
Choroby metaboliczne
Choroby metaboliczne

Choroby metaboliczne
Wrodzone choroby
metaboliczne
•
Są chorobami monogenowymi
•
Mutacje odpowiadają za wystąpienie zmian
patologicznych w produkcji białka (np. enzymu)
•
Brak lub zmiana aktywności tego białka
prowadzi do upośledzenia procesu
metabolicznego
•
Dziedziczone najczęściej w sposób autosomalny
recesywny

Choroby metaboliczne
.
z k w a s ic ą m e t a b o lic z n ą
b e z k w a s ic y m e ta b o lic z n e j
c h o ro b y m e ta b o lic z n e

Choroby metaboliczne
Choroby metaboliczne z
kwasicą metaboliczną
•
Kwasice organiczne (acydurie
organiczne)
•
Pierwotne kwasice mleczanowe
•
Niektóre glikogenozy wątrobowe
•
Choroba syropu klonowego
(zaburzenia dekarboksylacji
ketokwasów)

Choroby metaboliczne
.
Kwasice organiczne (acydurie organiczne):
- Bloki enzymatyczne metabolizmu
aminokwasów i tłuszczów
- W surowicy i w moczu obecne:
ketokwasy, hydroksykwasy, kwasy
dwukarboksylowe
- Rozpoznanie opiera się na badaniu
profilów kwasów organicznych w moczu
metodą GC-MS (chromatografia gazowa
sprzężona ze spektrometrią masową)

Choroby metaboliczne
.
Pierwotne kwasice mleczanowe:
- Deficyty enzymów: cyklu Krebsa,
przemiany pirogronianu
- Deficyty kompleksów łańcucha
oddechowego

Choroby metaboliczne
.
Glikogenozy wątrobowe:
- deficyty enzymatyczne syntezy i
rozkładu glikogenu
- glikogenoza I (niedobór glukozo-6-
fosfatazy) przebiega ze znaczną
kwasicą mleczanową

Choroby metaboliczne
Choroby metaboliczne bez
kwasicy
•
Zaburzenia przemiany
węglowodanów
- Galaktozemia (deficyt transferazy
galaktozo-1-fosforanu)
- Niektóre glikogenozy wątrobowe

Choroby metaboliczne
.
•
Zaburzenia przemiany
aminokwasów
- Fenyloketonuria (deficyt
hydroksylazy fenyloalaniny)
- Tyrozynemia
- Homocystynuria
- Hiperamonemia typu II (deficyt
transkarbamylazy ornitynowej)

Choroby metaboliczne
.
•
Zaburzenia oksydacji kwasów
tłuszczowych
•
Choroby lizosomalne
- Gangliozydozy
- Choroba Gaucher
- Choroba Niemann – Picka
- Mukopolisacharydozy

Choroby metaboliczne
Kiedy podejrzewać?
•
Zawsze w przypadku noworodka w
ciężkim stanie ogólnym
•
Pogarszanie się stanu początkowo
zdrowego noworodka
•
Hiperwentylacja, bez zmian
osłuchowych nad płucami i bez
odchyleń w rtg
•
Wtedy, gdy podejrzewamy posocznicę

Choroby metaboliczne
Objawy u noworodka
•
Wymioty, biegunka, brak apetytu
•
Zaburzenia świadomości
•
Trudności w ssaniu
•
Zaburzenia napięcia mięśniowego
•
Tachypnoe, niewydolność oddechowa
•
Drgawki
•
Powiększenie wątroby i śledziony

Choroby metaboliczne
Objawy u niemowląt i
dzieci starszych
•
Opóźnienie rozwoju umysłowego
•
Drgawki
•
Zapach mysi, zapach syropu klonowego
(karmelu)
•
Epizody wymiotów, kwasicy, zaburzeń
świadomości
•
Hepatomegalia, uszkodzenie czynności
wątroby
•
Kamica nerkowa

Choroby metaboliczne
.
•
Zaćma, zwichnięcie soczewki
•
Zwyrodnienie barwnikowe
siatkówki
•
Kardiomegalia
•
Krzywica witamino-D-oporna
•
Niedokrwistość, neutropenia
•
Dysmorfia twarzy
•
Zespoły wad wrodzonych
•
Wyprysk skórny, rybia łuska

Choroby metaboliczne
Jak rozpoznawać?
kw a s ica o rg a n ic z n a
a m o n ia k >N
kw a s ic a
kw a s ica o rg a n ic z n a
a m o n ia k w n o rm ie
kw a s ic a
a m ino a cyd o p a tie lu b g a la kto ze m ia
a m o n ia k w n o rm ie
p H p ra w id ło w e
d e fe kty c yklu m o c z n iko w e g o
a m o n ia k > N
p H p ra w id ło w e
o z n a c z yć a m o n ia k w su ro w icy
o z n a c z yć p H krw i
p o d e jrz e nie c h o ro b y m e ta b o lic z n e j

Choroby metaboliczne
.
•
Gazometria
•
Amoniak w surowicy
•
Glukoza w surowicy
•
Jonogram
•
Kwas mlekowy

Choroby metaboliczne
Określenie wrodzonej
choroby metabolicznej
•
Wykazanie w surowicy i/lub w moczu
nadmiaru substratu zablokowanej
reakcji
•
Stwierdzenie obecności
patologicznych metabolitów
(produktów reakcji alternatywnych)
•
Wykazanie braku produktu
zablokowanej reakcji
•
Wykazanie mutacji

Choroby metaboliczne
Testy diagnostyczne
•
Substancje redukujące w moczu (galaktozemia)
•
Chromatografia aminokwasów
(aminoacydopatie)
•
Mukopolisacharydy w moczu
(mukopolisacharydozy)
•
GC-MS (kwasice organiczne, zaburzenia beta-
oksydacji kwasów tłuszczowych)
•
Tandem MS
•
Aktywność enzymów w erytrocytach
(galaktozemia)
•
Metody genetyki molekularnej
•
inne

Choroby metaboliczne
Leczenie
•
Warunkiem powodzenia jest wczesne
ustalenie rozpoznania
•
Eliminacja substratu z pokarmów
•
Substytucja enzymu
•
Substytucja produktu (np. hormonu)
•
Dostarczanie kofaktora reakcji np.
witaminy lub pierwiastka śladowego
•
Pokrycie zapotrzebowania kalorycznego i
płynowego
•
Dializoterapia

Choroby metaboliczne
.
•
Metody genetyki molekularnej
(wprowadzenie do ustroju DNA
kodującego enzym)
•
Transplantacja narządu, w którym
zlokalizowana jest dana reakcja

Choroby metaboliczne
Wnioski dla klinicysty
•
Jeśli leczenia nie rozpocznie się
natychmiast, duża część wrodzonych
chorób metabolicznych kończy się zgonem
•
Zbyt późne rozpoznanie zagraża
nieodwracalnym uszkodzeniem mózgu
•
Jedynym sposobem wykrycia tych zaburzeń
jest stałe podejrzewanie ich obecności
•
Powodzenie większości strategii leczenia
zależy od jak najwcześniejszego
rozpoczęcia terapii

Choroby metaboliczne
Choroby metaboliczne
•
Zaburzenia metabolizmu węglowodanów
•
Zaburzenia metabolizmu aminokwasów
•
Kwasice organiczne
•
Defekty cyklu mocznikowego
•
Zaburzenia beta-oksydacji kwasów tłuszczowych
•
Hiperlipidemie
•
Choroby lizosomalne
•
Choroby peroksysomalne
•
Porfirie
•
Zaburzenia metabolizmu miedzi
•
Zaburzenia metabolizmu żelaza
•
Choroby mitochondrialne

Choroby metaboliczne
Zaburzenia metabolizmu
węglowodanów
•
Galaktoza
•
Galaktozemia klasyczna (deficyt urydylilotransferazy
galaktozo-1-fosforanu)
•
Deficyt galaktokinazy
•
Deficyt epimerazy urydylilodifosfogalaktozy
•
Glikogenoza typu XI
•
Fruktoza
•
Dziedziczna nietolerancja fruktozy
•
Deficyt fruktozo-1,6-difosfatazy
•
Glikogen
•
Glikogenozy

Choroby metaboliczne
Galaktozemia
•
Częstość występowania 1:60000
•
Drugi etap przemiany galaktozy (deficyt
urydylilotransferazy galaktozo-1-fosforanu)
•
Warianty alleliczne Duarte i Negro
•
W 70% mutacja Q188R
•
Wymioty, biegunka, powiększenie wątroby i
śledziony, żółtaczka zaburzenia
krzepnięcia, kwasica, hipoglikemia,
posocznica bakteryjna

Choroby metaboliczne
Galatozemia diagnostyka
•
Wzrost poziomu galaktozy w
moczu i surowicy
•
Brak aktywności enzymatycznej
urydylilotransferazy galaktozo-1-
fosforanu w erytrocytach,
fibroblastach i innych tkankach

Choroby metaboliczne
Galatozemia - leczenie
•
Preparaty mlekozastępcze sojowe
lub hydrolizaty kazeiny
bezlaktozowe
•
Powikłania późne mimo stosowania
diety:
•
Niedobór wzrostu
•
Zaburzenia rozwoju intelektualnego
•
Niewydolność hormonalna jajników

Choroby metaboliczne
Fruktozemia
•
Niedobór aldolazy B fruktozo-1-
fosforanu
•
Najczęstsze mutacje to A149P (67%) i
A174D (16%)
•
Objawy podobne do galaktozemii ale
później
•
Rozpoznanie: oznaczenie aktywności
w wątrobie
•
Leczenie: eliminacja z diety fruktozy

Choroby metaboliczne
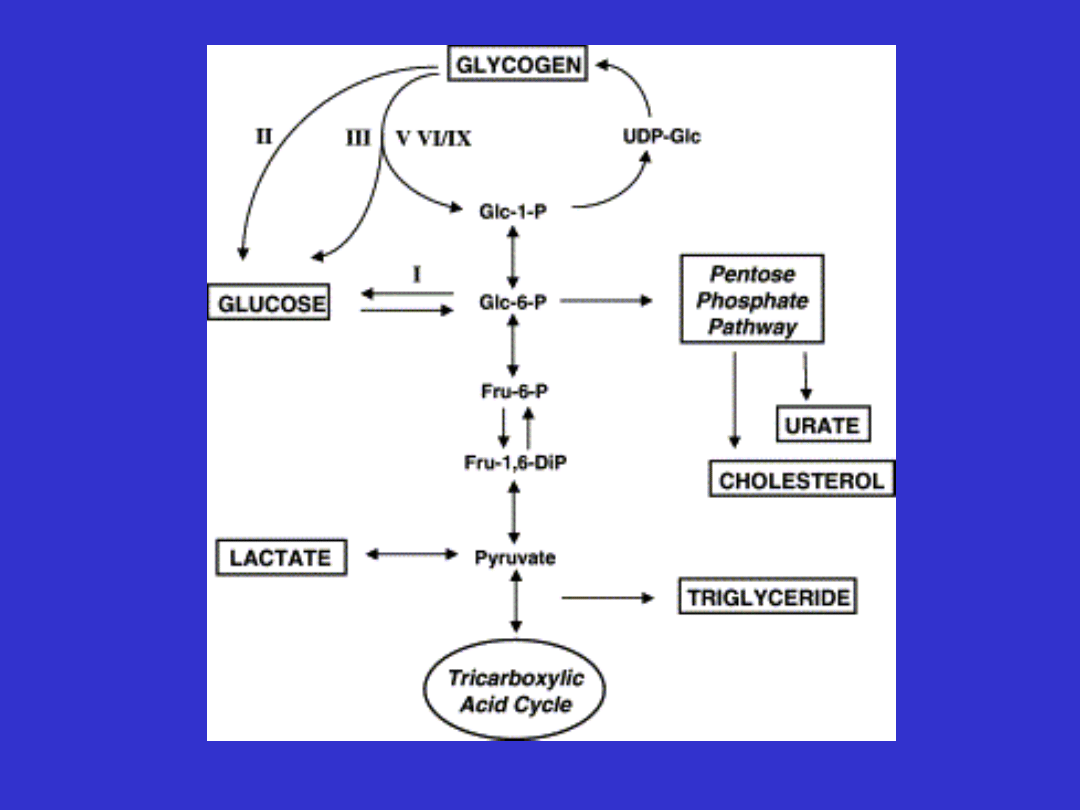
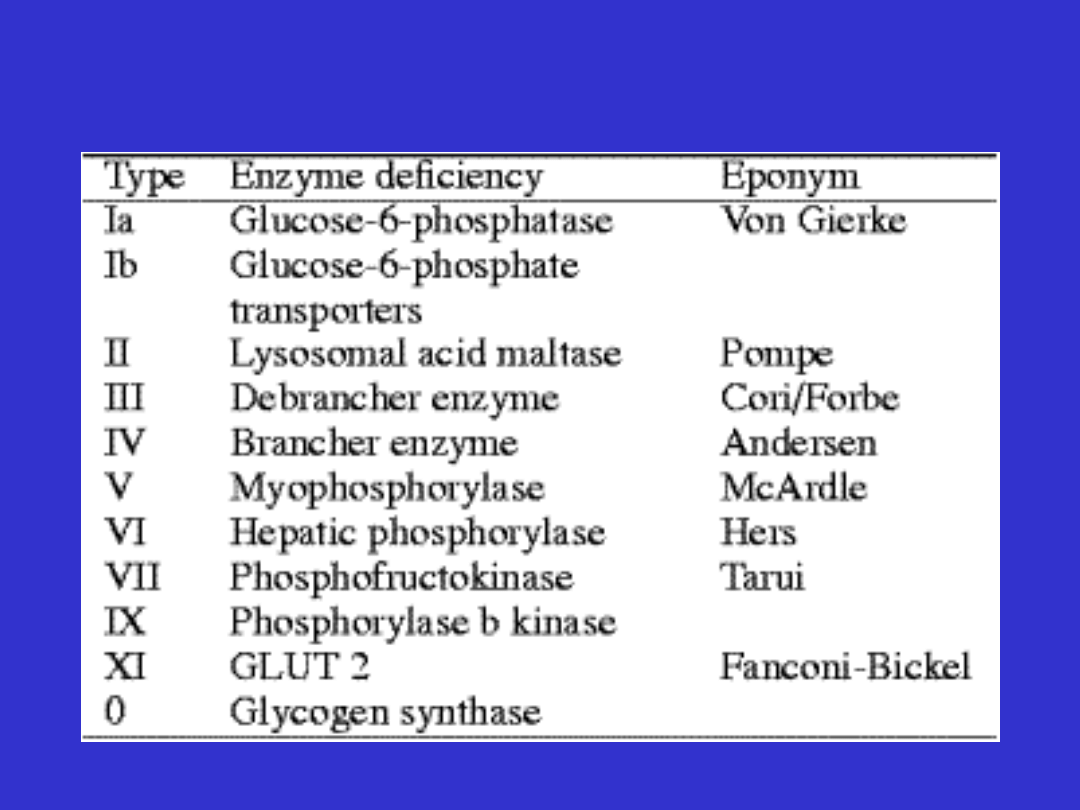
Glikogenozy
•
Choroby spichrzania glikogenu
•
Częstość ok. 1:40000
•
Dziedziczenie AR
•
Wyjątek GDS VI a recesywne sprzężone z
chromosomem X
•
Glikogenozy
•
Wątrobowe
•
Hipoglikemia (pomiędzy posiłkami) z wyjątkiem GDS
II, powiększenie wątroby, kwasica, hiperlipemia
•
Mięśniowe
•
Bez hipoglikemii objawy dotyczą tylko mięśni
Choroby metaboliczne
Choroby metaboliczne
Glikogenozy

Choroby metaboliczne
Glikogenozy - diagnostyka
•
Podanie glukagonu
•
Krzywa glukozowa płaska
•
Dożylne obciążenie glukozą
•
Zmniejszenie stężenie mleczanu
•
Podstawą rozpoznania jest badanie
aktywności enzymu w bioptacie
wątroby
Choroby metaboliczne

Choroby metaboliczne
Zaburzenia metabolizmu
aminokwasów
•
Fenyloalanina
•
Tyrozyna
•
Metionina
•
Leucyna, izoleucyna, walina
•
Glicyna

Choroby metaboliczne
Fenyloketonuria (OMIM
261600) (PKU)
•
Częstość występowania ok. 1:10000
•
Opisana po raz pierwszy w 1934 przez Asbörna
Föllinga
•
Objawy kliniczne:
•
Bezpośrednio po urodzeniu brak jakichkolwiek objawów
•
Postępujące i nieodwracalne opóźnienie rozwoju wyraźne
już w 1 roku życia (pierwsze objawy ok. 3 mies.)
•
Wymioty, wysypki skórne, jaśniejsza karnacja, „mysi”
zapach moczu
•
Małogłowie
•
Ostateczny iloraz inteligencji (IQ) 30 – 50
•
Często padaczka
Choroby metaboliczne

Choroby metaboliczne
Genetyka
•
Dziedziczenie AR
•
Gen kodujący hydroksylazę
fenyloalaniny (PAH) chromosom 12 w
regionie q22–q24.1.
•
90 kb 13 eksonów koduje 2.4 kb
mRNA
•
Dotychczas opisano ok. 460 różnych
mutacji
•
R408W najczęstsza w europie ok. 30%

Choroby metaboliczne
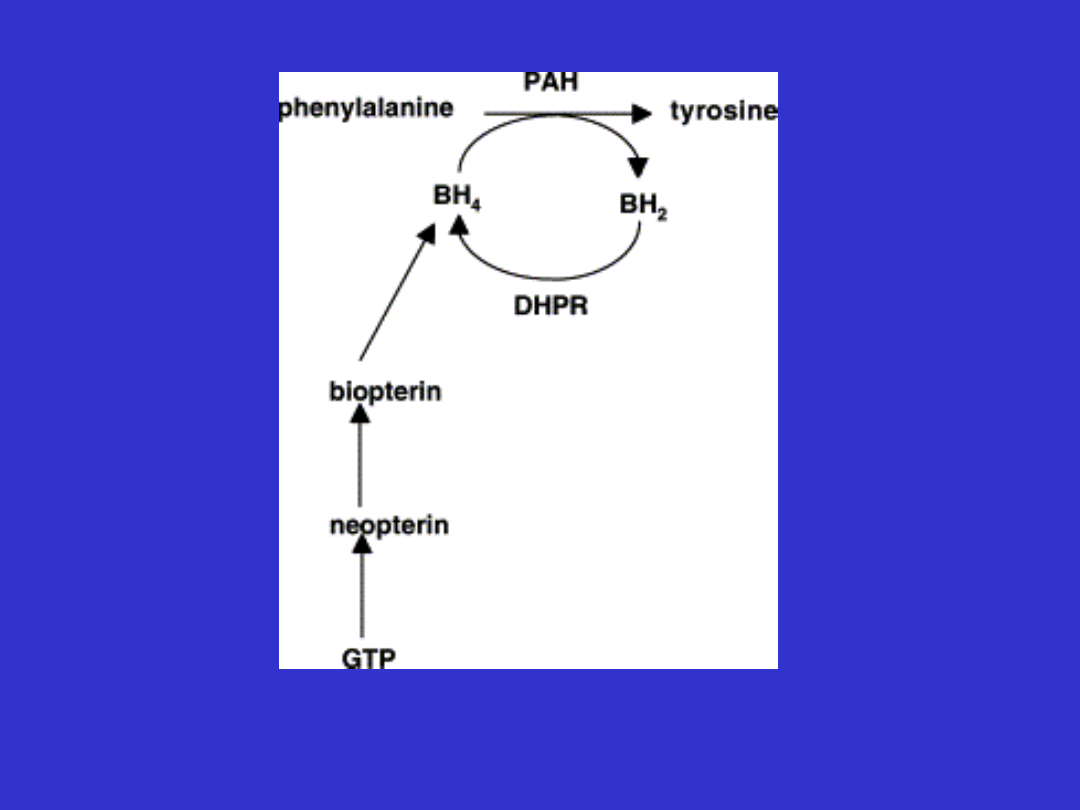
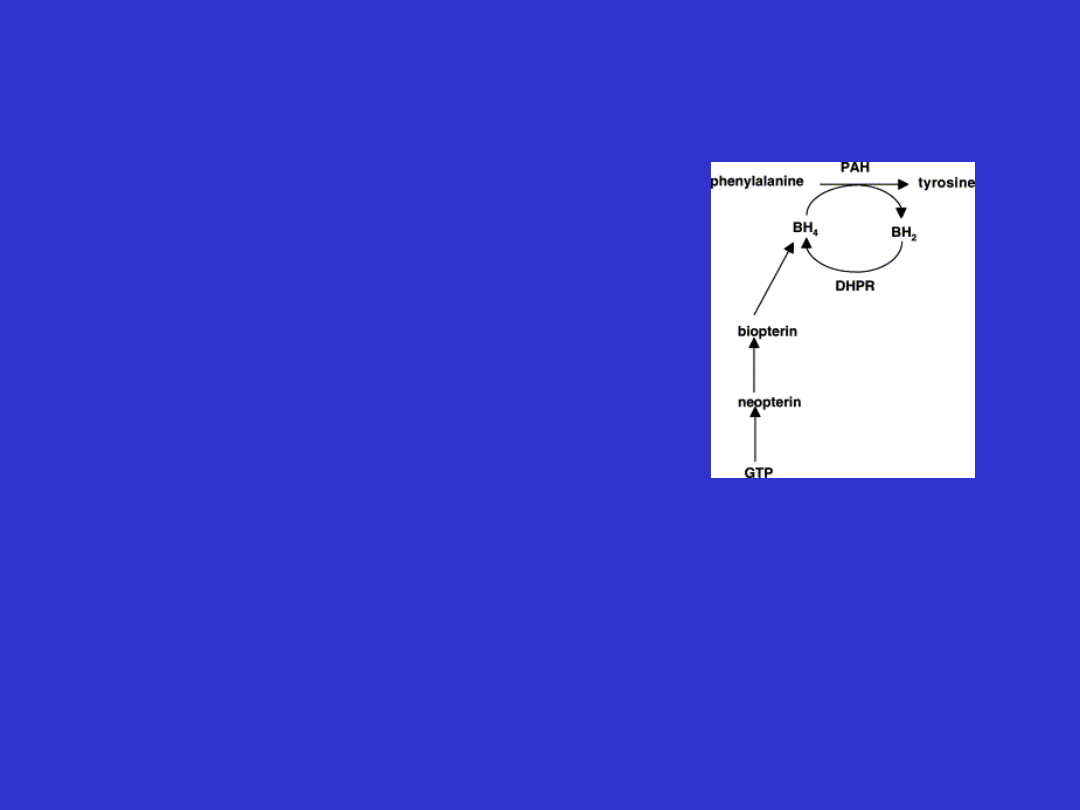
Reakcja hydroksylacji l-
fenyloalaniny do tyrozyny
•
Nadmiar fenyloalaniny jest
hydroksylowany do tyrozyny
•
Hydroksylaza fenyloalaniny jest
tetramerem
•
Aktywność enzymu jest regulowana
przez jego fosforylację i defosforylację
•
+fenyloalanina
•
- BH
4

Choroby metaboliczne
Rozpoznanie
•
Test Guthriego
•
Podwyższenie stężenia fenyloalaniny
w surowicy > 20 mg%
•
Badania molekularne
•
Rozpoznanie powinno być postawione
do 2 mż.
Choroby metaboliczne
Leczenie
•
Dieta eliminacyjna
•
Max. stężenie 3-7mg%
•
U kobiet w ciąży <10mg%
•
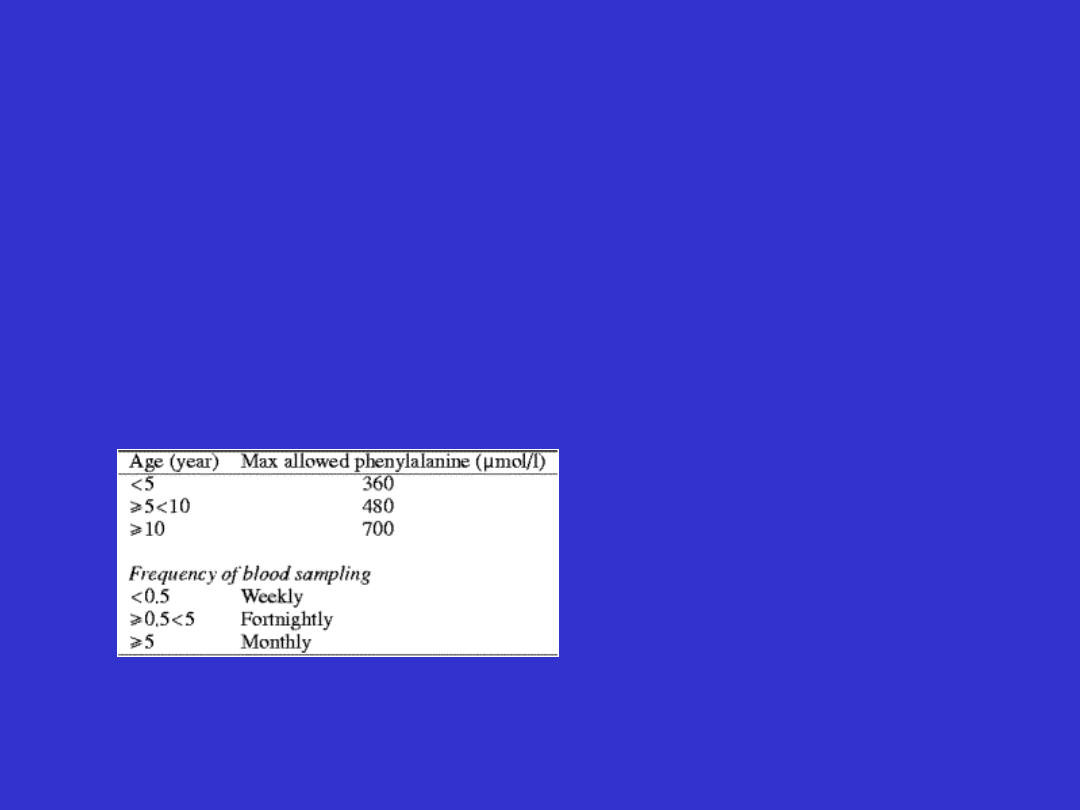
Kontrola poziomu fenyloalaniny
Choroby metaboliczne
Złośliwa
hiperfenyloalaninemia
•
Zaburzenia metabolizmu
tetrahydrobiopteryny BH
4
•
Deficyty enzymów
•
Reduktazy dihydropteryny
•
Cyklohydrolazy guanidynotrifosforanu
– GTP
•
Syntazy pirogronylo tetrahydropteryny
•
Reduktaza septiapteryny

Choroby metaboliczne
Późne powikłania
•
Niedobór vit. B
12
•
Niedobór:
•
Selenu
•
Żelaza
•
Wapnia
•
Kwasów tłuszczowych
•
Zmiany w MRI mózgu (istota biała)

Choroby metaboliczne
Tyrozyna
•
Ponad połowa powstaje z
fenyloalaniny
•
Jest niezbędna do
•
Syntezy białek
•
Hormonów tarczycy
•
Produkcji melaniny
•
Produkcji dopaminy i katecholamin

Choroby metaboliczne
Przejściowa tyrozynemia
noworodkowa
•
Przejściowe zwiększenie poziomu tyrozyny i
kwasu p-fenylohydroksypirogronowego w
surowicy i moczu
•
Niedojrzałość oksydazy PHPP
•
Wcześniaki i niemowlęta żywione sztucznie
•
>3g białka / kg / dobę
•
Niedobór vit. C
•
Mija po kilku tygodniach
•
Niekiedy powoduje niewielkie upośledzenie
rozwoju umysłowego

Choroby metaboliczne
Tyrozynemia typu I
(wątrobowo – nerkowa)
•
Deficyt hydroksylazy fumaryloacetooctanu
•
Gromadzenie bursztynyloacetonu
•
Diagostyka:
•
Wykrycie bursztynyloacetonu w surowicy i
moczu
•
Wzrost poziomu kwasu delta
aminolewulinowego w moczu
•
W ostrej fazie choroby wzrost poziomu alfa
fetoproteiny

Choroby metaboliczne
Tyrozynemia typu I
(wątrobowo – nerkowa)
objawy kliniczne
•
Postać ostra
•
W pierwszych miesiącach życia
•
Niewydolność wątroby, żółtaczka, hepatomegalia,
zaburzenia krzepnięcia, wodobrzusze
•
Uszkodzenie kanalika nerkowego
•
Nieprawidłowy obraz wątroby
•
Postać przewlekła
•
Przewlekła tubulopatia z krzywicą vit D
3
oporną
•
Neuropatia obwodowa
•
U 30% rozrost nowotworowy w wątrobie
(carcinoma hepatocellulare)

Choroby metaboliczne
Tyrozynemia typu I
(wątrobowo – nerkowa)
leczenie
•
Dieta
•
Redukcja do 500mg/dobę spożycia
fenyloalaniny i tyrozyny (łącznie)
•
U starszych dzieci 700-900 mg/dobę
•
Przeszczep wątroby
•
Zablokowanie przemian tyrozyny do
bursztynyloacetonu
•
NTBC (inhibitor oksydazy PHPP)
•
Może powodować powikłania w postaci tyrozynemii
typu II

Choroby metaboliczne
Tyrozynemia typu II
•
Defekt aminotransferazy tyrozyny
•
Objawy:
•
Oczne
•
Owrzodzenia rogówki
•
Skórne
•
Rogowacenie dłoni i stóp
•
Zwiększone wydalanie kwasu p-
fenylohydroksypirogronowego
•
Ograniczenie podaży tyrozyny w diecie
<600 mikromoli/l jest skutecznym
leczeniem

Choroby metaboliczne
Hiperglicynemia
nieketotyczna
•
Glicyna jest rozkładana do
amoniaku i CO
2
•
Objawy:
•
Hipotonia, drgawki, mioklonie
•
Podstawowe badania oraz GCMS
są w normie

Choroby metaboliczne
Choroba syropu
klonowego MSUD
•
Zaburzenia dekarboksylacji leucyny,
izoleucyny i waliny
•
Noworodek po urodzeniu nie wykazuje
objawów
•
W pierwszym tygodniu
•
Wiotkość, drżenia, senność, śpiączka
•
Hipoglikemia, ketony w moczu
•
Zgon
•
Leczenie : dieta eliminacyjna

Choroby metaboliczne
Deficyt trankarbamylazy
ornitynowej (OTC)
•
Hiperamonemia typu II
•
Dziedziczony recesywnie sprzężony z
chromosomem X
•
Objawy:
•
Wymioty, obniżenie napięcia
mięśniowego, drgawki śpiączka
zaburzenia oddychania
•
W postaci klasycznej rokowanie
niepomyślne

Choroby metaboliczne
.
Cykl mocznikowy:
Amoniak Mocznik

Choroby metaboliczne
cykl mocznikowy
cytrulina kwas
argininobursztynowy
mocznik
ornityna
arginina
amoniak

Choroby metaboliczne
Choroby lizosomalne
•
Spichrzanie w lizosomach
•
Dziedziczenie AR z wyjątkiem
choroby Fabry’ego i
mukopolisacharydozy II

Choroby metaboliczne
Choroby lizosomalne
•
Mukopolisacharydozy
•
Mukolipidozy
•
Lipidozy
•
Sulfatydozy
•
Gangliozydozy
•
Glikoproteinozy

Choroby metaboliczne
Choroby lizosomalne -
diagnostyka
•
Obraz kliniczny
•
Badania mikroskopowe
•
Badanie aktywności enzymatycznej
w leukocytach i fibroblastach skóry
•
Diagnostyka molekularna
•
Możliwa diagnostyka prenatalna:
biochemiczna i molekularna

Choroby metaboliczne
Choroby lizosomalne -
leczenie
•
Haematopoetic stem cell therapy
(HSCT)
•
Substrate reduction therapy (SRT)
•
Enzyme replacement therapy (ERT)

Choroby metaboliczne
Enzyme replacement therapy
(ERT)
•
Choroba Gaucher (lipidoza)
•
deficyt beta-glukozydazy
glukozyloceramidu
•
Postać I dorosłych
•
Hepatosplenomegalia, niedokrwistość, żółtaczka
•
Bez objawów neurologicznych
•
Ceredase i Cerezyme
•
Postać II niemowlęca
•
Hepatosplenomegalia i uszkodzenie OUN

Choroby metaboliczne
Enzyme replacement
therapy (ERT)
•
Choroba Fabry’ego
•
Deficyt alfa – galaktozydazy
•
Dziedziczenie resywne sprzężone z
chromosomem X
•
Objawy:
•
Spichrzanie trihesozyloceramidu w układzie
krążenia i nerkach
•
Na skórze teleangiektazje, obrzęki
•
Prawidłowy rozwój umysłowy
•
Leczenie:
•
Replagal i Fabrazyme

Choroby metaboliczne
Enzyme replacement therapy
(ERT)
•
Mukopolisachrydoza typu 1 (zespół Hurler)
•
Deficyt alfa-iduronidazy
•
Objawy:
•
Postępujące opóźnienie rozwoju
•
Pogrubienie rysów twarzy
•
Skrzywienie kręgosłupa
•
Hepatosplenomegalia
•
Niedosłuch
•
Leczenie:
•
Aldurazyme (bez objawów z OUN)

Choroby metaboliczne
Substrate reduction therapy
(SRT)
•
Choroba Gaucher typ I
•
Inhibitor syntazy glukozyloceramidu
Zavesca (Miglustat, N
butyldeoxynojirimycin, OGT 918)
•
Tay-Sach i Sandoff model mysi

Choroby metaboliczne
Choroby peroksysomalne
•
Nieprawidłowa synteza
peroksysomów
•
Zespół Zelwegera
•
Dysmorfia twarzy (wysokie czoło,
zmarszczka nakątna) hipotonia
mięśniowa, opóźnony rozwój, zaćma,
zwyrodnienie barwnikowe siatkówki
•
Wysoki poziom kwasówtłuszczowych o
bardzo długim łańcuchu (VLCFA)

Choroby metaboliczne
Choroby peroksysomalne
•
Adrenoleukodystrofia sprzężona z
chromosomem X
•
Adrenoleukodystrofia
noworodkowa
•
Niemowlęcy zespół Refsuma
•
Chondrodysplazja punkcikowata

Choroby metaboliczne
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
Wyszukiwarka
Podobne podstrony:
Choroby metaboliczne wrodzone
Padaczka we wrodzonych chorobach metabolicznych
CHOROBY METABOLICZNE KRÓW
choroby metaboliczne, MEDYCYNA VI rok, Medycyna rodzinna, medycyna rodzinna
CHOROBY METABOLICZNE
choroby metaboliczne stawów, fizjoterapia, AWF, III rok, Reumatologia
Choroby metaboliczne jako problem w rehabilitacji
Dna moczanowa, Uczelnia, interna, choroby metaboliczne
Żywienie krów mlecznych oraz żywieniowe metody zapobiegania chorobom metabolicznym
Pediatria choroby metaboliczne
Dna moczanowa, dietetyka, 3 rok, Żywienie Kliniczne, Choroby metaboliczne
Choroby metaboliczne kości, st. Rehabilitacja podręczniki
Choroby metaboliczne w pediatri
Leczenie chorób metabolicznych ostat 08 01 2008
Pediatria Choroby metaboliczne
CHOROBY-METABOLICZNE, studia pielęgniarstwo, pediatria
Choroby metaboliczne kości, fizjoterapia
więcej podobnych podstron