
Hormony trzustki i leki
przeciwcukrzycowe

• Część trzustki odpowiedzialna za czynność
wewnąrzwydzielniczą składa się z około miliona wysp
Langerhansa
• Wyspy składają się z co najmniej czterech typów komórek
produkujących hormony:
• insulina, hormon anaboliczny i odpowiedzialny za
magazynowanie
• polipeptyd amyloidu wysp trzustkowych (islet
amyloid polypeptide, IAPP, lub amylina), modulujący
apetyt, opróżnianie żołądka oraz wydzielanie insuliny i
glukagonu
• glukagon, czynnik podwyższający stężenie glukozy
poprzez mobilizację glikogenu
• somatostatyna, inhibitor wielu komórek wydzielniczych
• peptyd trzustkowy, małe białko poprawiające procesy
trawienia w nieznany dotąd sposób.

• Podwyższone stężenie glukozy w surowicy krwi
występujące w cukrzycy spowodowane jest
brakiem lub nieadekwatnym wydzielaniem
insuliny w trzustce, z lub bez towarzyszącego
upośledzenia działania insuliny
• Stany chorobowe leżące u podstaw cukrzycy
sklasyfikowano w czterech kategoriach:
• typ 1, cukrzyca insulinozależna;
• typ 2 cukrzyca nie zależna od insuliny;
• typ 3, inne przyczyny;
• typ 4, cukrzyca ciężarnych

Cukrzyca typu 1
• Cechą charakterystyczną cukrzycy typu 1 jest selektywne
zniszczenie wysp B (w Polsce nazywane ‘beta’) i ciężki lub
całkowity niedobór insuliny.
• U pacjentów z cukrzycą typu 1 niezbędne jest podawanie
insuliny.
• Typ 1 został podzielony na podtypy określające przyczyny:
immunologiczny i idiopatyczny.
• Postać immunologiczna jest najczęstszą formą cukrzycy typu
1. Większość pacjentów w momencie rozpoznania ma mniej
niż 30 lat, jednakże początek choroby może wystąpić w
każdym wieku.
• Cukrzyca typu 1 występuje we wszystkich grupach
etnicznych, najczęściej jednak wśród mieszkańców północnej
Europy i Sardynii.
• Podatność na chorobę wydaje się być związana z
dziedziczeniem wieloczynnikowym, choć tylko około 10-15%
pacjentów ma dodatni wywiad rodzinny.

Cukrzyca typu 2
• Cukrzyca typu 2 charakteryzuje się opornością tkanek na działanie
insuliny związaną z względnym niedoborem wydzielania insuliny.
• chory może mieć przewagę insulinooporności, bądź dysfunkcji
komórek B,
• Upośledzona funkcja insuliny wpływa także na gospodarkę
lipidową, powodując zwiększenie napływu wolnych kwasów
tłuszczowych, podwyższenie stężeń triglicerydów oraz obniżenie
poziomu lipoprotein o wysokiej gęstości (HDL).
• Pacjenci z cukrzycą typu 2 mogą zazwyczaj funkcjonować bez
podawania insuliny,
• Prawdopodobnie u 10-20% osób ze o zdiagnozowaną początkowo
cukrzycą typu 2 występują w istocie jednoczesnie typ 1 oraz 2, lub
powoli postępujący typ 1 i
• U osób z cukrzycą typu 2 nie występuje ketoza, jednak w wyniku
stresu, infekcji lub użycia leków zwiększających insulinooporność
(np. glikokortykosteroidów) wystąpić może kwasica ketonowa.
• U nieleczonych lub żle kontrolowanych pacjentów z cukrzycą typu
2 odwodnienie może doprowadzić do wystąpienia nieketonowej
śpiączki hiperosmolalnej będącej stanem zagrożenia życia. W
takim przypadku poziom glukozy we krwi wzrasta 6 – 20 krotnie w
stosunku do normy, czemu towarzyszą jakościowe zaburzenia
świadomości lub utrata przytomności. Niezbędna jest pilna pomoc
lekarska i nawodnienie.

Cukrzyca typu 3
• Rozpoznanie typu 3 odnosi się do
wielu innych, określonych przyczyn
podwyższenia glukozy we krwi jak
choroby pozatrzustkowe, stosowanie
leków, itp.

Cukrzyca typu 4
• Cukrzycę ciężarnych (Gestational diabetes, GDM)
definiuje się jako zaburzenie w poziomach
glikemii odnotowane po raz pierwszy w czasie
ciąży.
• W czasie ciąży łożysko oraz jego hormony
powodują insulinooporność najbardziej nasiloną w
trzecim trymestrze. Ryzyko cukrzycy powinno
zostać określone już na pierwszej kontrolnej
wizycie prenatalnej. U kobiet z wysokim ryzykiem
powinno się niezwłocznie wykonać badanie
kontrolne. U ciężarnych z niskim ryzykiem
badanie kontrolne można odłożyć do 24 – 28
tygodnia ciąży.
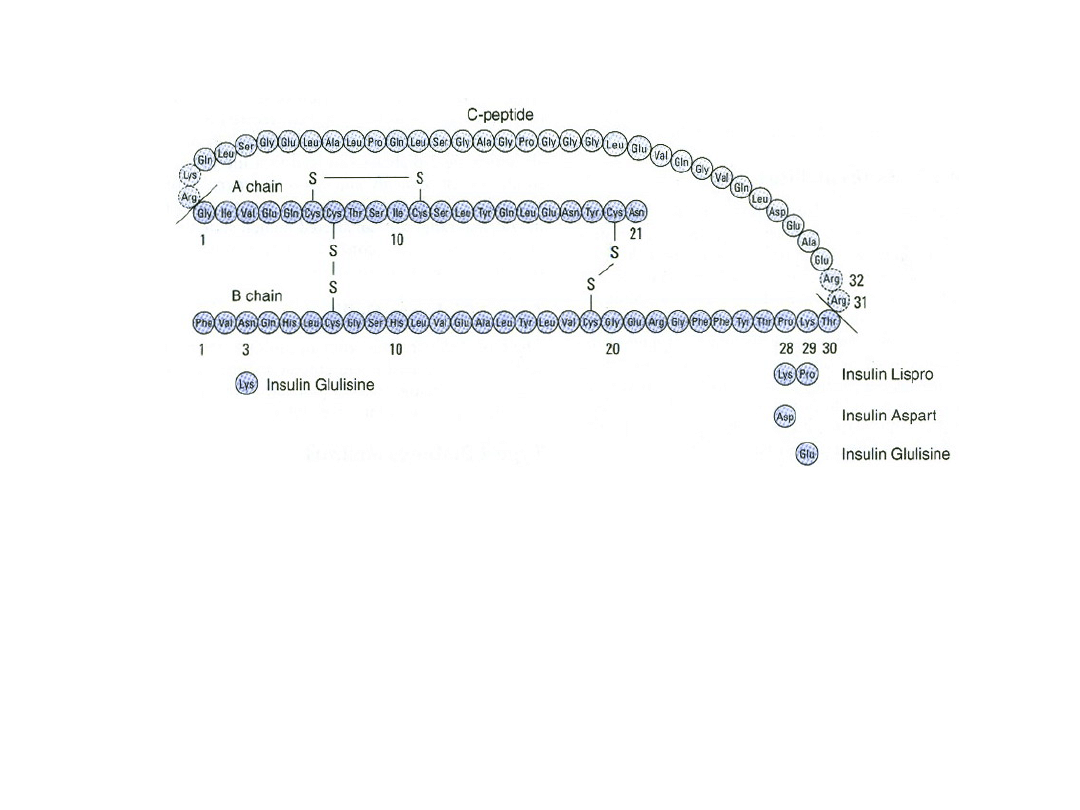
Struktura ludzkiej pro insuliny i kilku dostępnych analogów
insuliny. Insulinę pokazano jako zacienione (ciemniejszy kolor)
łańcuchy polipeptydowe A i B. Zaznaczono różnice w
łańcuchach A i B oraz modyfikacje aminokwasowe insulin
aspart, lispro, i glulizynowej.

INSULINA
• Insulina jest małym białkiem
• Zawiera 51 aminokwasów ułożonych w dwa
łańcuchy (A i B) połączone mostkami
dwusiarczkowymi; istnieją różnice gatunkowe
w składzie aminokwasowym obu łańcuchów.
• Proinsulina, cząsteczka białkowa składająca
się z pojedynczego, długiego łańcucha,
przetwarzana jest w aparacie Golgiego i
ładowana do ziarnistości, w których
hydrolizowana jest poprzez usunięcie 4
aminokwasów do insuliny i segmentu
resztkowego nazywanego C-peptydem.

• Insulina i C-peptyd są wydzielane w
odpowiedzi na wszystkie bodźce powodujące
wydzielanie insuliny w ilościach
ekwimolarnych; wydzielana jest również mała
ilość nieprzetworzonej lub częściowo
zhydrolizowanej proinsuliny.
• Proinsulina może mieć pewne niewielkie
działanie hipoglikemizujące, natomiast
funkcja fizjologiczna C-peptydu nie jest znana.
• Insulina magazynowana jest w ziarnistościach
komórek B w formie kryształów zawierających
dwa atomy cynku i sześć cząsteczek insuliny
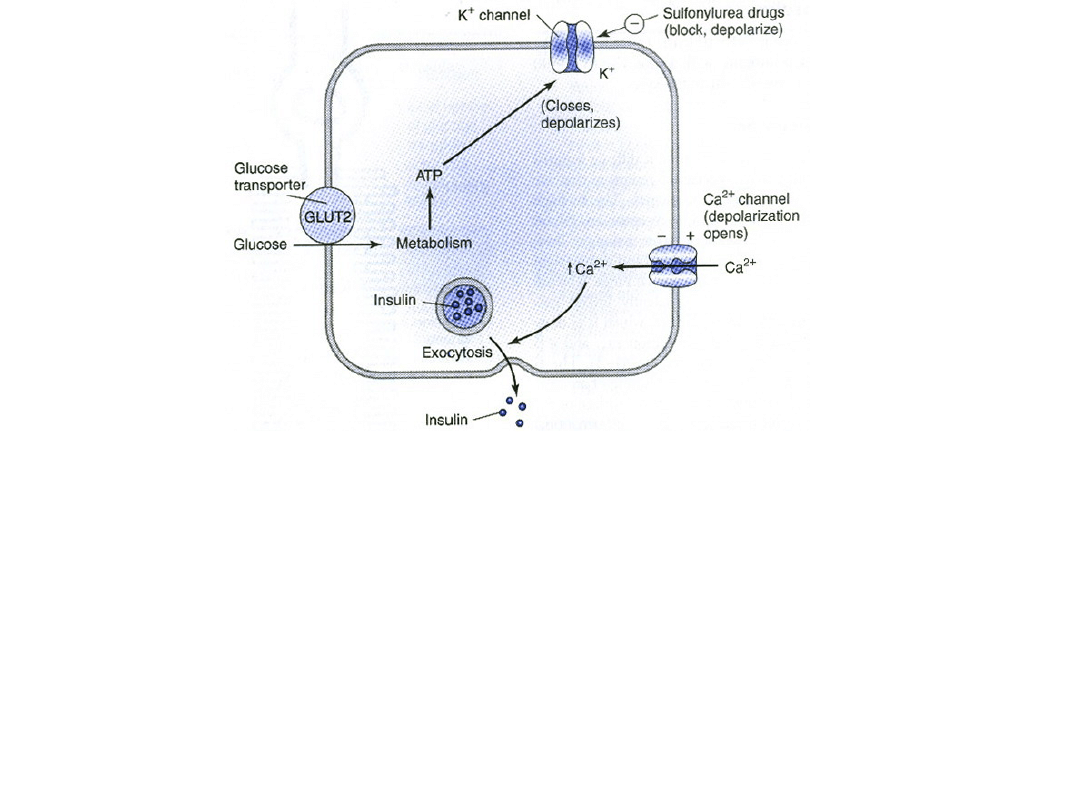
Jeden z modeli kontroli uwalniania insuliny z trzustkowych komórek B (beta)
przez glukozę i sulfonylomoczniki. W komórce będącej w spoczynku z
normalnymi (niskimi) poziomami ATP, potas dyfunduje zgodnie z gradientem
stężeń przez kanały potasowe bramkowane ATP utrzymując potencjał
błonowy na stałym w pełni spolaryzowanym, ujemnym poziomie. Wydzialanie
insuliny jest minimalne. W przypadku wzrostu stężenia glukozy zwiększa się
produkcja ATP powodująć zamknięcie kanałów potasowych i depolaryzację
komórki. Podobnie jak w tkance mięśniowej i nerwowej kanały wapniowe
otwierają się w odpowiedzi na depolaryzację pozwalając na napływ jonów
wapnia do komórki. Wzrost wewnątrzkomórkowego stężenia wapnia
powoduje zwiększenie wydzielania insuliny. Leki insulinotropowe zamykają
kanał potasowy zależny od ATP, depolaryzując błonę i tym samym
zwiększając wydzielanie insuliny w tym samym mechanizmie.

Wydzielanie insuliny
• Insulina jest wydzielana z trzustkowych komórek B w sposób
powolny (podstawowe wydzielanie) oraz w ilościach znacznie
większych w odpowiedzi na różne bodźce, zwłaszcza glukozę.
• inne bodźce stymulujące wydzielanie takie jak inne cukry (np.
mannoza), pewne aminokwasy (np. leucyna i arginina),
hormony takie jak polipeptyd-1 podobny do glukagonu
(glukagon-like polypeptide-1) i aktywność nerwu błędnego.
• Jeden z mechanizmów wydzielania insuliny w odpowiedzi na
bodziec: hiperglikemia powoduje zwiększenie
wewnątrzkomórkowych stężeń ATP, co powoduje zamknięcie
kanałów potasowych zależnych od ATP. Zmniejszony wypływ
potasu powoduje depolaryzację komórek B i otwarcie kanałów
wapniowych bramkowanych napięciem. Wywołane tym
zwiększone stężenie wewnątrzkomórkowego wapnia jest
czynnikiem inicjującym sekrecję hormonu. Leki zwiększające
wydzielanie insuliny (sulfonylomoczniki, meglanidy i D-
fenyloalanina) wykorzystują niektóre etapy tego mechanizmu.

Rozkład insuliny
• Wątroba i nerki są dwoma najważniejszymi
organami usuwającymi insulinę z krwiobiegu.
• Wątroba zazwyczaj usuwa około 60% insuliny
uwolnionej z trzustki z racji swojej lokalizacji na
zakończeniu żyły wrotnej,n
• nerki usuwają około 35-40% endogennej insuliny.
• U pacjentów cukrzycowych otrzymujących
insulinę stosunek ten jest odwrócony - do 60%
insuliny egzogennej jest usuwane przez nerki, a
maksymalnie 30-40% przez wątrobę.
• Czas półtrwania insuliny w krwiobiegu to 3 – 5
minut.

Receptor insulinowy
• Po dotarciu do krwiobiegu, insulina dyfunduje do tkanek
gdzie wiąże się ze swoistym receptorem znajdujący się
na błonach komórkowych większości tkanek.
• Receptor insulinowy składa się z dwóch połączonych
kowalencyjnie heterodimerów, z których każdy zawiera
podjednostkę α, w całości zewnątrzkomórkową,
zawierającą miejsce rozpoznające agonistę i
podjednostkę β, penetrującą błonę komórkową
• Podjednostka β zawiera kinazę tyrozynową. Połączenie
insuliny z podjednostką α na zewnętrznej powierzchni
komórki aktywuje receptor i poprzez zmiany
konformacyjne zbliża do siebie pętle katalityczne
podjednostki β po stronie cytoplazmatycznej.
• Ułatwia to wzajemną fosforylację reszt tyrozynowych na
podjednostkach β i kieruje aktywność kinazy tyrozynowej
na białka cytoplazmatyczne.
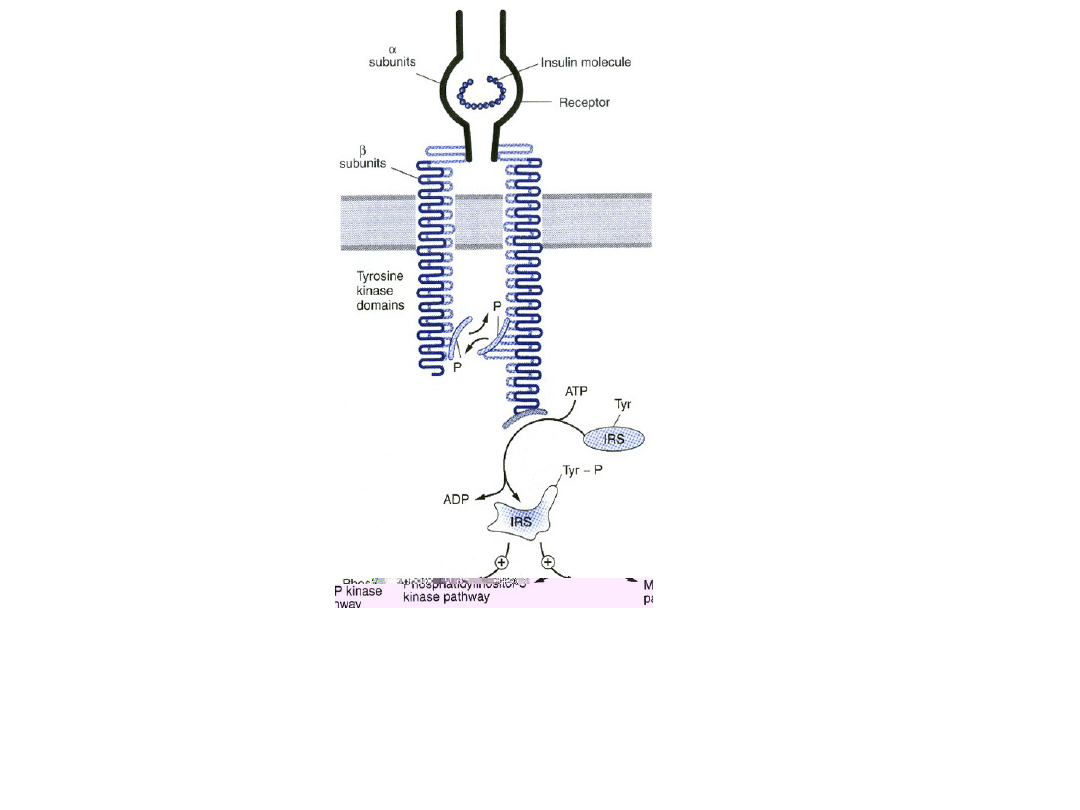
Schemat hetero dimera receptora insulinowego w
stanie zaktywowanym. IRS – substrat receptora
insulinowego; P – fosforan; tyr –tyrozyna
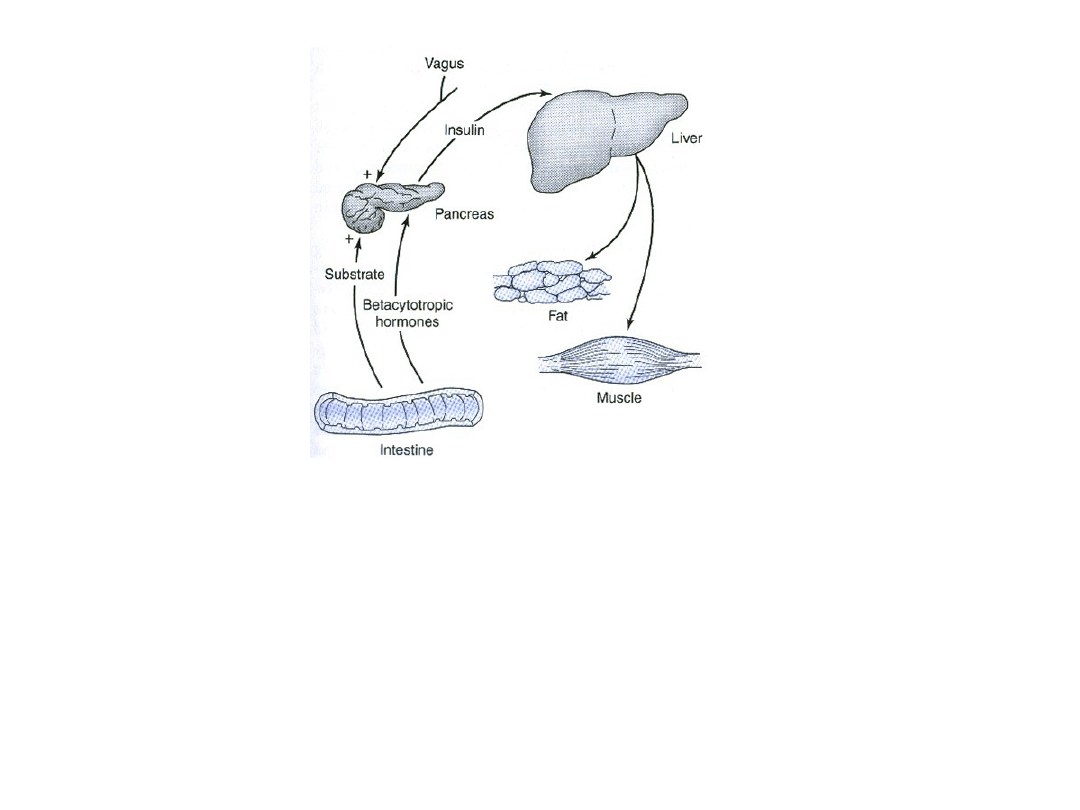
Insulina w swoich głównych tkankach docelowych (wątroba,
tkanka tłuszczowa i mięśnie) nasila syntezę (z krążących
substancji odżywczych) i magazynowanie glikogenu,
triglicerydów i białek. Wydzielanie insuliny z trzustki nasilane
jest w odpowiedzi na podwyższenie stężenia glukozy we krwi,
hormony inkretynowe, stymulacje nerwu błędnego i inne

Preparaty insuliny
• Insuliny stosowane w lecznictwie
różnią się od siebie technikami
produkcji rekombinowanego DNA,
sekwencją aminokwasową,
stężeniem, rozpuszczalnością i
szybkością wystąpienia działania
biologcznego.

PODSTAWOWE TYPY I CZAS
DZIAŁANIA PREPARATÓW INSULINY
• Dostępne są cztery główne typy preparatów insuliny:
• (1) ultra-szybko działające, z bardzo szybkim początkiem i
krótkim czasem działania;
• (2) krótko-działające z szybkim początkiem działania;
• (3) o pośrednim czasie działania;
• (4) długo-działające, z długim początkiem działania
• Insuliny ultra-szybkodziałające i krótko-działające
podawane we wstrzyknięciach sporządzane są w formie
przezroczystych roztworów o neutralnym pH i zawierają
małe ilości cynku dla polepszenia stabilności i wydłużenia
okresu przydatności.
• Insuliny NPH o pośrednim czasie działania podawane we
wstrzyknięciach zmodyfikowano w celu przedłużenia czasu
działania - sporządzane są w formie mętnych roztworów o
neutralnym pH z protaminą w buforze fosforanowym
(neutralna insulina protaminowa Hagedorna [NPH]).
• Insulina glarginowa i insulina detemir są insulinami
rozpuszczalnymi o długim czasie działania.

• Celem terapii podawaną podskórnie
insuliną jest zastąpienie normalnego
podstawowego wydzielania insuliny (w
nocy, na czczo i pomiędzy posiłkami), oraz
zwiększonego wydzielania związanego z
posiłkami (bolus albo pik insulinowy).
• Obecnie stosowane schematy zazwyczaj
wykorzystują długo-działające insuliny do
naśladowania podstawowego wydzielania
i ultra-szybko działające do pokrycia
zapotrzebowania związanego z posiłkami.
Intensywna insulinoterapia dąży do
przywrócenia bliskich normie glikemii w
ciągu całego dnia z ograniczeniem ryzyka
hipoglikemii.

Działania endokrynne
insuliny
• Wpływ na wątrobę:
• Odwracanie procesów katabolicznych powodowanych
przez niedobór insuliny
• Hamuje glikogenolizę
• Hamuje przemianę kwasów tłuszczowych i
aminokwasów do keto kwasów
• Hamuje przemianę aminokwasów na glukozę
• Działania anaboliczne
• Nasila magazynowanie glukozy jako glikogenu
(indukuje aktywność glukokinazy i syntazy glikogenu,
hamuje fosforylazę)
• Nasila syntezę triglicerydów oraz tworzenie lipoprotein
o bardzo małej gęstości (LDL)

• Wpływ na mięśnie
• Zwiększona synteza białek
• Zwiększony transport aminokwasów
• Zwiększona produkcja białek w
rybosomach
• Zwiększona produkcja glikogenu
• Zwiększony transport glukozy
• Indukuje syntezę glikogenu i hamuje
fosforylazę

• Wpływ na tkankę tłuszczową
• Zwiększone magazynowanie triglicerydów
• Indukcja i aktywacja lipazy lipoproteinowej
powodująca hydrolizę triglicerydów z
lipoprotein
• Zwiększony napływ glukozy dostarcza
fosforanu glicerolu co umożliwia estryfikację
kwasów tłuszczowych dostarczonych przez
lipoproteiny
• Hamowanie lipazy wewnątrzkomórkowej.

• Dokładne odtworzenie normalnego profilu glikemii
jest technicznie niemożliwe z powodu ograniczeń
związanych z podskórnym podawaniem insuliny.
• Najbardziej złożony schemat insulinowy dostarcza
ultra-szybko działającą insulinę w postaci ciągłego
wlewu podskórnego przy użyciu pompy insulinowej;
• alternatywne intensywne schematy insulinowe
określane jako schematy wielokrotnych wstrzyknięć
(multiple daily injections, MDI) wykorzystują
insulinę długo-działającą oraz wielokrotne
wstrzyknięcia insuliny ultra-szybko działającej.
• Klasyczna (konwencjonalna) insulinoterapia składa
się obecnie z dwóch dawek mieszanek insulinowych
zawierających insulinę ultra-szybko lub krótko-
działającą w połączeniu z insuliną o pośrednim
czasie działania.

Insuliny ultra-szybko
działające
• Dostępne są trzy analogi insuliny przeznaczone do stosowania
podskórnego: lispro, aspart, glulisine i jeden preparat insuliny
ultra-szybko działającej stosowanej wziewnie – rekombinowana
insulina ludzka do inhalacji (human insulin recombinant inhaled).
• Insuliny ultra-szybko działające pozwalają na najbardziej
fizjologiczne odwzorowanie poposiłkowego wydzielania insuliny,
ponieważ mają bardzo szybki początek działania i wcześnie
osiągają działanie szczytowe, co lepiej niż zwykła insulina
naśladuje normalne poposiłkowe wydzielanie insuliny, można je
stosować bezpośrednio przed posiłkiem bez wcześniejszego
kłopotliwego pomiaru glikemii. Ich czas działania rzadko jest
dłuższy niż 3-5 godzin, co redukuje ryzyko późnej poposiłkowej
hipoglikemii (wyjątkiem jest insulina podawana wziewnie, która
może działać nawet 6-7 godz.).
• Insuliny ultra-szybko działające stosowane we wstrzyknięciach
mają najmniejsze wahania wchłaniania (w przybliżeniu 5%) ze
wszystkich dostępnych preparatów insuliny (porównując do 25%
dla normalnej insuliny i 25-50% dla insulin o pośrednim czasie
działania i długodziałających).
• Są insulinami preferowanymi do stosowania w pompach
insulinowych podających insulinę podskórnie.

Insulina lispro
• pierwszy dostępny na rynku analog insuliny będący
monomerem wytwarzany jest metodami rekombinacji
genetycznej – zamieniono miejscami dwa aminokwasy
blisko końca karboksylowego łańcucha B
• Zamiana tych aminokwasów nie zmienia wiązania
insuliny lispro z receptorem insulinowym, czasu jej
półtrwania w krwiobiegu czy immunogenności, które są
podobne jak w przypadku naturalnej insuliny ludzkiej
• Aby zwiększyć czas przydatności do użycia insuliny
przechowywanej w ampułkach, insulina lispro jest
stabilizowana przy użyciu krezolowego środka
konserwującego co powoduje tworzenie heksametrów.
• Po wstrzyknięciu podskórnym lek szybko dysocjuje do
monomerów i jest gwałtownie wchłaniany z początkiem
działania po około 5-15 minutach i szczytową siłą
działania po niecałej godzinie. Czas wystąpienia
szczytowej siły działania jest względnie stały,
niezależnie od dawki.

Insulina aspart
• stworzona została przez zamianę proliny w
pozycji B28 na ujemnie naładowany kwas
asparginowy. Modyfikacja ta zmniejsza
normalne oddziaływanie monomer-monomer
przez ProB28 i GlyB23, i w związku z tym
hamuje spontaniczną agregację.
• Wchłanianie oraz profil aktywności podobny
jest do insuliny lispro (bardziej powtarzalny niż
w przypadku normalnej insuliny), z kolei siła
wiązania, aktywność, mitogenność i
immunogenność jest porównywalna z
normalną insuliną.

Insulina glulizynowa
• produkowana jest przez podstawienie
asparaginianu w miejsce lizyny w pozycji B3 i
kwasu glutaminowego w miejsce lizyny B29.
• wchłanianie, działanie i charakterystyka
immunologiczna są podobne do innych ultra-
szybko działających insulin podawanych
podskórnie.
• Przy dużych dawkach połączenie glulizyny z
receptorem może powodować różnice w
ścieżce aktywacji IRS-2 w porównaniu z
insuliną ludzką. Znaczenie kliniczne tych różnic
jest dotąd nieznane.

Preparat
Pochodzenie gatunkowe
Stężenia
Insuliny ultra-szybko działające
Insulina Lispro, Humalog (Lilly)
Analog insuliny ludzkiej
U100
Insulina Aspart, Novolog (NovoNordisk)
Analog insuliny ludzkiej
U100
Insulina Glulizynowa, Apidra (Aventis)
Analog insuliny ludzkiej
U100
Insuliny krótko-działające
Regular Novolin R (NovoNordisk)
Insulina ludzka
U100
Regular Humulin R (Lilly)
Insulina ludzka
U100, U500
Velosulin BR (NovoNordisk)
2
Insulina ludzka
U100
Regular, Exubera (Pfizer)
Insulina ludzka
Proszek do inhalacji 1, 3, 6mg
Insuliny o pośrednim czasie działania
NPH Humulin N (Lilly)
Insulina ludzka
U100
NPH Novolin N (NovoNordisk)
Insulina ludzka
U100
Mieszanki insulinowe (%NPH/%normalnej)
Novolin 70/30 (NovoNordisk)
Insulina ludzka
U100
Humulin 70/30 (Lilly)
Insulina ludzka
U100
50/50 NPL,Lispro (Lilly)
Analog insuliny ludzkiej
U100
75/25 NPL,Lispro (Lilly)
Analog insuliny ludzkiej
U100
70/30 NPA, Aspart (NovoNordisk)
Analog insuliny ludzkiej
U100
Insuliny długo-działające
Insulina detemir, Levemir (NovoNordisk)
Analog insuliny ludzkiej
U100
Insulina glarginowa, Lantus (Aventis/Hoechst Marion
Roussel)
Analog insuliny ludzkiej
U100

Insulina krótko-
działająca
• Naturalna insulina jest krótko-działającą, rozpuszczalną insuliną
cynkową wytwarzana na drodze inżynierii genetycznej, co pozwala na
uzyskanie cząsteczki identycznej jak w insulinie ludzkiej.
• Jej początek działania pojawia się po około 30 minutach a szczytowa
siła działania pomiędzy 2 a 3 godziną od wstrzyknięcia podskórnego a
czas działania wynosi zwykle 5 – 8 godzin.
• W wysokich stężeniach, na przykład w ampułce, cząsteczki naturalnej
insuliny spontanicznie łączą się ze sobą tworząc dimery stabilizujące
się wokół atomów cynku w heksamerach. Skłonność naturalnej insuliny
do tworzenia heksametrów jest przyczyną jej wolniejszego początku
działania i późniejszego działania szczytowego.
• Po podaniu podskórnym heksamery insuliny są zbyt duże i masywne
by przechodzić przez śródbłonek naczyń i przenikać do krwiobiegu.
Podana insulina jest stopniowo rozcieńczana przez płyn
śródmiąższowy, przez co jej stężenie spada a heksamery rozpadają się
na dimery a następnie monomery. Powoduje to trzyfazowe
wchłanianie wstrzykniętej insuliny, najszybsze w fazie ostatniej
(monomerycznej).
• Opóźnione wchłanianie powoduje niedopasowanie zapotrzebowania na
insulinę z jej dostępnością.

• W przypadku podania naturalnej insuliny podczas posiłku,
poziom glukozy we krwi rośnie szybciej niż stężenie
insuliny z następową wczesną hiperglikemią poposiłkową i
zwiększonym ryzykiem późnej poposiłkowej hipoglikemii.
• Naturalna insulina powinna być podana na 30-45 minut
przed posiłkiem aby zminimalizować to niedopasowanie.
• Jak w przypadku wszystkich starszych preparatów
insulinowych czas początku działania, długość działania i
nasilenie działania szczytowego wzrasta wraz z wielkością
zastosowanej dawki.
• własności farmakokinetyczne i farmakodynamiczne
małych dawek insuliny naturalnej i protaminowej (NPH)
znacznie różnią się od własności dawek dużych.
• Do podawania dożylnego stosuje się wyłącznie
rozpuszczalne insuliny drobnocząsteczkowe, ponieważ
rozcieńczenie powoduje gwałtowny rozpad heksamerów
do monomerów.
• Jest to szczególnie użyteczne w terapii dożylnej takich
stanów jak kwasica ketonowa oraz w przypadku
znacznych wahań zapotrzebowania na insulinę, jak po
zabiegach chirurgicznych czy w ostrych infekcjach.

Insuliny o pośrednim czasie
działania i insuliny długodziałające
• Neutralna insulina protaminowa lub
insulina izofanowa (neutral
protamine Hagehorn,NPH, isophane
insulin) – insulina izofanowa jest insuliną
o pośrednim czasie działania, której
wchłanianie i początek działania są
opóźnione poprzez połączenie
odpowiednich ilości insuliny i protaminy,
tak aby żadna z nich nie występowała w
formie niezwiązanej

•
Protamina jest mieszaniną sześciu głównych i kilku
mniej ważnych cząsteczek izolowanych z nasienia
pstrąga tęczowego. Są one zasadowymi, bogatymi w
argininę peptydami
•
Do wytworzenia kompleksu izofanowego (takiego w
którym żaden ze składników nie ma wolnych miejsc
wiążących) niezbędne jest połączenie protaminy z
insuliną w stosunku wagowym około 1:10, co odpowiada
w przybliżeniu jednej cząsteczce protaminy na sześć
jednostek insuliny.
•
Po podaniu podskórnym tkankowe enzymy
proteolityczne rozkładają protaminę co pozwala na
wchłanianie insuliny.
•
Insulina izofanowa zaczyna działać po około 2 – 5
godzinach i ma czas działania 4 – 12 godzin; zwykle jest
mieszana z insuliną naturalną, lispro, aspart lub
glulizynową i podawana dwa do czterech razy dziennie
w leczeniu cukrzycy typu 1.
•
Dawka określa profil aktywności; małe dawki mają
szybsze szczyty działania i krótkie czasy półtrwania a
duże dawki odwrotnie.

Insulina glarginowa
• insulina glarginowa jest rozpuszczalnym ‘bezszczytowym’
ultra-długo-działającym analogiem insuliny.
• Forma ta została stworzona dla zapewnienia długotrwałego,
powtarzalnego, wygodnego odtwarzania podstawowego
wydzielania insuliny. Dodanie dwóch cząsteczek argininy na
końcu karboksylowym łańcucha B oraz wymiana glicyny na
asparaginę w pozycji A21 spowodowała stworzenie analogu
rozpuszczalnego w kwaśnym pH ale wytrącającego się w
bardziej obojętnym pH organizmu po podaniu podskórnym.
• Pojedyncze cząsteczki insuliny powoli odłączają się od
wytrąconego kryształu i zapewniają niskie, stałe stężenie
krążącej insuliny.
• Insulina glarginowa ma powolny początek działania (1-1.5
godziny) i osiąga maksymalny efekt po 4 -6 godzinach.
Maksymalna aktywność jest utrzymywana przez 11 – 24
godzin lub nawet dłużej.
• Glarginę podaje się zazwyczaj raz dziennie, choć niektórzy
bardzo wrażliwi na insulinę pacjenci odnoszą korzyści z
podzielenia dawki (dwa razy na dobę).

• Dla utrzymania rozpuszczalności roztwór
insuliny glarginowej ma zazwyczaj kwaśny
odczyn i w związku z tym nie powinien być
mieszany z innymi insulinami.
• Wchłanianie insuliny glarginowej wydaje
się niezależne od miejsca wstrzyknięcia, a
immunogenność w badaniach na
zwierzętach była mniejsza niż insuliny
naturalnej.

Insulina detemir
• jest to ostatnio stworzony długo-działający analog insuliny.
• Usunięto krańcową treoninę z pozycji B30 i przyłączono
kwas tetradekanowy (myristic acid, tetradecanic acid,
kwas tłuszczowy o czternastu węglach) do krańcowej
lizyny w pozycji B29.
• Te modyfikacje przedłużają dostępność wstrzykniętego
leku poprzez zwiększenie zarówno samoistnej agregacji w
miejscu wstrzyknięcia podskórnego jak i odwracalnego
wiązania z albuminami.
• Insulina detemir ma najbardziej powtarzalne efekty wśród
insulin o pośrednim czasie działania i długo-działających, a
jej zastosowanie wiąże się z mniejszą częstością
hipoglikemii niż po insulinie izofanowej NPH.
• Insulina detemir ma zależny od dawki początek działania
po 1 – 2 godzinach i czas działania dłuższy niż 24 godziny.
• Podawana jest dwa razy dziennie aby zapewnić
odpowiednio stały poziom naśladujący wydzielanie
podstawowe.

Mieszanki insulinowe
• ponieważ insuliny izofanowe o pośrednim czasie działania wymagają
kilku godzin do osiągnięcia właściwych poziomów terapeutycznych,
ich użycie u pacjentów z cukrzycą typu 1 wymaga dodanie insuliny
krótko-działającej lub ultra-szybko-działającej przed posiłkami.
• są one zazwyczaj łączone przed wstrzyknięciem w tej samej
strzykawce. Insuliny aspart, lispro i glulizynowa mogą być doraźnie
mieszane (t.j. tuż przed podaniem) z insuliną izofanową NPH bez
wpływania na ich gwałtowne wchłanianie.
• insuliny o pośrednim czasie działania będące izofanowymi
kompleksami protaminy z insuliną lispro lub insuliną aspart.
Oznaczono je jako NPL (neutral protamin lispro, neutralna insulina
protaminowa lispro) i NPA (neutral protamine aspart, neutralna
insulina protaminowa aspart) – ich czas działania jest identyczny jak
insuliny izofanowej NPH.
• Mają one tą pozytywną cechę, że pozwalają na przygotowanie
stabilnych mieszanek złożonych z insuliny NPL z insuliną lispro oraz
insuliny NPA z insuliną aspart,
• FDA (Food and Drug Administration, Agencja do Spraw Leków i
Żywności) dopuściła do stosowania gotowe mieszanki 50%/50% i
75%/25% insulin NPL/lispro i 70%/30% insulin NPA/aspart.
• Insuliny glarginowa i detemir muszą być podawane w oddzielnych
wstrzyknięciach, ponieważ nie są one mieszalne z żadnymi innymi
insulinami ani doraźnie, ani w gotowych mieszankach.

WYTWARZANIE INSULINY
• Insuliny ludzkie – Masowa produkcja
insulin ludzkich i ich analogów
technikami rekombinacji DNA polega na
wstawieniu ludzkiego bądź
zmodyfikowanego genu kodującego
proinsulinę do komórek Esherichia coli
lub drożdży i przetworzeniu
wyizolowanej proinsuliny dla otrzymania
insuliny ludzkiej bądź analogu.

Sposoby podawania
insuliny
• Klasyczną metodą podawania
insuliny jest wstrzyknięcie podskórne
przy użyciu jednorazowej igły i
strzykawki. W ostatnich trzech
dekadach duży nacisk położono na
udoskonalanie innych sposobów
podawania, czego rezultatem jest
dostępna już insulina wziewna.

PODRĘCZNE WSTRZYKIWACZE
(PENY INSULINOWE)
• Aby ułatwić wielokrotne wstrzyknięcia insuliny,
zwłaszcza przy intensywnej insulinoterapii,
wprowadzono podręczne wstrzykiwacze podobne
do długopisu (w Polsce nazywane penami
insulinowymi)
• Zawierają wkłady z insuliną i wymienialne igły.
• Niektóre preparaty insulinowe są dostępne we
wstrzykiwaczach jednorazowych - insulina
naturalna, insulina lispro, insulina aspart, insulina
glulizynowa, insulina glarginowa, insulina detemir i
kilka mieszanek insuliny NPH z insuliną naturalną,
lispro i aspart

URZĄDZENIA DO CIĄGŁEGO,
PODSKÓRNEGO PODAWANIA
INSULINY (POMPY INSULINOWE)
• Urządzenia do ciągłego, podskórnego podawania insuliny
są zewnętrznymi pompami o otwartej pętli służącymi do
podawania insuliny
• zawierają możliwą do zaprogramowania przez użytkownika
pompę dostarczającą zindywidualizowaną podstawową
dawkę insuliny i bolusy insulinowe w oparciu o wyniki
samokontroli poziomów glikemii
• Zwykle 24 godzinna, podstawowa szybkość podawania jest
w miarę stała, jednakże aby sprostać krótkoterminowym
zmianom w zapotrzebowaniu możliwa jest czasowa zmiana.
• Podstawowa szybkość podawania może wymagać obniżenia
na kilka godzin na przykład z powodu zwiększonej
wrażliwości na insulinę związanej z wysiłkiem fizycznym.

• Ilości podawane w bolusie często są zróżnicowane i
służą do korekty wysokich poziomów glikemii i
pokrycia zapotrzebowania na insulinę związanego z
posiłkami w oparciu o zawartość węglowodanową
spożytego pokarmu i towarzyszącą aktywność
• Pompa, która zawiera zbiornik insuliny, układ
programujący, klawiaturę i wyświetlacz ma wielkość
zbliżoną do pagera.
• Umieszczana jest zwykle na pasku lub w kieszeni, a
insulina podawana jest przez rurkę połączoną z
zestawem wkłutym podskórnie
• Brzuch jest preferowanym miejscem wkłucia,
jednakże wykorzystuje się również boki i uda
• Zbiornik insuliny, rurka łącząca i zestaw do wkłucia
muszą być wymieniane co 2 – 3 dni w warunkach
jałowych
• jest uważane za najbardziej fizjologiczną metodę
substytucji insuliny.

• Zastosowanie tych urządzeń zalecane jest
• u osób, które nie były w stanie uzyskać
docelowych wartości glikemii na schematach
wielokrotnych wstrzyknięć
• kiedy wymagana jest doskonała kontrola glikemii,
jak na przykład w ciąży
• Ich stosowanie wymaga odpowiedzialnego
zaangażowania i poświęcenia ze strony pacjenta
• Velosulin (insulina klasyczna) oraz insulina
aspart, lispro i glulizynowa dopuszczono do użycia
w pompach insulinowych
• Preferowane są jednak insuliny aspart, lispro i
glulizynowa, z uwagi na ich korzystne właściwości
farmakokinetyczne pozwalające na dobrą kontrolę
glikemii bez zwiększania ryzyka hipoglikemii

INSULINA WZIEWNA
• FDA dopuściła wziewny preparat insuliny wytworzony
poprzez sproszkowanie i wytworzenie aerozolu insuliny
ludzkiej. Insulina ta jest gwałtownie wchłaniana do
krwiobiegu przez ściany pęcherzyków płucnych.
• Ma szybki początek działania i szczytowe stężenia
insuliny (po 30 minutach) podobne do insulin lispro,
aspart i glulizynowej, a z drugiej strony szczytowy efekt
działania (2 – 2.5 godz.) i czas działania (6 – 8 godz.)
podobne do insuliny naturalnej.
• podawana w celu pokrycia zapotrzebowania związanego
z posiłkami i korekty wysokich poziomów glukozy we
krwi
• nie podawana do pokrywania zapotrzebowania
podstawowego.
• Z podanej wziewnie dawki insuliny (która waha się od 1
do 6 mg) wchłania się mniej niż 10%.

Powikłania leczenia
insuliną

HIPOGLIKEMIA
• Mechanizmy powstawania i
diagnostyka –
• Mogą wynikać z opóźnienia posiłku,
• niewłaściwej ilości spożytych
węglowodanów,
• nieoczekiwanego wysiłku fizycznego
• zbyt dużej dawki insuliny w porównaniu
do bieżącego zapotrzebowania.

• Gwałtownie wystąpienie hipoglikemii
powoduje:
• wystąpienie objawów nadaktywności
układu autonomicznego, zarówno
współczulnego (tachykardia, kołatanie
serca, pocenie, drżenia) i
przywspółczulnego (nudności, głód),
które nieleczone może pogłębiać się
do drgawek i śpiączki.

• U osób narażonych na częste epizody
hipoglikemii podczas ścisłej kontroli glikemii,
autonomiczne objawy hipoglikemii mogą być
mniej nasilone lub nawet nieobecne. Ten stan
określa się mianem ‘nieświadomość
hipoglikemii’.
• U pacjentów z uporczywą, nieleczoną
hipoglikemią pojawić się mogą objawy
nadmiaru insuliny – dezorientacja, osłabienie,
dziwne zachowania, śpiączka, drgawki
• Każdy pacjent chory na cukrzycę leczony
hipoglikemiczne powinien być zaopatrzony w
identyfikator (bransoletkę, naszyjnik, kartę
identyfikacyjną w portfelu czy torebce) oraz
źródło szybko wchłanianej glukozy.

Leczenie hipoglikemii
• Wszystkie objawy hipoglikemii ustępują po podaniu
glukozy
• Aby przyspieszyć wchłanianie powinno się podać
cukier prosty lub glukozę, najlepiej w formie płynnej.
• W celu leczenia niewielkich hipoglikemii u pacjentów
przytomnych mogących przełykać stosuje się
tabletki z dekstrozą, żel glukozowy, lub każdy inny
napój czy pokarm zawierający cukier.
• W przypadku cięższej hipoglikemii powodującej
utratę przytomności bądź zamroczenie leczeniem z
wyboru jest podanie dożylne 20 – 50 ml 50%
roztworu glukozy w czasie 2 – 3 minut.

• Jeśli nie ma możliwości podania dożylnego,
podanie podskórne lub domięśniowe 1 mg
glukagonu w ciągu 15 minut przywraca
przytomność
• Jeśli pacjent jest zamroczony, a glukagon
niedostępny, można podać dopoliczkowo
niewielką ilość miodu bądź syropu.
• Ogólnie przeciwwskazane jest karmienie doustne
u pacjentów nieprzytomnych.
• W każdym przypadku poważnie zaburzonej
świadomości powinno się wezwać pogotowie.

IMMUNOPATOLOGIA
TERAPII INSULINOWEJ
• U
chorych na cukrzycę w czasie leczenia insuliną może powstać
przynajmniej pięć klas przeciwciał przeciwinsulinowych: IgA, IgD, IgE,
IgG i IgM. Istnieją 2 główne typy zaburzeń immunologicznych u tych
pacjentów:
• Alergia insulinowa – postać natychmiastowa nadwrażliwości jest
rzadkim stanem w którym miejscowa lub uogólniona pokrzywka
wynika z uwolnienia histaminy z tkankowych komórek tucznych
pobudzonych przez przeciwciała przeciwinsulinowe klasy IgE. W
poważnych przypadkach może wystąpić anafilaksja.
• Immunologiczna oporność insulinowa – Małe, pomijalne stężenia
przeciwciał przeciwinsulinowych klasy IgG neutralizujących działania
insuliny występują u większości leczonych insuliną pacjentów.
Rzadko, zazwyczaj w związku z innymi procesami
autoimmunologicznymi takimi jak toczeń rumieniowaty, pojawiają
się większe stężenia przeciwciał przeciwinsulinowych powodujące
insulinooporność.

LIPODYSTROFIA W
MIEJSCU WSTRZYKNIĘCIA
• Stosowanie starszych zwierzęcych preparatów
insulinowych czasem powodowało atrofię tkanki
podskórnej w miejscu wstrzyknięcia. Powikłanie to jest
uwarunkowane immunologicznie i w zasadzie nie
występuje w związku z powstaniem insulin ludzkich i
ich analogów o obojętnym pH. Wstrzyknięcie nowszych
preparatów bezpośrednio do zanikłego obszaru
powoduje przywrócenie normalnych kształtów.
• Problemem pozostaje hipertrofia tkanki podskórnej
występująca przy powtarzanym podawaniu w to samo
miejsce. Można temu zapobiegać poprzez unikanie
tego miejsca lub korygować poprzez liposukcję

DOUSTNE LEKI
PRZECIWCUKRZYCOWE
• są obecnie cztery kategorie doustnych
leków przeciwcukrzycowych:
• środki nasilające wydzielanie insuliny -
insulinotropowe (sulfonylomoczniki,
glinidy (meglitinidy) i pochodne D-
fenyloalaniny)
• biguanidy
• glitazony (tiazolidinediony)
• inhibitory α-glukozydazy

LEKI ZWIĘKSZAJĄCE
WYDZIELANIE INSULINY
(INSULINOTROPOWE):
• SULFONYLOMOCZNIKI
• Mechanizm działania
• Głównym działaniem sulfonylomoczników
jest zwiększenie uwalniania insuliny z
trzustki
• ponadto dwa dodatkowe mechanizmy
działania – redukcja osoczowych poziomów
glukagonu i zamykanie kanałów
potasowych w tkankach pozatrzustkowych.

OBNIŻENIE
OSOCZOWYCH STĘŻEŃ
GLUKAGONU
• Długotrwałe podawanie sulfonylomoczników
w cukrzycy typu 2 obniża osoczowe poziomy
glukagonu, co może się przyczyniać do
działania hipoglikemizującego tych leków.
Mechanizm tego hamującego efektu
sulfonylomoczników na poziomy glukagonu
jest niejasny, ale wydaje się być związany z
hamowaniem pośrednim spowodowanym
zwiększonym wydzielaniem insuliny i
somatostatyny hamujących wydzielanie
komórek A.

Sulfonylomoczniki
pierwszej generacji
• Tolbutamid jest lekiem dobrze wchłanianym ale
gwałtownie metabolizowanym w wątrobie. Jego
czas działania jest stosunkowo krótki (czas
półtrwania wynosi 4 – 5 godzin)
• powinien być podawany w dawkach podzielonych.
• jest najbezpieczniejszym sulfonylomocznikiem u
diabetyków w wieku podeszłym.
• Rzadko zgłaszano przedłużającą się hipoglikemię,
najczęściej u pacjentów otrzymujących leki
hamujące metabolizm tolbutamidu (dikumarol,
fenylbutazon, niektóre sulfonamidy).

• Chlorpropamid ma czas półtrwania 32 godziny i jest powoli
metabolizowany w wątrobie do produktów zachowujących
pewną aktywność biologiczną
• około 20 – 30% leku jest wydalana z moczem w formie
niezmienionej
• wykazuje interakcje z wymienionymi wcześniej lekami, których
eliminacja zależy od wątrobowego katabolizmu oksydacyjnego
• jest przeciwwskazany u pacjentów z niewydolnością wątroby
czy nerek.
• jest przeciwwskazany u chorych w wieku podeszłym z uwagi na
częstsze przedłużone reakcje hipoglikemiczne występujące w
tej grupie chorych.
• Do innych działań niepożądanych należą uderzenia gorąca
związane z przekrwieniem predyspozycją genetyczną i
hiponatremia z rozcieńczenia. U mniej niż 1% pacjentów
występują zaburzenia hematologiczne (przejściowa leukopenia,
trombocytopenia) po spożyciu alkoholu u osób z predyspozycją
genetyczną i hiponatremia z rozcieńczenia. U mniej niż 1%
pacjentów występują zaburzenia hematologiczne (przejściowa
leukopenia, trombocytopenia)

• Tolazamid jest równie silny jak
chlorpropamid, ale ma krótszy czas
działania.
• jest wchłaniany znacznie wolniej niż inne
sulfonylomoczniki więc jego wpływ na
poziom glukozy we krwi pojawia się
dopiero po kilku godzinach.
• czas półtrwania wynosi 7 godzin.
• jest metabolizowany do kilku związków
zachowujących działanie hipoglikemiczne.

Sulfonylomoczniki
drugiej generacji
• Gliburyd jest metabolizowany w wątrobie do związków o bardzo
małej aktywności hipoglikemicznej.
• jest dostępny w postaci tabletek ‘mikronizowanych’ (Glynase
PresTab) w kilku różnych dawkach. Istnieją jednak wątpliwości
dotyczące ich biorównoważności z postaciami
niemikronizowanymi, i w związku z tym FDA zaleca ścisłą
kontrolę i dostosowanie dawki przy przechodzeniu z tradycyjnych
preparatów gliburydu czy innych sulfonylomoczników.
• Poza swoją tendencją do powodowania hipoglikemii gliburyd
• Istnieją rzadkie doniesienia o występowaniu uderzeń gorąca po
spożyciu alkoholu; dodatkowo lek powoduje nieznaczne
zwiększenie klirensu wody. Gliburyd jest przeciwwskazany u
pacjentów z upośledzeniem funkcji wątroby oraz z
niewydolnością nerek.

•
Glipizyd ma najkrótszy czas półtrwania (2 – 4 godzin)
spośród leków tej grupy.
•
powinien być podawany na 30 minut przed śniadaniem,
ponieważ jego wchłanianie jest spowalniane przy
stosowaniu razem z pokarmem.
•
Preparat o przedłużonym uwalnianiu (Glucotrol XL)
zapewnia 24-godzinne działanie po jednorazowej,
porannej dawce
•
Z powodu krótszego czasu półtrwania tradycyjne postaci
glipizydu znacznie rzadziej niż gliburyd powodują
poważną hipoglikemię.
•
Co najmniej 90% glipizydu jest metabolizowana w
wątrobie do nieczynnych metabolitów a 10% jest
wydalane przez nerki w formie niezmienionej.
•
Leczenie glipizydem jest więc przeciwwskazane u chorych
ze znacznym upośledzeniem funkcji wątroby czy nerek z
uwagi na wyższe ryzyko hipoglikemii.

• Glimepiryd został dopuszczony do
stosowania raz na dobę w monoterapii
oraz w połączeniu z insuliną.
• Działanie hipoglikemiczne osiągane jest
po najmniejszej spośród
sulfonylomoczników dawce.
• Ma długi czas działania (czas półtrwania
5 godzin) co pozwala na stosowanie raz
na dobę
• Glimepiryd jest w całości metabolizowany
w wątrobie do nieaktywnych metabolitów

Wtórna nieskuteczność i
tachyfilaksja na
sulfonylomoczniki
• Wtórna nieskuteczność, t.j. niepowodzenie w
utrzymaniu dobrej odpowiedzi na leczenie
sulfonylomocznikami w czasie długotrwałego
stosowania pozostaje niepokojącym
problemem w leczeniu pacjentów z cukrzycą
typu 2. Do wystąpienia wtórnej nieskuteczności
mogą przyczyniać się postępująca utrata
komórek B trzustki, ograniczenie aktywności
fizycznej, obniżenie masy mięśniowej oraz
zwiększenie ekotopowego odkładania tłuszczu
w przewlekłej cukrzycy typu 2.

LEKI ZWIĘKSZAJĄCE WYDZIELANIE
INSULINY (INSULINOTROPOWE):
GLINIDY
• Repaglinid będący pierwszym lekiem z tej
grupy
• Glinidy - modulują wydzielanie insuliny z
komórek B poprzez wpływ na kanały
potasowe
• podobieństwo do sulfonylomoczników w
budowie części aktywnej – glinidy mają
dwa miejsca wiążące wspólne z
sulfonylomocznikami oraz jedno unikatowe

Repaglinid
• bardzo szybki początek działania, ze szczytowym stężeniem
i szczytowym działaniem w ciągu godziny od podania;
• czas działania wynosi 5 – 8 godzin
;
• jest zalecany w celu kontroli hiperglikemii poposiłkowej
• istnieje ryzyko hipoglikemii w przypadku opóźnienia posiłku
lub nieodpowiedniej zawartości węglowodanowej posiłku
• powinien być używany ostrożnie u chorych z upośledzeniem
funkcji wątroby czy nerek
• w monoterapii lub w połączeniu z biguanidami
• W cząsteczce repaglanidu nie ma siarki, może on być więc
stosowany u chorych z cukrzycą typu 2 uczulonych na
siarkę lub sulfonylomoczniki.

LEKI ZWIĘKSZAJĄCE WYDZIELANIE
INSULINY (INSULINO TROPOWE):
POCHODNE D-FENYLOALANINY
• Nateglinid - pochodna D-fenyloalaniny jest najnowszym
lekiem insulinotropowym
• powoduje bardzo gwałtowne i przejściowe uwalnianie
insuliny z komórek B poprzez zamknięcie zależnych od ATP
kanałów potasowych
• Dodatkowo przywraca on częściowo wstępne wydzielanie
insuliny w odpowiedzi na dożylny test obciążenia glukozą
• Stosowany w leczeniu chorych z izolowaną poposiłkową
hipoglikemią
• jest skuteczny w monoterapii lub w skojarzeniu ze środkiem
doustnym o mechanizmie działania nie obejmującym
nasilania wydzielania insuliny (jak na przykład metformina)

• Nateglinid podaje się tuż przed posiłkami.
• Wchłania się w ciągu 20 minut od podania
doustnego osiągając szczytowe stężenie po
niecałej godzinie
• Całkowity czas działania jest mniejszy niż 4
godziny.
• nasila wydzielanie insuliny w odpowiedzi na
obciążenie glukozą
• Częstotliwość występowania hipoglikemii jest
najniższa ze wszystkich leków insulinotropowych
• korzystną cechą jest bezpieczeństwo stosowania
u chorych ze znacznie ograniczoną funkcją nerek

BIGUANIDY
• Mechanizm działania
• Nie wyjaśniono w pełni mechanizmu działania
biguanidów
• Ich działanie hipoglikemiczne nie zależy od obecności
funkcjonujących komórek B.
• Obecnie sugerowane mechanizmy działania obejmują:
• (1) zmniejszoną glukneogenezę w wątrobie i nerkach;
• (2) spowolnienie wchłaniania glukozy, połączone ze
zwiększoną przemianą glukozy do mleczanu w
enterocytach;
• (3) bezpośrednie nasilanie glikogenolizy ze
zwiększonym usuwaniem glukozy z krwi;
• (4) obniżanie osoczowych poziomów glukagonu

Metabolizm i wydalanie
• Metformina -czas półtrwania 1.5 – 3 godzin,
nie wiąże się z białkami osocza
• nie jest metabolizowana i jest wydalana przez
nerki w formie niezmienionej Konsekwencją
zablokowania glukoneogenezy przez
metforminę jest upośledzanie wątrobowego
metabolizmu kwasu mlekowego.
• U pacjentów z niewydolnością nerek dochodzi
do kumulacji biguanidów i zwiększa się ryzyko
kwasicy mleczanowej

Zastosowanie kliniczne
• Biguanidy przepisuje się najczęściej chorym, u których
hiperglikemia jest spowodowana osłabieniem działania
insuliny (zespół oporności na insulinę)
• metformina jest środkiem oszczędzającym insulinę i nie
powoduje przybierania na wadze
• leczenie obniża ryzyko choroby naczyniowej zarówno małych
jak i dużych naczyń (mikroangiopatię i makroangiopatię)
• Biguanidy można również stosować w skojarzeniu z lekami
insulinotropowymi lub glitazonami u chorych z cukrzycą typu
2, u których monoterapia jest niewystarczająca.
• Metformina jest również skuteczna w zapobieganiu cukrzycy
typu 2
• metformina nie zapobiegała wystąpieniu cukrzycy u osób
starszych i szczuplejszych

Toksyczność
• zaburzenia żołądkowo-jelitowe (brak apetytu,
nudności, wymioty, bóle brzucha, biegunka),
• zmniejsza się wchłanianie witaminy B
12
• Kwasica mleczanowa zdarza się rzadziej po
metforminie niż po fenforminie jeśli nie ma
niedotlenienia, niewydolności nerek czy wątroby.
• Biguanidy są przeciwwskazane u pacjentów z chorobą
nerek, alkoholizmem, chorobą wątroby czy chorobami
predysponującymi do niedotlenienia tkanek (np.
przewlekła niewydolność sercowo-płucna), z uwagi na
zwiększone przy tych schorzeniach ryzyko kwasicy
mleczanowej powodowanej przez biguanidy.

TIAZOLIDINEDIONY
(GLITAZONY)
•
zmniejszają oporność na insulinę
•
głównym działaniem jest regulacja genów
odpowiedzialnych za metabolizm glukozy i tłuszczów
oraz różnicowanie adipocytów.
•
Dostępne obecnie glitazony nie mają jednakowego
działania
•
działają na adipocyty, miocyty i hepatocyty
•
mają istotny wpływ na śródbłonek naczyń, układ
odpornościowy, jajniki i komórki guzów nowotworowych
•
U osób z cukrzycą głównym miejscem działania
glitazonów jest tkanka tłuszczowa gdzie leki te
zwiększają wychwyt i zużycie glukozy, oraz modulują
wydzielanie hormonów lipidowych lub cytokin oraz
innych białek zaangażowanych w regulację procesów
energetycznych
•
Glitazony regulują apoptozę i różnicowanie adipocytów

• Dostępne są dwa glitazony: pioglitazon i
rosiglitazon
• Pioglitazon działa zarówno na receptor PPAR-
α, jak i PPAR-γ. Wchłania się w ciągu 2 godzin
od podania; pomimo, że pokarm może
spowalniać wchłanianie, biodostępność nie
zmienia się
• Leczenie pioglitazonem zmniejsza śmiertelność
i częstość występowani incydentów sercowo-
naczyniowych (zwału mięśnia sercowego i
udaru)
• Pioglitazon został dopuszczony do stosowania w
cukrzycy typu 2 zarówno w monoterapii, jak i w
połączeniu z metforminą, sulfonylomocznikami
oraz insuliną

• Rosiglitazon jest szybko wchłaniany i w
wysokim stopniu wiąże się z białkami osocza.
• Metabolizowany jest do związków o minimalnej
aktywności
• Podawany jest raz lub dwa razy na dobę
• ma działania niepożądane podobne do
wszystkig glitazonów
• nie wykazuje jakichkolwiek istotnych interakcji
lekowych
• Lek dopuszczono do stosowania w cukrzycy
typu 2 zarówno w monoterapii jak i w
skojarzeniu z biguanidem i sulfonylomocznikiem
oraz z insuliną

• Z uwagi na mechanizm działania obejmujący
regulację ekspresji genów, ich początek
działania jest powolny, a ustępowanie działania
trwa tygodniami a nawet miesiącami
• Leczenie skojarzone z sulfonylomocznikami lub
insuliną może prowadzić do występowania
hipoglikemii i wymagać dostosowania dawki.
• Długotrwałe leczenie jest związane ze spadkiem
stężeń triglicerydów i niewielkim wzrostem
poziomów poziomów cholesterolu frakcji HDL
oraz LDL.

• Działaniem niepożądanym wspólnym dla obu
glitazonów jest retencja płynów objawiająca się
niewielką niedokrwistością i obrzękami
obwodowymi
• nie powinny być stosowane w ciąży, w obecności
zaawansowanej choroby wątroby (większe niż 2.5-
krotne podwyższenie ALAT) oraz w razie
współistniejącej niewydolności serca.
• U kobiet z zahamowaniem owulacji może nastąpić
jej wznowienie, dlatego należy poinformować o
zwiększeniu ryzyka zajścia w ciążę
• mają korzystne działanie w zapobieganiu
cukrzycy typu 2

INHIBITORY ALFA-
GLUKOZYDAZY
• Tylko cukry proste, jak glukoza czy fruktoza, mogą zostać
wchłonięte ze światła jelita do krwi
• Przed wchłonięciem w dwunastnicy i jelicie cienkim
skrobie, oligosacharydy i dwucukry muszą zostać
rozłożone na cukry proste. Rozkład ten zachodzi pod
wpływem enzymów jelitowych jak trzustkowa α-amylaza
oraz α-glukozydazy związanej z kosmkami enterocytów.
• Akarboza i miglitol są kompetycyjnymi inhibitorami
jeltowych α-glukozydaz i redukują rozkład i wchłanianie
spożytych skrobii i dwucukrów
• Miglitol różni się strukturą od akarbozy i jest
sześciokrotnie silniejszym inhibitorem inwertazy
(sacharazy)

• Konsekwencją zahamowania enzymów jest
zmniejszenie trawienia w górnej części jelita i
przesunięcie trawienia (i co za tym idzie
wchłaniania) spożytych skrobii i dwucukrów do
dystalnej części jelita cienkiego, redukując w ten
sposób poposiłkową glike
• są dopuszczone do stosowania w cukrzycy typu 2 w
monoterapii oraz w połączeniu z
sulfonylomocznikami (addytywny efekt na glikemię)
• Zarówno akarboza jak i miglitol są stosowane tuż
przed spożyciem pierwszej porcji posiłku
• Znacznie nasilone działania niepożądane
obejmują wzdęcia, biegunkę oraz bóle brzucha

• Hipoglikemia zazwyczaj nie jest problemem przy
monoterapii czy w skojarzeniu z biguanidem, może
pojawić się przy jednoczesnym stosowaniu z
sulfonylomocznikiem.
• Hipoglikemie powinno się leczyć glukozą (dekstroza) a
nie sacharozą, której rozkład może być zablokowany.
• Leki te są przeciwwskazane u pacjentów z chorobami
zapalnymi jelit i innymi schorzeniami jelit, których
przebieg może się pogarszać się w związku z obecnością
gazu i wzdęciami.
• zarówno akarboza jak i miglitol są wydalane przez nerki,
nie powinny być przepisywane chorym z upośledzeniem
funkcji nerek.
• Stosowanie akarbozy wiązało się z odwracalnym
podwyższeniem enzymów wątrobowych i dlatego
powinna być ostrożnie stosowana u osób z chorobami
wątroby.

• stosowanie inhibitorów α-glukozydazy u
chorych w stanie przedcukrzycowym
skutecznie zapobiegało wystąpieniu licznych
nowych przypadków cukrzycy typu 2
• pomagało przywrócić funkcję komórek B i
dodatkowo redukowało występowanie
choroby sercowo-naczyniowej i nadciśnienia
• Leczenie akarbozą redukowało również ilość
incydentów secowo-naczyniowych u chorych
na cukrzycę.
• Cukrzyca i zapobieganie chorobie sercowo
naczyniowej być może stanie się kolejnym
wskazaniem do stosowania tej klasy leków.

PRAMLINTID
• Pramlintid, syntetyczny analog amyliny, będący środkiem
cukrzycowym stosowanym podskórnie modulującym
poposiłkowe poziomy glikemii
• zatwierdzony do użycia przed posiłkami u chorych z
cukrzycą typu 1 i 2
• stosowany jako dodatek do insuliny u chorych, którzy nie
osiągają pożądanych glikemii poposiłkowych
• w nieznany sposób hamuje wydzielanie glukagonu,
• opóźnia opróżnianie żołądka oraz ma efekt zmniejszający
apetyt poprzez wpływ na ośrodkowy układ nerwowy
• Po podaniu podkórnym jest gwałtownie wchłaniany;
szczytowe stężenia pojawiają się po 20 minutach a czas
działania nie przekracza 150 minut.

• jest metabolizowany w nerce i wydalany, jednakże
nawet znaczne obniżenie klirensu kreatyniny nie
zmienia jego biodostępności
• Najlepsze wchłanianie osiąga się po podaniu w brzuch
lub udo; podanie w ramię jest bardziej zawodne
• powinien być wstrzykiwany bezpośrednio przed
jedzeniem;
• Z uwagi na ryzyko hipoglikemii dawki podawanych
jednocześnie insulin ultra-szybko-działających i krótko-
działających powinny zostać zredukowane o połowę lub
więcej.
• Pramlintid powinien być zawsze wstrzykiwany
oddzielnie przy pomocy oddzielnej strzykawki; nie
może być mieszany z insuliną.
• Najczęstszymi działaniami niepożądanymi pramlintidu
jest hipoglikemia i zaburzenia żołądkowo-jelitowe jak
nudności, wymioty czy brak apetytu.

EKSENATID
• Będący syntetycznym analogiem polipeptydu 1 podobnego
do glukagonu (glukagon-like-polipeptide-1; GLP-1)
• jest pierwszym dostępnym lekiem z grupy analogów
hormonów inkretynowych
• został dopuszczony do stosowania podskórnego jako
dodatek u chorych z cukrzycą typu 2 nie osiągających
optymalnych poziomów glikemii przy leczeniu metforminą
czy sulfonylomocznikami.
• Powoduje zwiększenie wydzielania insuliny w odpowiedzi na
hiperglikemię, zmniejszenie poposiłkowego wydzielania
glukagonu w nieznanym dotąd mechanizmie, opóźnienie
opróżniania żołądka i ośrodkową utratę apetytu (związaną z
działaniem na układ nerwowy).
• Zwiększenie wydzielania insuliny wydaje się być częściowo
rezultatem zwiększenia masy komórek B (beta)

•
Eksenatid jest wchłaniany jednakowo z miejsc
wstrzyknięcia na udzie, brzuchu czy ramieniu,
osiąga szczytowe stężenia po około 2 godzinach,
ma czas działania do 10 godzin
•
jest wstrzykiwany podskórnie na 60minut przed
posiłkiem;
•
Przy dodawaniu eksentidu do istniejącego
leczenia sulfonylomocznikiem, dawka środka
doustnego może wymagać redukcji w celu
zapobiegania hipoglikemii.
•
Najpoważniejszymi działaniami niepożądanymi
są nudności, wymioty i biegunka

SITAGLIPTIN
•
Sitagliptin jest inhibitorem peptydazy dipeptydylowej 1
(dipeptidyl peptidase-4; DPP-4), enzymu rozkładającego
hormony inkretynowe i inne białka
•
W badaniach klinicznych fazy 2 i 3 donoszono, że
sitagliptin ma biodostępność około 80% i czas półtrwania
8 – 14 godzin.
•
Obserwowano kontrolę hiperglikemii i obniżenie w
poziomach HBA
1c
przy doustnym stosowaniu dawki 100mg
raz na dobę.
•
Epizody hipoglikemii zdarzały się rzadko, a lek ułatwiał
redukcję masy ciała.
•
Leczenie sitagliptinem może odbywać się w skojarzeniu z
metforminą, glitazonami lub sulfonylomocznikami.

Leczenie skojarzone środkami
doustnymi i wstrzykiwanymi
podskórnie
• LECZENIE SKOJARZONE W LECZENIU
CUKRZYCY TYPU 2
• Leczenie skojarzone eksenatidem –
Eksenatid został dopuszczony do użycia u
osób nieosiągających zadowalających
glikemii przy leczeniu biguanidami,
sulfonylomocznikami lub oboma tymi
grupami. Przy stosowaniu eksenatidu z
insuliną lub lekiem insulinotropowym
istnieje ryzyko hipoglikemii.

• Leczenie skojarzone pramilntidem –
Pramlintid został dopuszczony do skojarzonego
stosowania w czasie posiłku u chorych z
cukrzycą typu 2 nieosiągających
zadowalających glikemii w trakcie leczenia
insuliną, metforminą lub sulfonylomocznikiem.
Leczenie skojarzone powoduje znaczną redukcję
wczesnych hiperglikemii poposiłkowych; dawki
insuliny podawane do posiłków oraz dawki
sulfonylomocznika powinny zostać obniżone w
celu zapobiegnia hipoglikemii.

• Leczenie skojarzone z insuliną – Zaleca
się podawanie długodziałającej insuliny
wieczorem, jako dodatek do leczenia
doustnego u pacjentów z cukrzycą typu 2
nieodpowiadających na dawki maksymalne
leków doustnych. W praktyce klinicznej
stosuje się leczenie skojarzone insuliną
oraz sulfonylomocznikami, glinidami,
pochodnymi D-fenyloalaniny, biguanidami,
glitazonami oraz inhibitorami α-glikozydaz.

LECZENIE SKOJARZONE
W CUKRZYCY TYPU 1
• Leczenie skojarzone z pramlintidem
–
Pramlintid został dopuszczony do
skojarzonego stosowania przed posiłkami u
chorych z cukrzycą typu 1 mających złą
kontrolę glikemii pomimo stosowania
optymalnej insulinoterapii. Dodanie
pramlintidu powodowało istotna redukcję
częstości poposiłkowej hiperglikemii;
zazwyczaj niezbędne jest obniżenie dawek
insuliny stosowanych do posiłku.

• Leczenie skojarzone z lekami
doustnymi – Nie ma wskazań do
łączenia insuliny z lekami
insulinotropowymi (sulfonylomoczników,
glinidów, pochodnych D-fenyloalaniny) u
chorych z cukrzycą typu 1.
• Chorzy z dietami bardzo bogatymi w
skrobię mogą odnieść korzyść z
dołączenia inhibitora α-glikozydazy

GLUKAGON
•
Budowa chemiczna i metabolizm
•
Glukagon jest produkowany przez komórki A (alfa) wysp
trzustkowych Langerhansa
•
Glukagon jest peptydem zbudowanym z pojedynczego
ważącego 3485 łańcucha 29 aminokwasów (budowa jest
identyczna u wszystkich ssaków).
•
Glukagon jest bardzo szybko rozkładany w wątrobie,
nerkach, osoczu oraz w miejscach wiązania z receptorem.
•
Z uwagi na gwałtowny rozkład w osoczu, przy pobieraniu
próbek na oznaczenie poziomów glukagonu wymagane
jest schłodzenie próbówek i dodanie inhibitorów enzymów
proteolitycznych.
•
Czas półtrwania glukagonu w osoczu jest podobny do
insuliny i wynosi 3 – 6 minut.

• EFEKTY METABOLICZNE
• Pierwszych sześć aminokwasów po stronie aminowej
cząsteczki glukagonu wiąże się specyficznie z receptorem
w komórkach wątrobowych.
• Prowadzi to do aktywacji cyklazy adenylanowej i
zwiększenia produkcji cAMP, co powoduje zwiększenie
katabolizmu zmagazynowanego glikogenu, zwiększenie
glukoneogenezy oraz ketogenezy.
• Natychmiastowym efektem infuzji glukagonu jest wzrost
stężenia glukozy w surowicy kosztem zmniejszenia ilości
zmagazynowanego w wątrobie glikogenu.
• Nie obserwuje się wpływu na glikogen zawarty w
mięśniach, prawdopodobnie z uwagi na nieobecność
receptora glukagonowego w tkance mięśniowej.
• Ilości glukagonu stosowane w leczeniu powodują ponadto
uwolnienie insuliny z niezmienionych komórek B trzustki,
uwolnienie katecholamin z guza chromochłonnego oraz
kalcytoniny z komórek rakowych w rdzeniu.

• EFEKTY SERCOWE
• Glukagon ma silny wpływ inotropowo i chronotropowo
dodatni na serce, spowodowany zwiększeniem
stężenia cAMP, podobnie jak opisano powyżej.
Powoduje więc efekt podobny do działania agonisty
receptora β-adrenergicznego bez obecności
funkcjonujących receptorów β.
• WPŁYW NA MIĘŚNIÓWKĘ GŁADKĄ
• Duże dawki glukagonu powodują nasilony rozkurcz
jelit. W przeciwieństwie do efektów opisanych
powyżej, to działanie może być spowodowane innymi
mechanizmami niż aktywacja cyklazy adenylanowej.

Zastosowania kliniczne
• CIĘŻKA HIPOGLIKEMIA
• Głównym zastosowaniem glukagonu jest leczenie
ratunkowe ciężkich stanów hipoglikemicznych u
pacjentów z cukrzycą typu 1, w przypadku utraty
przytomności wykluczającej zastosowanie podania
węglowodanów doustnie i braku możliwości podania
glukozy dożylnie.
• Dostępny obecnie glukagon produkowany jest
metodą inżynierii genetycznej i sprzedawany w
ampułkostrzykawkach do podawania podskórnego w
dawce 1mg
• Stworzono również aplikatory donosowe, jednakże nie
doczekały się one jeszcze dopuszczenia przez FDA.

• DIAGNOSTYKA
ENDOKRYNOLOGICZNA
• Kilka testów wykorzystuje glukagon do
diagnostyki endokrynologicznej.
• U pacjentów z cukrzycą typu 1 wykonując
standardowy test trzustkowej rezerwy
wydzielniczej komórek B stosuje się glukagon w
1mg dożylnym bolusie. Z uwagi na fakt, że u
osób stosujących insulinę pojawiają się
przeciwciała przeciwinsulinowe wpływające na
oznaczenia insuliny, do pomiaru sekrecji
insuliny wykorzystuje się oznaczenia C-peptydu.

• ZATRUCIA BETA-BLOKERAMI
• Glukagon używany jest czasem w celu odwrócenia
sercowych efektów przedawkowania β-blokerów z
uwagi na zdolność podwyższania stężenia cAMP w
sercu. Nie jest jednakże klinicznie skuteczny w leczeniu
niewydolności serca.
• BADANIA RADIOLOGICZNE JELIT
• Glukagon był szeroko stosowany w radiologii jako
środek pomocniczy w badaniach rentgenowskich jelit z
uwagi na swoją zdolność do powodowania rozkurczu
jelit.
• Działania niepożądane
• Po zastosowaniu glukagonu mogą się pojawić
przejściowe nudności, a rzadziej wymioty. Zazwyczaj są
to reakcje łagodne, a glukagon nie ma innych
poważnych działań niepożądanych.

ISLET AMYLOID POLYPEPTIDE
(IAPP, AMYLINA)
• Amylina jest 37-aminokwasowym peptydem wyizolowanym ze złogów
amyloidowych wysp trzustkowych pobranych od pacjentów z długotrwałą
cukrzycą typu 2 lub insulinomą. Jest ona produkowana jest w komórkach
B trzustki, pakowana do ziarnistości w stężeniu 1 -2 % stężenia insuliny i
wydzielana wspólnie w sposób pulsacyjny oraz w odpowiedzi na bodziec.
Wydzielana jest w przybliżeniu 1 cząsteczka amyliny na 10 cząsteczek
insuliny. Krąży w formie glikowanej (aktywnej) i nieglikowanej
(nieaktywnej) w stężeniach około 4 – 25 pmoli/l, a wydalana jest w
większości przez nerki. Amylina wydaje się być członkiem rodziny
peptydów neuroregulacyjnych, posiada 46% homologii z peptydem
związanym z genem kalcytoniny - calcytonin gene related peptide; CGRP.
Rolą fizjologiczną amyliny może być modulacja wydzielania insuliny
poprzez negatywne sprzężenie zwrotne na wydzielanie insuliny. W
dawkach farmakologicznych amylina zmniejsza sekrecję glukagonu,
opóźnia opróżnianie żołądka przez mechanizm związany z nerwem
błędnym oraz zmniejsza apetyt poprzez działanie na ośrodkowy układ
nerwowy. Analog amyliny pramlintid (patrz wyżej) różni się od amyliny
substytucją proliny w pozycjach 25, 28 i 29. modyfikacje te zwiększają
jego rozpuszczalność i znoszą skłonność do autoagregacji.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
Wyszukiwarka
Podobne podstrony:
hormony płciowe, leki przeciwhormonalne leki antykoncepcyjne
W15 SL W01 Hormony jako leki insulina i doustne leki przeciwcukrzycowe (Paulina)
88 Leki przeciwreumatyczne część 2
Opioidowe leki przeciwbólowe 2
Leki przeciwdepresyjne
(65) Leki przeciwreumatyczne (Część 1)
2011 Leki przeciwgrzybicze Kopiaid 27453 ppt
79 Doustne leki przeciwcukrzycowe
Leki przeciwwirusowe 4
Leki przeciwbakteryjne i przeciwwirusowe
Trójpierścieniowe leki przeciwdepresyjne
więcej podobnych podstron