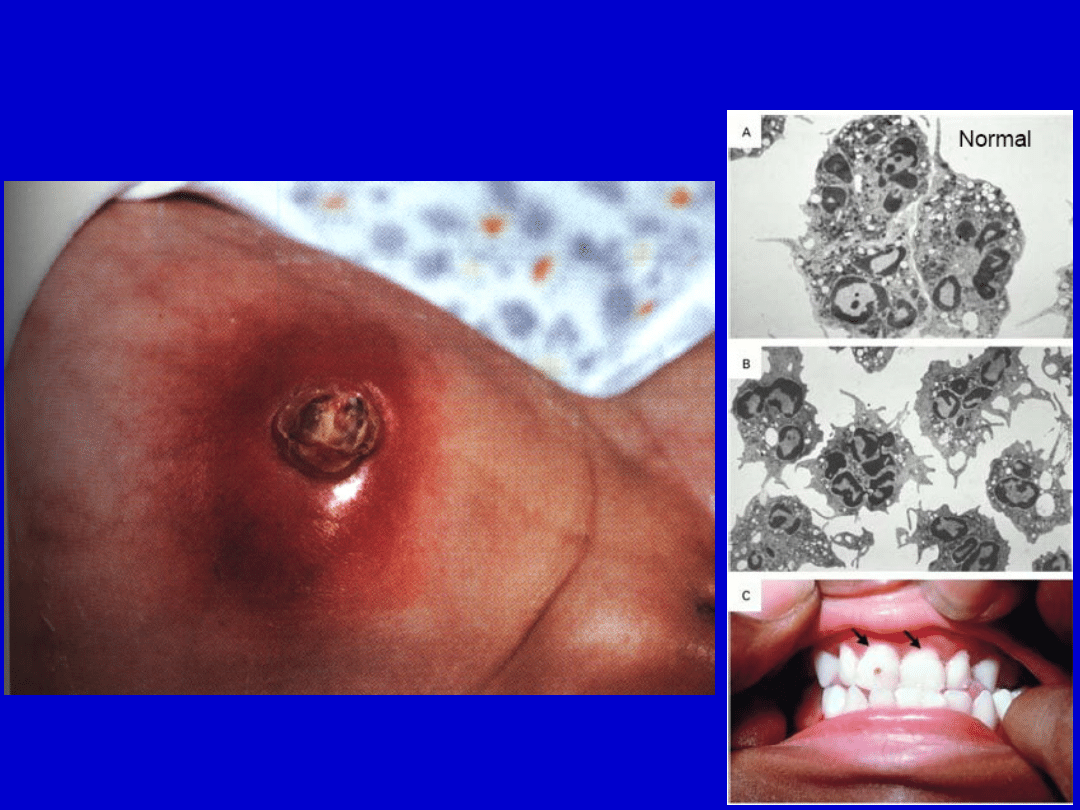
Infekcje w przewlekłej chorobie
ziarniakowej (CGD)
Przewlekła choroba ziarniakowa wynika z wadliwego działania
oksydazy NADPH
Mutacje
jednostek
wchodzących
w skład
kompleksu
oksydazy NADPH

Niedobory adhezji leukocytów
(LAD)
Charakteryzują się nawracającymi zakażeniami
bakteryjnymi
Częste objawy to: paradentoza, nawracające infekcje
błony śluzowej, skóry, jelit i dróg oddechowych,
zapalenia tkanki okołozębowej, z wczesnym
wypadaniem zębów
Najczęściej spotykanym zespołem niedoboru cząsteczek
adhezyjnych jest tak zwany typ 1
(
LAD-1
), w którym
przyczyną niedoboru jest mutacja w łańcuchu
β2
(
CD18
).
Liczba granulocytów we krwi jest zwiększona nawet przy
braku zakażenia, ale nie potrafią one przylegać do
śródbłonka ani tworzyć konglomeratów.
Drugi typ niedoboru (
LAD-2
) związany jest z
defektem
syntezy fukozy z GDP mannozy
. Uniemożliwia to
powstawanie ligandów dla selektyn, nie ma również
fukozylacji innych glikoprotein. Obok objawów typowych
dla LAD-1, LAD-2 charakteryzuje się występowaniem
zaburzeń wzrostu i opóźnienia umysłowego
Niedobory adhezji leukocytów
(LAD)

Defekty fagocytozy
Defekt
Choroba /
mechanizm
Różnicowanie komórki
macierzystej (wczesne etapy
rozwoju)
Neutropenia: zbyt mała liczba
neutrofilii
Brak adhezji do śródbłonka
oraz migracji
Niedobór adhezji leukocytów
(LAD), spowodowany utratą
ekspresji (poprzez mutację
specyficznych genów) ważnej
cząsteczki adhezyjnej, CD 18
(LFA)*
Nieprawidłowa fagocytoza
Zespół Chediaka-Higashiego:
brak fuzji fagososmu z
lizosomami
Defekt w zabijaniu
wewnątrzkomórkowym
Przewlekła choroba
ziarniakowa: defekt w genach
kodujących oksydazę NADPH,
biorącą udział
w zabijaniu zależnym od tlenu
w fagolizosomach
Defekt receptorów dla IFN-γ
lub IL-12
Infekcje prątkami: brak
aktywacji oksydazy NADPH
Brakujący składnik
Choroby spowodowane
infekcją
Czynniki regulatorowe
Inhibitor C1q
Obrzęk naczynio-ruchowy
dziedziczny (ciągła aktywacja
dopełniacza)
Czynnik przyspieszający rozkład
DAF (CD55)
Napadowa nocna
hemoglobulinuria (liza
erytrocytów)
Składowe dopełniacza
C1, C2 lub C4
Choroba kompleksów
immunologicznych (niezdolność
do usunięcia kompleksów Ag-
Ab); niedobór C2 związany z SLE
C3
Najpoważniejsze: nawracające
infekcje bakteriami
ropotwórczymi
Niedobory MAC składowych
dopełniacza (C5-8)
Zakażenia meningokokowe
Niedobory dopełniacza
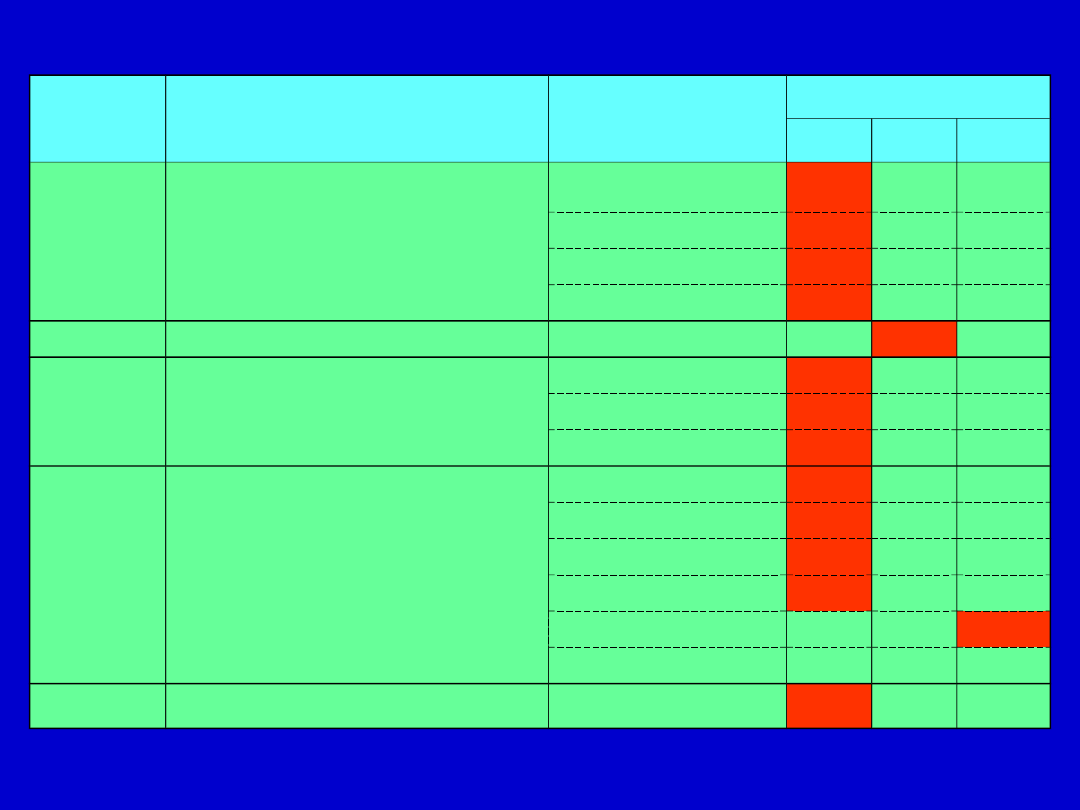
GRUPA
TYP ZMIAN
DEFEKT
DZIEDZICZENIE
AR
AD
XL
I
Niedobór kompleksów
immunologicznych
C1q
C1s lub C1r + C1s
C2
C4
II
Obrzęk naczynioruchowy
C1 inh
III
Nawracające zakażenia ropne
C3
Czynnik H
Czynnik I
IV
Nawracające zakażenia
bakteryjne (Neisseria)
C5
C6
C7
C8
Properdyna
Czynnik D
?
?
?
V
Bezobjawowe
C9
Dziedziczne niedobory ludzkiego dopełniacza
AR
– dziedziczenie autosomalnie recesywne
AD
– autosomalnie dominujące
XL
– sprzężone z chromosomem X recesywne

Przewlekła choroba ziarniakowa sprzężona
z chromosomem X (CGD)
(defekt
fagocytozy)
Zespół Wiskott-Aldricha (WAS)
(defekt
limfocytów T)
Cięzki złożony niedobór odporności sprzężony
z chromosomem X (XSCID)
(defekt limfocytów
T)
- mutacja genu IL-2 Rγ
Agammaglobulinemia sprzężona z
chromosomem X (X-LA) –
(defekt limfocytów
B)
- mutacja genu cytoplazmatycznej kinazy
białkowej limf. B (BTK), onkogen
Niedobór odporności z hiper IgM sprzężony
z chromosomem X (HIGM)
(defekt limfocytów
B)
- brak przełączania klas Ig
- mutacja genu kodującego ligand dla CD40
Mutacje genów
odpowiedzialn
e za pierwotne
niedobory
odporności
sprzężone z
chromosomem
X

KONIEC

Zespół Chediaka-Higashiego
Jest to zespół dziedziczony autosomalnie
recesywnie
Charakteryzuje się nawracającymi
zakażeniami, zwłaszcza gronkowcowymi i
paciorkowcowymi
oraz zaburzeniami neurologicznymi.
Chorzy mają łagodną neutropenię, z
charakterystycznymi
komórkami o olbrzymich
lizosomach
. Brak jest natomiast ziaren
azurofilnych
w neutrofilach (ulegają one fuzji z ziarnami
wtórnymi). Prawdopodobną przyczyną
niedoboru odporności jest opóźniona fuzja
tych olbrzymich lizosomów
z fagosomami.
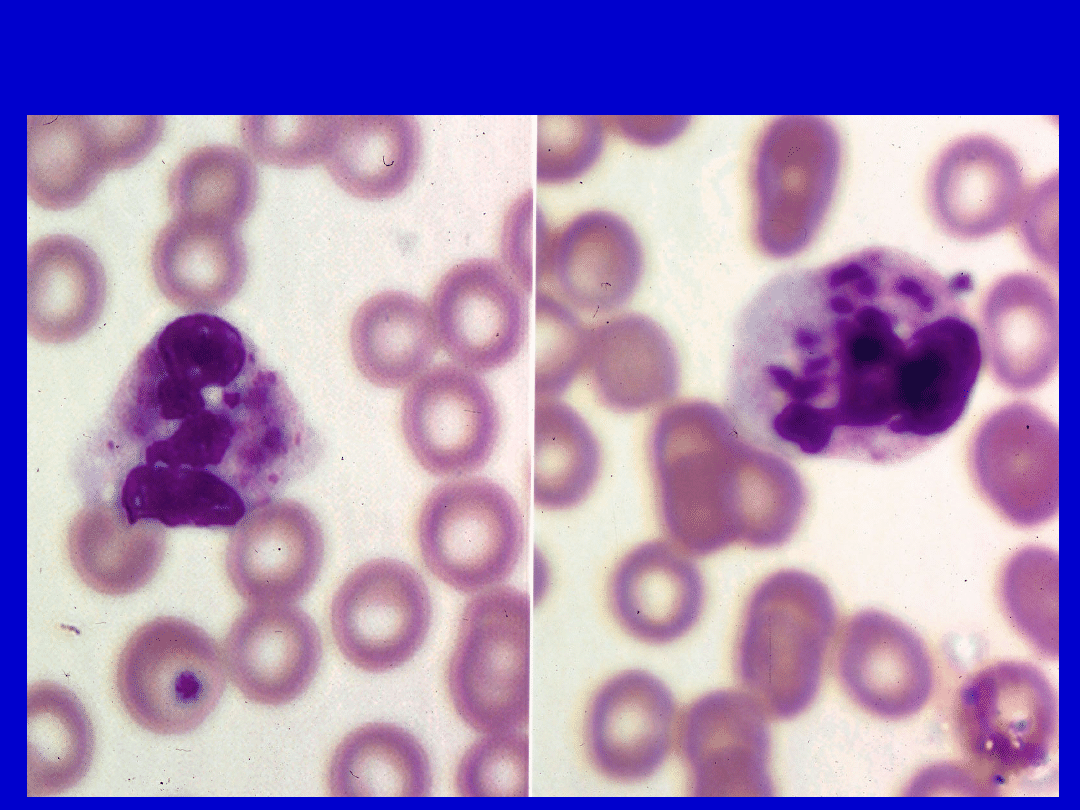
Zespół Chediaka-Higashiego
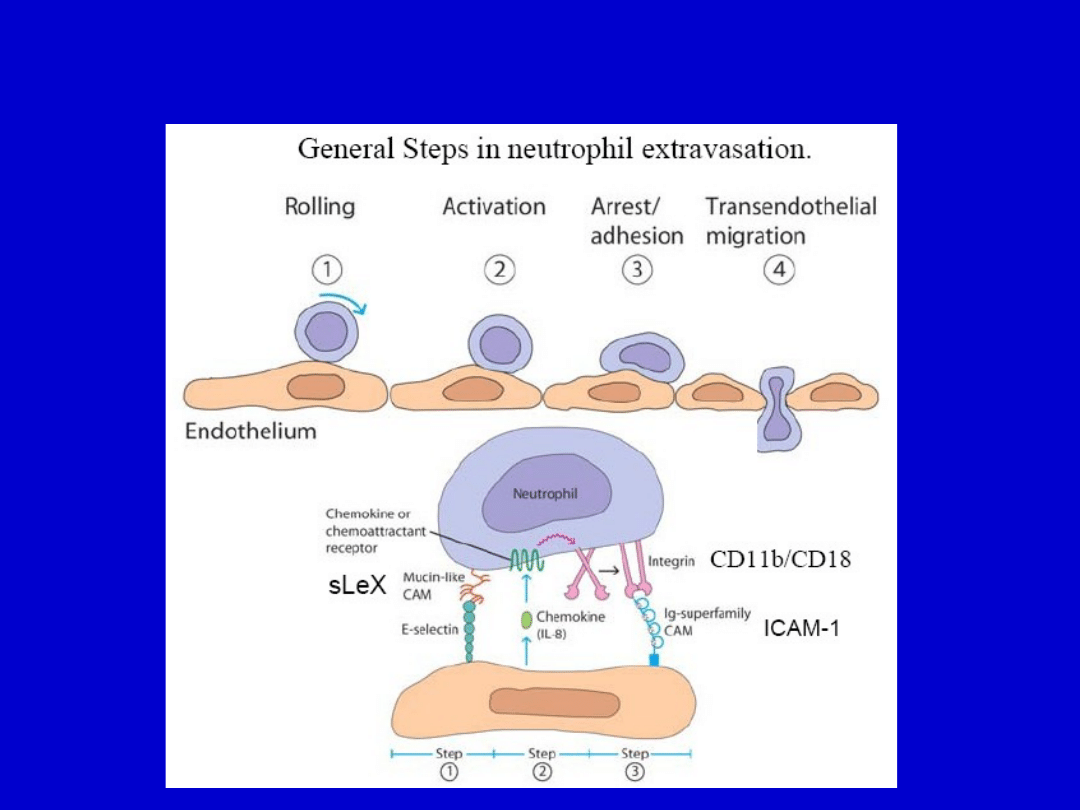
Główne etapy wynaczynienia neutrofili

Element ukł.
odpornościowe
go
Główny patogen
Miejsce
działania
Niedobór
Limfocyty T
bakterie, wirusy,
pierwotniaki, grzyby
niespecyficzne
SCID, zespół DiGeorga
Limfocyty B
Pneumococcus,
Streptococcus,
Haemophilus
płuca, skóra,
centralny układ
nerwowy
niedobór IgG i IgM
enterobakterie,
wirusy
nos, oczy
niedobór IgA
Fagocyty
Staphylococcus,
Klebsiella,
Pseudomonas
płuca, skóra,
węzły
limfatyczne
przewlekła choroba
ziarniakowa (CGD)
Dopełniacz
Neisseria,
Haemophilus,
Pneumococcus,
Streptococcus
Centralny układ
nerwowy,
płuca, skóra
C3, czynnika I, H i C
Charakterystyczne infekcje
w pierwotnych niedoborach
odporności
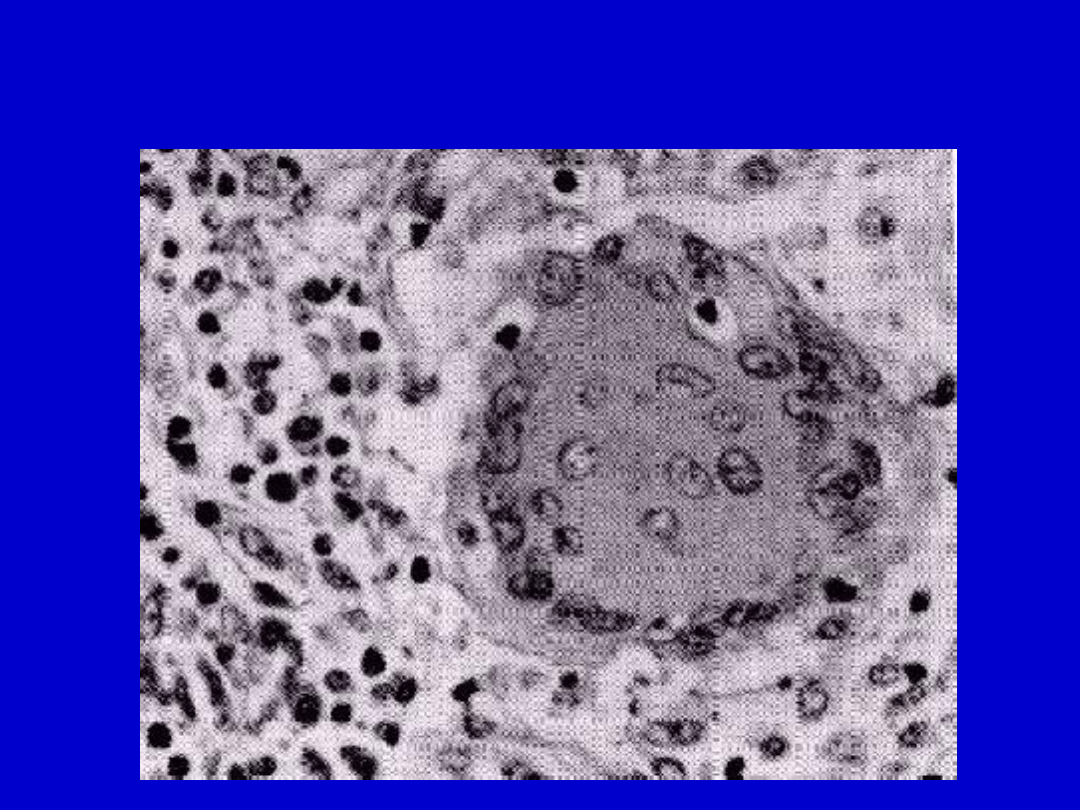
Ziarniak charakteystyczny dla przewlekłej
choroby ziarniakowej
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
Wyszukiwarka
Podobne podstrony:
Niedobory odpornosci wersja dluzsza
TEMAT 10 GLOBALIZACJA, + DOKUMENTY, Politologia 1 pwsz wykaz zajec, Nauka o polityce wykłady
Imm Cw 3 Wt rne niedobory odpornosci
00 Niedobory odpornoscioweid 1 Nieznany (2)
03 0000 014 02 Leczenie pierwotnych niedoborow odpornosci u dzieci immunoglobulinami
Temat 9-10 ISPS 09-2009, Zarządzanie bezpieczną eksploatacją statku -Zdanowicz
ETYKA referat temat 10 r 4, Administracja, I ROK, Etyka
Temat 9-10 ISM -09-2009, Zarządzanie bezpieczną eksploatacją statku -Zdanowicz
Niedobory odporności
09 pierwotne niedobory odporności
WT RNE NIEDOBORY ODPORNO CI
Niedobory odporności
więcej podobnych podstron