

DZIEDZICZENIE JEDNOGENOWE
SPRZĘŻONE Z PŁCIĄ
AUTOSOMALNE
•
DOMINUJĄCE
•
RECESYWNE
•
KODOMINUJĄCE
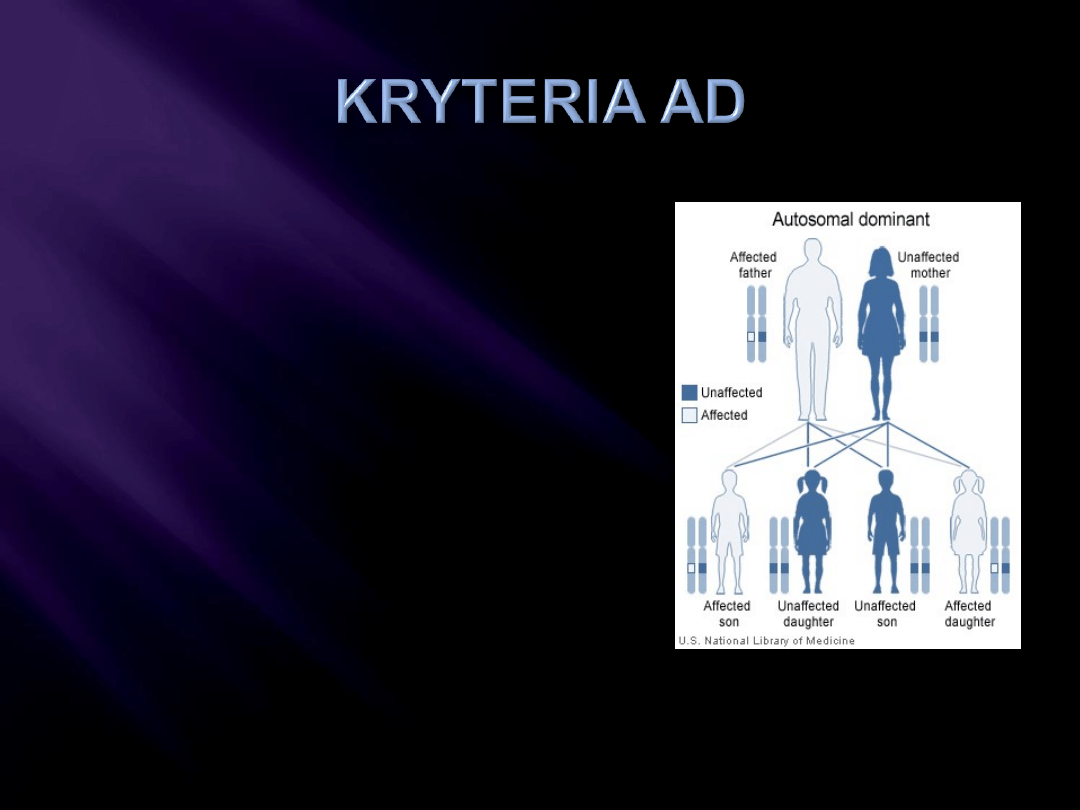
Pionowy wzór rodowodu
Objawy choroby
u heterozygot
- Aa
Jednakowa częstość
objawów K/M
Ryzyko odziedziczenia
zmiany – 50%

ZMIENNA EKSPRESJA
ZMNIEJSZONA PENETRACJA
PENETRACJA = CHORZY / NOSICIELE
MOZAIKOWATOŚĆ GERMINALNA =
zmiany tylko w części komórek gonad
=>
zdrowi rodzice => chore dzieci

Brak lub uszkodzenie LDL-R (IIa) , apoB-
100 (IIb)
Podwyższony poziom LDL-Ch
Częstość występowania:
Postać hetero - 1:500
Postać homo – 1: 1 000 000 (większość
popul.)
TC mg/dl
LDL-Ch mg/dl
norma
< 200 - 250
< 135 - 155
heterozygoty
250 – 500
200 - 400
homozygoty
> 700
> 500

Glikoproteina na pow. kom – 839 AA
Składa się z 5 podjednostek:
domena wiążąca ligand
d. homologiczna z EGF
domena 3
domena 4
odcinek wewnątrz cytoplazmy

Krótkie ramię chromosomu 19 (19p13.2)
~45 kb
18 exonów i 17 intronów

> 200
Większość to mutacje punktowe oraz
niewielkie del lub ins
Występują w każdym exonie,
ale najwięcej w 4
Polish

Syntetyzowana w wątrobie
Masa cząsteczki ok. 550 kD
Stanowi rdzeń lipoprotein o niskiej
gęstości
Na jej pow. Miejsce wiążące dla LDL-R
Odpowiedzialna za komórkowy
metabolizm LDL

Na krótkim ramieniu chromosomu 2
(2p24-p23)
ApoB-100 – 4536 AA , mRNA > 16kb

Mogą powodować:
Zaburzenia syntezy białka
(hypobetalipoproteinemia)
Osłabienie wiązania Apob-100 z LDL-R
(FH-FDB)
23 różne mutacje – tylko 2 => FH-
FDB
ApoB-3500 Arg-Glu (b.
rozpowszechniona)
ApoB-3531 Arg-Cys (rzadka mutacja)

DANE RODOWODOWO-KLINICZNE
WYNIKI TESTÓW BIOCHEMICZNYCH
TESTY DNA (LDL-R I ApoB-100)

HETEROZYGOTY
od momentu
urodzenia mają
2X wyższe stężenie TC (350-550 mg/dl),
żółtaki ścięgniste (tendon xantothomas)
zwykle po 20,
s. Achillesa, prostowniki ręki
nasilona miażdżyca po 30 roku życia,
IHD – po 40 r.ż.

HOMOZYGOTY
od momentu urodzenia
mają
niezwykle wysokie stężenie TC (700-1200
mg/dl),
żółtaki w skórze (cutaneous xanthomas) od 3
r.ż.
miażdżyca jest b. ciężka, rozsiana i
obejmuje tętnice wieńcowe, szyjne, biodrowe,
udowe oraz początkowy odcinek aorty.
miażdżyca n. wieńcowych, i zawał mięśnia
sercowego, zwykle przed 20 r.ż..
Rzadko dożywają 20 r.ż.

u wszystkich -
zmiana stylu życia
(dieta, ruch)
rozpocząć stosowanie leków
zmniejszających stężenie LDL-C u
młodych dorosłych
statyny
stanowią leki pierwszego
wyboru (jednocześnie rozpocząć dietę
leczniczą)
żywice jonowymienne
(w skojarzeniu
ze statynami, jeśli to konieczne)
jeśli to konieczne, rozważyć
leczenie
trójlekowe
(statyna + żywica
jonowymienna + kwas nikotynowy

leczenie za pomocą diety nieskuteczne
żywice jonowymienne nieskuteczne
kwas nikotynowy mało skuteczny
statyny mogą być umiarkowanie
skuteczne u niektórych chorych
zespolenie omijające jelito kręte
nieskuteczne
przeszczep wątroby skuteczny, ale
niepraktyczny
obecnie stosuje się LDL-aferezę
(usuwa VLDL i LDL)
W przyszłości – terapia genowa ?

POSTAĆ DZIECIĘCA – ARPKD
POSTAĆ DOROSŁYCH – ADPKD
o bardzo znacznej penetracji
MOŻE WYJĄTKOWO UJAWNIĆ SIĘ U
DZIECI !
W bardzo rzadkich sytuacjach wykrywana w
badaniach prenatalnych.

CZĘSTOŚĆ 1:1000
Najczęstsza spośród dziedzicznych
nefropatii
Czwarta przyczyna PNN 7-15%
Zwykle Late-onset
Zaburzenie wielonarządowe
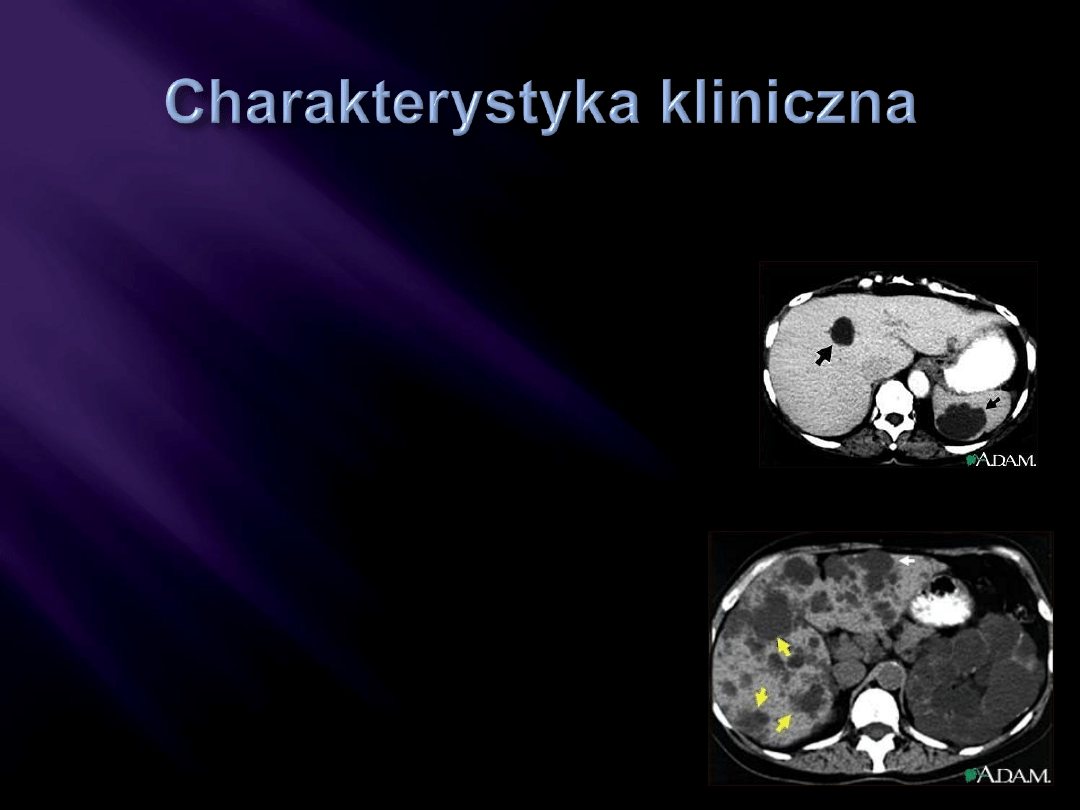
zmiany torbielowate dotyczące obu
nerek (Zwykle obu nerek)
zmiany torbielowate w innych narządach:
wątroba, trzustka, śledziona, płuca
pęcherzyki nasienne,
pajęczynówka;
zaburzenia naczyniowe:
tętniaki wewnątrzczaszkowe- tt.podstawy mózgu –
12%
poszerzenie pnia aorty,
rozwarstwienie aorty piersiowej;
wypadanie płatka zastawki mitralnej;
przepukliny brzuszne

Manifestacja nerkowa ADPKD zawiera
zaburzenia funkcji:
nadciśnienie
ból nerek
niewydolność.
średnio 50% chorych z ADPKD - schyłkowa NN
do 60 r.ż.
Zmiany policystyczne wątroby = najcz.
pozanerkowa manifestacja.
20% w III dekadzie do 75% po 60 r.ż.
Mitral valve prolapse to najczęstsza wada
zastawkowa do 25% nosicieli
Zmienność narządowa i ciężkość zmian różni się
u członków tej samej rodziny
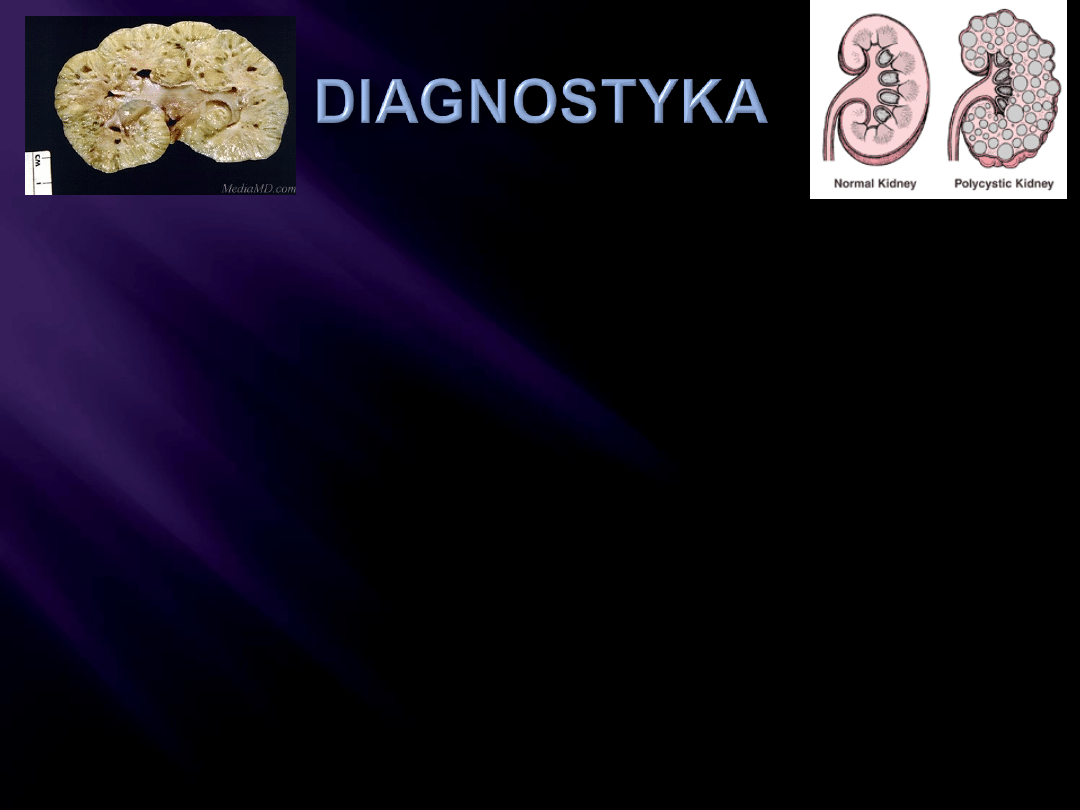
Wywiad rodzinny
Testy laboratoryjne
Zmiany w obrazowaniu nerek – USG/CT
Kryteria dgn u osób z 50% ryzykiem:
Co najmniej 2 jedno- lub obustronne torbiele
u młodszych niż 30 lat;
2 torbiele w każdej nerce u 30-59 latków;
4 torbiele w każdej nerce > 60 r.ż.

CT
of the abdomen without and with contrast
enhancement
Standardized blood pressure
Measurement of blood lipid concentrations
because
hyperlipidemia is a correctable risk factor for progressive
renal disease, including ADPKD
Urine studies
to detect the presence of
microalbuminuria or proteinuria, which in the presence
of severe renal cystic disease indicates a worse prognosis
and mandates strict control of the blood pressure
Echocardiography
in persons with heart murmurs or
systolic clicks possibly resulting from valvular heart
disease, mitral valve prolapse or congenital cardiac
abnormalities
Echocardiography or cardiac MRI
to screen persons
at high risk because of a family history of thoracic aortic
dissections
Head MRA or CT angiography
to screen persons at
high risk because of a family history of intracranial
aneurysms

Dwa geny związane są z ADPKD:
PKD1, dotyczy 85% chorych
PKD2, dotyczy 15% chorych
PKD1 16p13.3-p13.1 Polycystin-1
PKD2 4q21-q23
Polycystin-2
mnóstwo pojedynczych mutacji
pseudogeny

Nadciśnienie
Nie ustalono stałego schematu leczenia
(USA)
Z uwagi na rolę układu R-A w
patogenezie nadciśnienia w ADPKD,
zaleca się stosowanie:
ACE inhibitors
Antagonistów receptora angiotensyny II.
zwiększają przepływ nerkowy,
Mało efektów ubocznych,
Redukują proliferację mięśniówki naczyń

Ból
Leki nienarkotyczne – często
stosowane. Ostrożnie – efekt
nefrotoksyczny
TPLD- pomocne w leczeniu
długotrwałych zespołów bólowych
Leki narkotyczne – zarezerwowane dla
ostrych epizodów
Blokada zwojów trzewnych - czasowa
terapia
Odnerwienie nerek – u 1 chorego
[
].

Krwawienia do torbieli i ich nadmierny
wzrost
Kamica nerkowa
Zakażenia torbieli
Niewydolność nerek
Zmiany w wątrobie
Naczyniaki i tętniaki wewnątrzczaszkowe
Rozwarstwienia aorty

Średni czas przeżycia od pierwszych
objawów NN wynosi ok. 10 lat
Czynniki przyspieszające NN
Nawracające zakażenia układu moczowego
Źle kontrolowane nadciśnienie
Główna przyczyna zgonu obok
niewydolności narządowej – pęknięcie
tętniaka

Częstość 1:2000
Choroba dziedziczona w sposób AD
Z pełną, zależną od wieku penetracją
genu
Wiek
penetracja
30
10%
40
30%
50
60%
60
85%
70
95%

Gen huntingtyny 4p16.3
Duży gen – 210 kb , 67 exonów
Mutacja polega na wystąpieniu nadmiernej
ilości powtórzeń trójnukleotydowej sekwencji
CAG na końcu 5’ genu (...CAGCAGCAG...)
>36
Zdrowi średnio 10-29 powtórzeń
Chorzy 36-120 (średnio 45-55)
Zmiana od matki – ta sama liczba CAG
Zmiana od ojca –
WZROST LICZBY
POWTÓRZEŃ !!!
Antycypacja!

Hiperkinetyczne zaburzenia ruchu –
mimowolne ruchy pląsawicze
Postępujący zespół otępienny
Zaburzenia psychiczne -
niewytłumaczalne
zmiany nastroju
Zaburzenia mowy
Cherlactwo fizyczne
W końcowej fazie – sztywność
pozapiramidowa

Postać młodzieńcza (Westphala)
3% (przed 15 rokiem życia)
Od początku choroby dominuje sztywność
Pojawiają się napady padaczkowe
Objawia się ona początkowo trudnościami w
nauce, czasami drobnymi zmianami pisma czy
pewną ociężałością ruchów.

Przebieg choroby postępujący
Nieuchronnie prowadzi do śmierci
P. typowa – 15-20 lat
P. młodzieńcza – 10 lat

CT , MRI, PET
Znacznego stopnia zanik jądra
ogoniastego z wyraźnym poszerzeniem
układu komór i zanikiem kory
Zmiany neuropatologiczne pojawiają się
z opóźnieniem w stosunku do objawów
klinicznych

Brak leczenia przyczynowego
Leczenie objawowe
Neuroleptyki (haloperidol, pimozyd)
Zmniejszają objawy ruchowe bez wpływu na
zmiany otepienne
Lewodopa lub agonisci rec.
Dopaminowego
Leczenie stanów terminalnych i p. młodzieńczej
kontrola omamów i urojeń oraz napadów
gniewu i złości pojawiających się czasami
w przebiegu choroby.
Również zwalczanie depresji i lęku

Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
Wyszukiwarka
Podobne podstrony:
dziedziczenie AD AR diagnostyka kurs 2012
dziedziczenie AD AR diagnostyka kurs 2012
Wyk ad 5 6(1)
Rodowody, dziedziczenie, imprinting
uk ad pokarmowy
dziedziczenie chorob jednogenowych
Wyk ad II
Tkanki wyk ad 1
dziedziny wychowania(1)
Ekonomika Transportu wyk+ad 1
Kolonialne dziedzictwo
Wyk ad Fizyka 2
16 Dziedziczenie przeciwtestamentowe i obliczanie zachowkuid 16754 ppt
Wyk ad 04
więcej podobnych podstron