
Choroby układu
pozapiramidowego

Choroby układu
pozapiramidowego
Zespół sztywności akinetycznej
zespół Parkinsona
Zespoły hiperkinetyczno-hipotoniczne
hemibalizm, atetoza, zespoły dystoniczne itd.

Główne zespoły parkinsonowskie
1.
Pierwotny parkinsonizm
Choroba Parkinsona- sporadyczna lub dziedziczna.
2.
Wtórne:
- zatrucie CO i Mn.
- polekowy: środki przeciwwymiotne- metoklopramid; neuroleptyki:
pochodne
fenotiazyny, butyrofenony; rezerpina.
- poanoksyjny.
- naczyniowy (stan zatokowy).
- MPTP (1-metylo-4-fenylo-1,2,3,6-tetrahydropirydyna).
- guzy.
- wodogłowie normotensyjne (zespół Hakima).
- czerwienica prawdziwa.
- uraz (bokserzy).
- pozapalny.

Główne zespoły parkinsonowskie cd.
3.
Parkinsonizm plus
- zwyrodnienie korowo-podstawne CBD (Corticobasal Degeneration).
- zespoły otępienne: choroba Alzheimera, otępienie z ciałami
Lewy’ego, otępienie
czołowo-skroniowe.
- zanik wieloukładowy MSA (Multiple System Atrophy).
- postępujące porażenie nadjądrowe PSP (Progressive Supranuclear
Palsy).
4.
Dziedziczne choroby zwyrodnieniowe
- choroba Hallervordena-Spatza.
- choroba Wilsona.
- neuroakantocytoza.

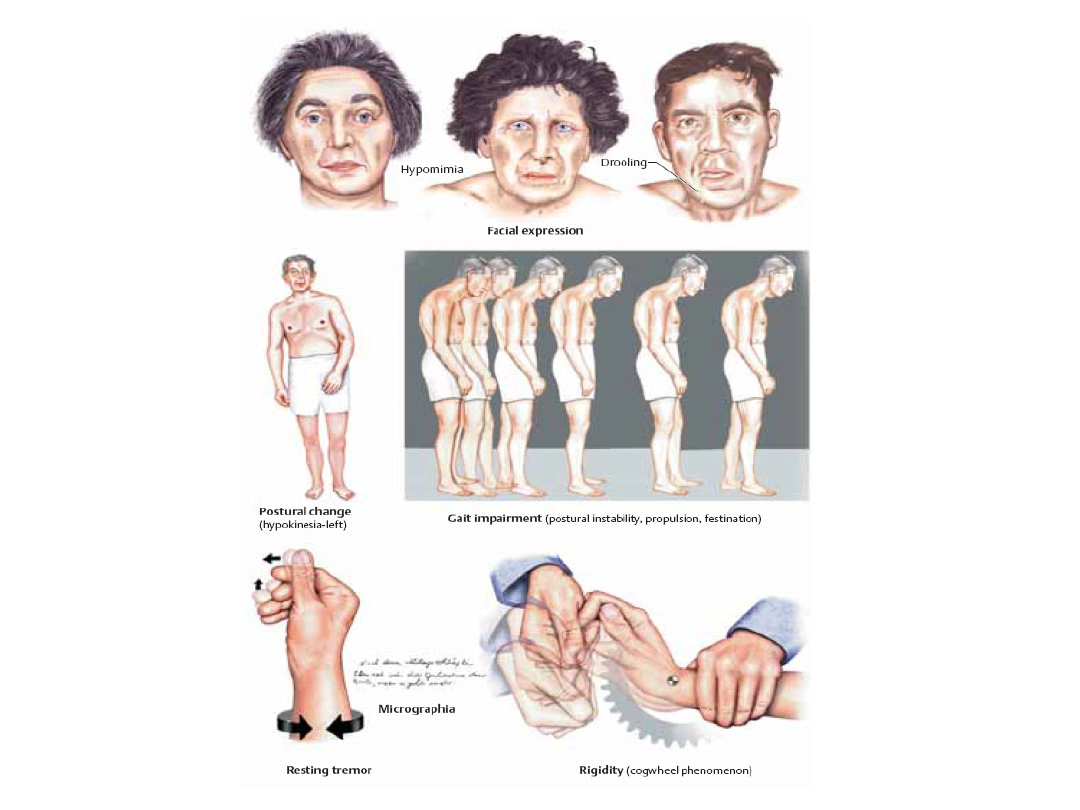
Objawy kliniczne
Objawy dodatnie:
1. sztywność mięśniowa
2. drżenie spoczynkowe
3. pochylona sylwetka
Objawy ujemne:
1. spowolnienie ruchowe
2. upośledzenie automatyzmów ruchowych
3. objaw przymrożenia
Objawy dodatnie:
1. sztywność mięśniowa
2. drżenie spoczynkowe
3. pochylona sylwetka
Objawy ujemne:
1. spowolnienie ruchowe
2. upośledzenie automatyzmów ruchowych
3. objaw przymrożenia

Wzmożenie napięcia mięśniowego:
- Charakterystyczna postawa.
- Sztywność – objaw „rury ołowianej”.
- Wzrost napięcia mięśni antagonistycznych -
objaw „koła zębatego”.
Drżenie spoczynkowe
- „Kręcenie pigułek”, „liczenie pieniędzy”.
- Zanika w czasie: wykonywania ruchów (w
drżeniu samoistnym nasila się), napinania
mięśni, w skrajnych ustawieniach, w czasie snu.
- Typowe, ale nie obligatoryjne.

Spowolnienie ruchowe,
upośledzenie automatyzmów
ruchowych
• Hipokinezja akinezja – twarz maskowata,
mikrografia.
• Zniesienie fizjologicznych odruchów, chód
drobnymi kroczkami (démarche á petits pas).
• Pro-, latero- i retropulsja (objaw niekorzystny
rokowniczo).
• Utrata odruchu postawnego (padanie).
• Mowa monotonna, słabo artykułowana, ściszona.

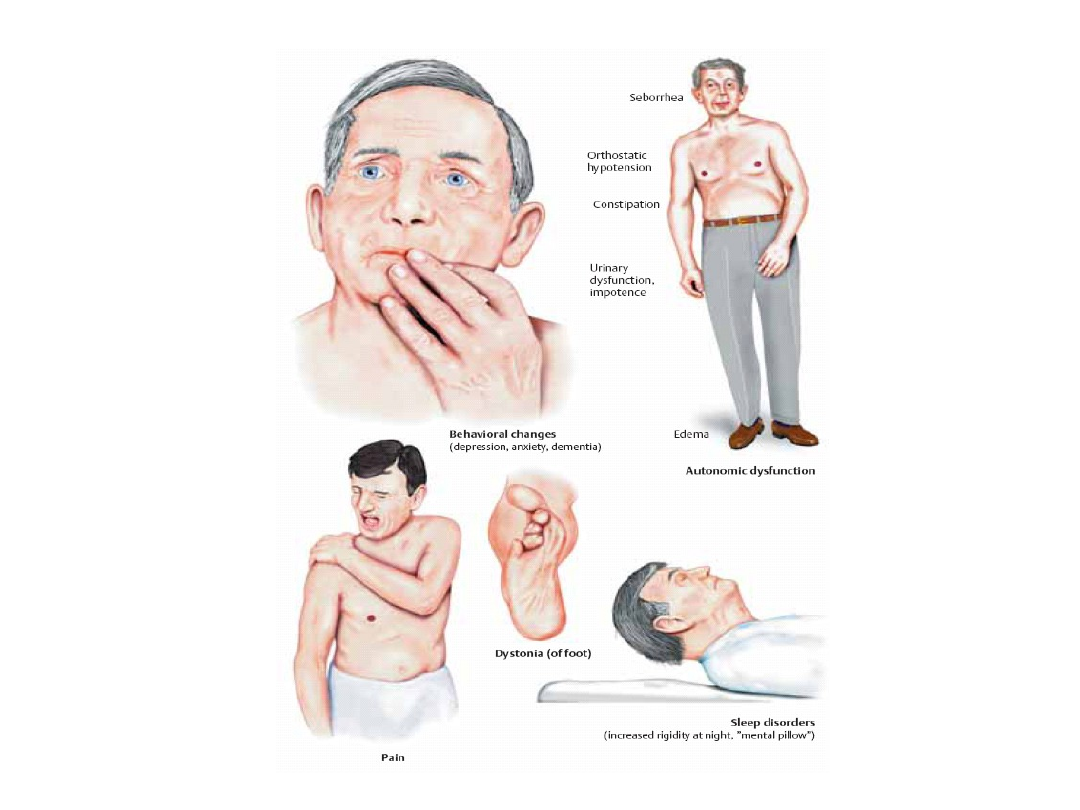
Objawy wegetatywne
• Łojotok twarzy.
• Ślinotok.
• Napady pocenia.
• Zaburzenia zwieraczy (pęcherz hiperaktywny).
Objawy psychiczne
• Labilność nastroju.
• Nadwrażliwość.
• Osłabienie pamięci.
• Spowolnienie myślenia.
• Zmiany psychoorganiczne.

Choroba Parkinsona-
Pierwotny idiopatyczny
parkinsonizm
• Opisana przez Parkinsona w 1817 r. (drżączka
poraźna).
• Jest jedną z najczęstszych chorób układu
nerwowego
(ok. 1,5/1000).
• W Polsce ok. 50-55 tys. chorych, częstość
występowania wzrasta wraz z wiekiem
(osoby >65 r.ż. – 1,5% populacji).
• Dziedziczona AD, rzadziej AR.
• M>K, wiek: 50-60 lat.

Choroba Parkinsona-
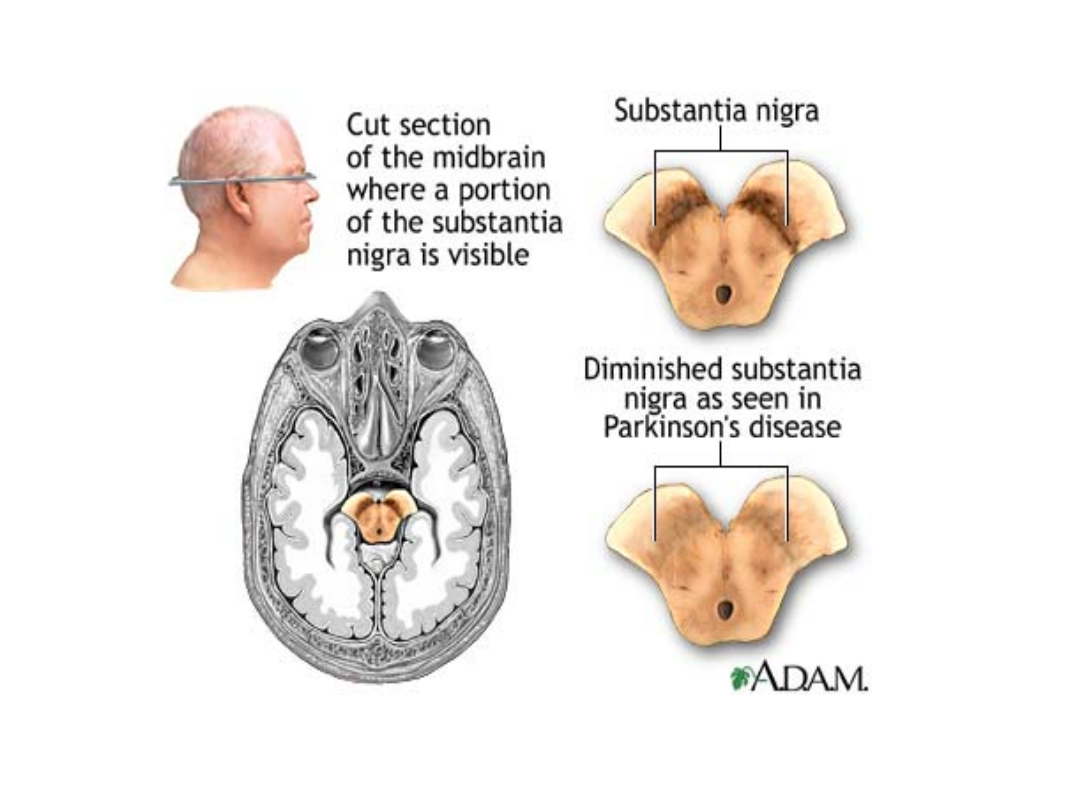
neuropatologia
• Postępujący zanik komórek istoty czarnej i
miejsca sinawego zawierających melaninę:
makroskopowo
- zblednięcie
mikroskopowo
- wtręty komórkowe
(ciałka Lewy’ego, rok 1920).
• Objawy kliniczne pojawiają się, gdy zniszczenie
dotyczy ok. 60% neuronów dopaminergicznych a
stężenie dopaminy w prążkowiu obniża się o ok.
80%.

Dlaczego komórki istoty czarnej
ulegają zwyrodnieniu?
• Uwarunkowania genetyczne (indukcja apoptozy,
przyspieszone starzenie się).
• PARK1-alfa-synukleina – forma ta jest powodowana przez
mutację w genie SNCA.
• PARK2-parkina (E2-zależna ligaza E3 ubikwityno-białkowa)
odmiana powstała w wyniku mutacji w genie kodującym
białko Parking –AR
młodzieńczy parkinsonizm.
• Czynniki toksyczne (endo- i egzogenne).
• Neuroinfekcje.
• Stres oksydacyjny.
• Zmniejszenie liczby czynników wzrostu.
• Suma działania kilku powyższych czynników.

Choroba Parkinsona -
rozpoznanie
• Spowolnienie ruchowe oraz co najmniej 1 z
następujących objawów:
-sztywność mięśniowa.
-drżenie spoczynkowe (4-6 Hz).
-zaburzenia postawy nie dające się wytłumaczyć
pierwotnymi zaburzeniami wzrokowymi,
błędnikowymi, móżdżkowymi lub czucia głębokiego.
• Kryteria wspomagające: jednostronny początek,
postępujący charakter objawów, bardzo dobra
reakcja na L-dopę, utrzymująca się przez co najmniej
5 lat, ewolucja objawów przez co najmniej 10 lat.

Choroba Parkinsona -
diagnostyka
-
TK lub RM głowy- diagnostyka różnicowa.
-DaTSCAN.
-Przezczaszkowe USG oceniające echogeniczność
istoty czarnej.
-PET (pozytonowa tomografia emisyjna).
-SPECT(tomografia emisyjna pojedynczego fotonu).

Jak działa DaTCSAN(Joflupane
123 I)?
Preparat DaTSCAN ma postać roztworu do
wstrzykiwań zawierającego substancję czynną
joflupan (
123
I).
- Joflupan jest radiofarmaceutykiem. Zawiera
substancję o nazwie ioflupan, która została
wyznakowana
123
I (jodem-123) radioaktywną
postacią pierwiastka chemicznego jodu.
- DaTSCAN obrazuje stan czynnościowy
presynaptycznego układu dopaminergicznego.
-Wiąże się z wysokim powinowactwem do nośnika
dopaminy-DAT, zlokalizowanego w błonie
presynaptycznych, nigrostriatalnych zakończeń
nerwowych.
-Pozwala na uzyskanie obrazu ciała prążkowiowego
w postaci obszarów o kształcie symetrycznych
rożków o takiej samej intensywności gromadzenia
się znacznika u osób zdrowych.
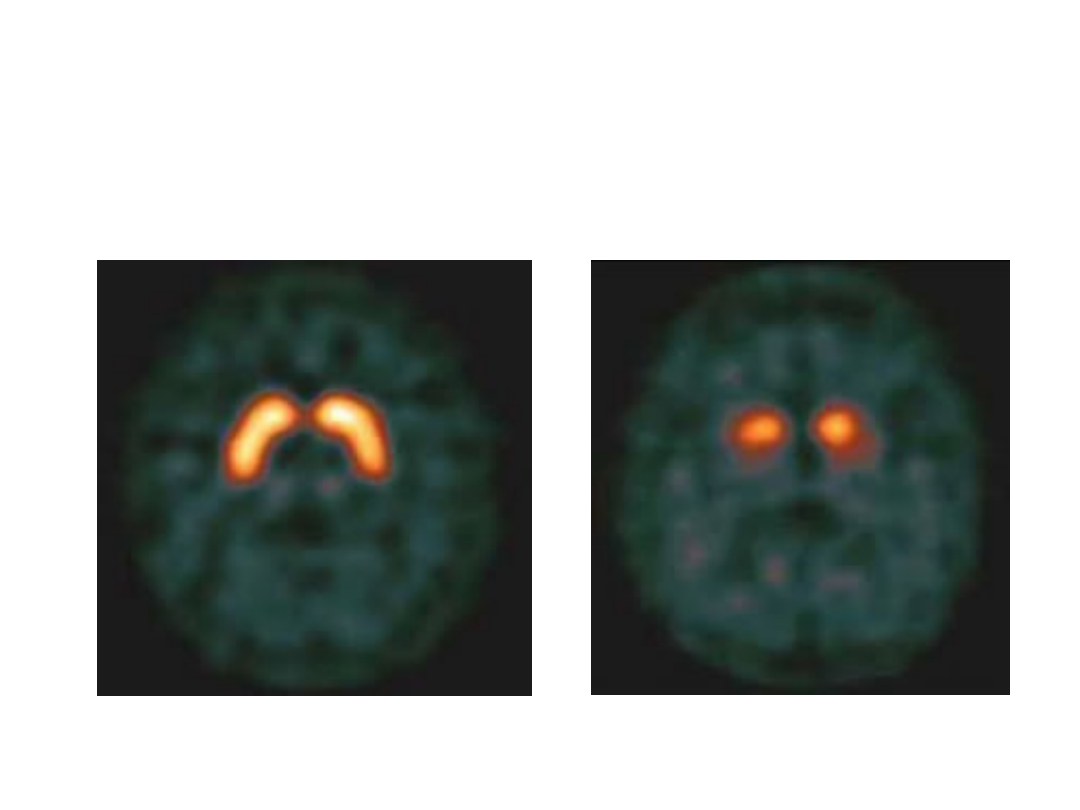
DaTCSAN

Choroba Parkinsona –
leczenie farmakologiczne
1. Preparaty L-dopy.
2. Agoniści receptorów dopaminergicznych.
3. Inhibitory monoaminooksydazy i inhibitory
katecholo-O-metylotransferazy.
4. Leki antycholinergiczne.
5. Inne: np. hamujące aktywność neuronów
glutaminergicznych- amantadyna.

L-dopa
• Lewodopa - metaboliczny prekursor dopaminy
(L-dihydroksyfenyloalanina)
+
• Inhibitor dekarboksylazy :
benserazyd (Madopar)
karbidopa (Nakom, Sinemet)
Powikłania ruchowe po L-dopie:
- Fluktuacje ruchowe: objaw wyczerpania dawki „wearing off’,
nagłe wyłączenia „off”, objaw przełączania „on-off.”
- Dyskinezy: dyskinezy pląsawicze i dystoniczne szczytu dawki,
końca dawki.

Agoniści dopaminy
•Bromokryptyna, kabergolina, pergolid, lizuryd-
pochodne ergotaminy.
Objawy uboczne: zaczerwienienie skóry, zwłóknienie
zaotrzewnowe, opłucnej i osierdzia, zastawek serca.
•Pramipeksol, ropinirol- pochodne
nieergotaminowe
Objawy uboczne: senność, obrzęki.
Wszystkie leki działające agonistycznie na receptor
dopaminowy mogą powodować halucynacje i
hipotonię ortostatyczną.

Leki antycholinergiczne
• Triheksyfenidyl (Parkopan), Pridinol, Biperiden
(Akineton)
Objawy uboczne: zaburzenia pamięci, halucynacje, psychozy.
Inne leki
• Amantadyna (Viregyt) (stymuluje rec. D, zwiększa
uwalnianie dopaminy, blokuje rec. NMDA)
Objawy uboczne: livedo reticularis, obrzęki, halucynacje
wzrokowe.

• Selegilina, rasagilina – inhibitor MAO-B
enzymu rozkładającego dopaminę- zwiększa się
stężenie dopaminy w mózgu, wykazuje działanie
neuroprotekcyjne.
Włączamy:
-Leczenie wczesnego okresu choroby.
-Pomocniczo w standardowym leczeniu
(umożliwia obniżenie stężeń L- dopy).
Selektywny inhibitor
monoaminooksydazy
typu B (MAO-B)

Choroba Parkinsona – leczenie
operacyjne
Zabiegi stosowane w leczeniu choroby Parkinsona można podzielić
na 2 typy:
•
Zabiegi uszkadzające (wywołanie mikrouszkodzenia) w obrębie
gałki bladej (palidotomia), jądra niskowzgórzowego
(subtalamotomia)- gdy jest wzmożone napięcie mięśniowe,
fluktuacje ruchowe, dyskinezy, oraz zabiegi w obrębie wzgórza
(talamotomia)- gdy dominuje drżenie.
•
Wszczepienie stymulatora i założenie elektrody na stałe do tych
samych struktur, a zabiegi te określa się jako głęboką
stymulację mózgu (tzw. DBS – Deep Brain Stimulation). W
metodzie tej stymulator, wszczepiony w powłoki skórne klatki
piersiowej poprzez przeprowadzoną podskórnie elektrodę, z
końcówką umieszczoną w mózgu, wysyłając prąd o określonej
częstotliwości, powoduje także wyłączenie funkcji określonej
struktury. Metoda wydaje się lepsza z uwagi na możliwość
ingerencji i zmiany parametrów stymulacji zależnie od potrzeb.
Stymulacja jądra niskowzgórzowego – zmniejszenie drżenia,
bradykinezji i sztywności mięśni, pozwala na zredukowanie
dawki L-dopy.

Choroba Parkinsona -
rokowanie
• Skuteczne początkowo leczenie opóźnia ostatnie
stadia choroby (inwalidztwo) tylko o kilka lat.
• Średni okres trwania choroby wynosi 7,4 lat.
• Główne przyczyny zgonu (ok. 70 r.ż.):
- choroby serca
- zapalenie płuc

Zanik wieloukładowy - multiple system
atrophy (MSA).
• Heterogenna grupa chorób charakteryzujących się współwystępowaniem cech
zespołu parkinsonowskiego, móżdżkowego i piramidowego, a także zaburzeń
wegetatywnych.
• Obecnie wyróżnia się dwie podstawowe formy zaniku wielopostaciowego:
Postać przebiegającą z dominującymi objawami parkinsonowskimi
(MSA-P).
Postać przebiegającą z dominującymi objawami móżdżkowymi (MSA-C).
• Zanik neuronów w prążkowiu, istocie czarnej, gałce bladej, móżdżku, komórkach
rogu przedniego rdzenia kręgowego i drodze korowo-rdzeniowej.
• 10% wszystkich zespołów parkinsonowskich.
• Parkinsonizm jako jedyny objaw-zwyrodnienie prążkowiowo-czarne.
• Zaburzenia autonomiczne-zespół Shy-Dragera.
• Ataksja- Zanik oliwkowo-mostowo-móżdżkowy.
• Zanik mięśni - parkinsonizm-amiotrofia (z towarzyszącymi objawami
rdzeniowego zaniku mięśni).
• PET z fluorodeoksyglukozą-obniżenie metabolizmu w prążkowiu i płatach
czołowych.
• Leczenie L-dopą przynosi niewielką poprawę, leki antycholinergiczne są
umiarkowanie skuteczne.

Postępujące porażenie nadjądrowe –
Steel, Richardson, Olszewski
syndrom, PSP.
• Zanik śródmózgowia, gałki bladej, jądra
niskowzgórzowego, odbarwienie istoty czarnej, zanik kory
przedczołowej i przedśrodkowej.
• Patologia białka tau.
• Zespół hipertoniczno-hipokinetyczny, rzadko drżenie,
często zaburzenia równowagi i upadki, przeważa
sztywność mięśni osiowych, sylwetka nadmiernie
wyprostowana, twarz-wyraz zdziwienia lub zaniepokojenia;
dyzartria, dysfagia, przymrożenie.
• Objawy oczne- porażenie spojrzenia początkowo w dół,
kurcz powiek, apraksja powiek.
• Otępienie podkorowe, labilność emocjonalna.
• Szybki przebieg, zgon po 6-10 latach.
• MRI głowy – zanik mostu, śródmózgowia i przedniej części
płatów skroniowych –
„dziób kolibra”.

Drżenie samoistne
• 0,1-5% populacji.
• Drżenie pozycyjne, może dotyczyć także głowy.
• 4-12 Hz.
• Często występuje u innych członków rodziny.
• Częstość występowania wzrasta wraz z wiekiem.
• Brak objawów móżdżkowych i
pozapiramidowych.
• Poprawa po alkoholu.
• W leczeniu - propranolol, primidon, gabapentyna,
benzodiazepiny (alprazolam, klonazepam),
topiramat, stymulacja wzgórza.

Choroba Wilsona –
zwyrodnienie wątrobowo-soczewkowe.
• Wrodzone zaburzenie metabolizmu miedzi; 30:1000000.
• Dziedziczenie AR chromosom 13 gen dla ATP-azy typu P.
• Zmniejszone wydalanie miedzi do żółci i zaburzenie syntezy
ceruloplazminy osoczowej – akumulacja miedzi w wątrobie
prowadzące do uszkodzenia wątroby oraz w mózgu (jądra
podstawy), nerkach i rogówce (pierścień Kaysera-Fleischera).
• Objawy: początek 11-25 lat; najczęstsze wczesne objawy to
drżenie i sztywność, utrwalony grymas ust, ruchy dystoniczne,
dyzartria, dysfagia, objawy psychotyczne, napady padaczkowe
(pojawiają się po rozpoczęciu leczenia).
• MRI głowy – poszerzenie komór oraz rozlany zanik kory mózgu,
móżdżku i pnia mózgu, u chorych z objawami niewydolności
wątroby wzmożenie sygnału w T1 w jądrach podstawy.
• Niskie stężenie ceruloplazminy w osoczu i zwiększone
wydalanie miedzi z moczem.
• Leczenie: penicylamina, preparaty cynku.

Pląsawica
• Pozapiramidowy zespół hipotoniczno-
hiperkinetyczny.
• W niektórych postaciach choroby może wystąpić
sztywność mięśniowa (rigor).
• Ruchy pląsawicze - mniej lub bardziej obszerne
ruchy mimowolne, obejmujące przede wszystkim
dosiebne odcinki kończyn, tułów, a także głowę,
szyję, mięśnie języka i krtani.
• Niecelowe, chaotyczne, często gwałtowne,
nakładają się na ruchy dowolne, dezorganizują
ich przebieg (teatralna postać dziwacznego
pląsu).

Pląsawica przewlekle postępująca.
Choroba Huntingtona.
• W Europie z częstością 4-8/100000 mieszkańców.
• Rozpoczyna się zazwyczaj w wieku dorosłym (4 - 5 dekada życia).
• Rzadko (ok. 10% zachorowań) występuje w wieku młodzieńczym,
przybierając wówczas
hipertoniczno-hipokinetyczną postać
Westphala.
• Warunkowanie genetyczne, dziedziczenie AD.
• Mutacja genu kodującego białko huntingtynę, na krótkim
ramieniu chromosomu 4, ekspansja trójki nukleotydów CAG >36.
• W prążkowiu i istocie czarnej zmniejsza się ilość GABA.
• Objawy kliniczne: ruchy pląsawicze, narastające otępienie oraz
zaburzenia osobowości.
• W późnym okresie choroby oraz w postaci Westphala występuje
sztywność mięśniowa.
• Niekiedy obserwuje się zaburzenia afektu, depresję z
tendencjami samobójczymi, urojenia i inne objawy psychotyczne.
• W badaniach obrazowych (TK, MRI głowy) - zanik jąder
ogoniastych (obraz poszerzonych komór bocznych przypomina
skrzydła motyla).
• Badanie PET wykazuje obniżenie metabolizmu w jądrze
ogoniastym i skorupie.

Pląsawica przewlekle
postępująca cd.
• W leczeniu stosuje się:
- neuroleptyki blokujące receptory dopaminergiczne
(haloperidol, perfenazyna),
- leki wypłukujące dopaminę z zakończeń
presynaptycznych (rezerpina,
tetrabenazyna),
- ewentualnie preparaty o działaniu
przeciwdepresyjnym.
• U chorych z dominującą sztywnością (postać
Westphala) podaje się leki z grupy agonistów receptora
dopaminergicznego i lewodopę.

Pląsawica Sydenhama
(chorea minor, rheumatica,
infectiosa).
• Rzadkie schorzenie okresu dziecięcego, następstwo
infekcji paciorkowcowej.
• Przeważnie w wieku szkolnym, częściej u dziewcząt.
• Patomechanizm - reakcja autoimmunologiczna,
powstawanie przeciwciał reagujących z antygenami
komórek jąder ogoniastego i niskowzgórzowego.
• W badaniach obrazowych - powiększenie jądra
ogoniastego, gałki bladej i skorupy, wywołane
stanem zapalnym.
• Pierwsze objawy - często zaburzenia zachowania z
nadmierną pobudliwością i niepokojem ruchowym.
• Ruchy pląsawicze - przeważające w kończynach
górnych, napięcie mięśniowe obniżone.
• Zaburzenia emocjonalne, męczliwość.
• W leczeniu farmakologicznym - niesterydowe leki
przeciwzapalne, kortykosterydy oraz antybiotyki i
leki uspokajające.

Inne pląsawice
• Pląsawica może występować także: u kobiet w
ciąży (chorea gravidarum), jako powikłanie
leczenia estrogenami, w podeszłym wieku
(pląsawica starcza), w przebiegu naczyniowego
uszkodzenia mózgu, tocznia układowego oraz
zespołu antyfosfolipidowego.
• Łagodna pląsawica dziedziczna.
• Neuroakantocytoza
, w której poza ruchami
pląsawiczymi występują tiki, dystonia języka i
napady padaczkowe, a we krwi stwierdza się
obecność tzw. akantocytów, czyli zniekształconych
erytrocytów, z kolczystymi wypustkami.

Tiki
• Szybkie, mimowolne skurcze pojedynczych mięśni lub ich grup,
powtarzające się rytmicznie w krótkich odstępach czasu.
• Mogą mieć charakter ruchowy lub głosowy, prosty (np. mruganie,
unoszenie ramion, chrząkanie) lub złożony-skoordynowane ruchy
mimowolne, często poprzedzone nieprzyjemnym odczuciem,
ustępującym wraz z wykonywaniem czynności:
np. samookaleczanie się, podskakiwanie, wypowiadanie słów lub
zdań.
• Jeżeli tiki przebiegają szybko, są krótkotrwałe - określa się je
jako kloniczne, w przypadku dłużej utrzymującego się skurczu
mięśni – tiki dystoniczne.
Zespół Gillesa de la Tourette’a
• Występuje częściej u mężczyzn.
• Rozpoczyna się w okresie dziecięcym ok. 5rż, po okresie
dojrzewania nasilenie objawów wyraźnie się zmniejsza.
• Tiki mają charakter ruchowy i głosowy, niekiedy z mimowolnym
wykonywaniem nieprzyzwoitych ruchów (kopropraksja),
powtarzaniem wulgarnych słów (koprolalia).
• Tiki nasilają się w czasie emocji, mogą pojawiać się natręctwa i
nadpobudliwość ruchowa.
• Leczenie polega na podawaniu neuroleptyków (haloperidol,
pimozyd, risperidon), klonidyny, tetrabenazyny, klonazepamu,
blokerów kanału wapniowego, leków przeciwdepresyjnych i
psychoterapii.

Dystonia
• Przetrwałe powtarzające się skurcze mięśni
powodujące skręcające ruchy różnych części ciała lub
nieprawidłową postawę.
• Nieprawidłowe napięcie mięśni powoduje zaburzenia
postawy lub ruchu, prowadząc niekiedy do
powstawania utrwalonych, patologicznych pozycji
ciała.
• Ruchy dystoniczne wyzwalane są zazwyczaj przez
ruchy dowolne „task-specific dystonia”.
• Zmęczenie, stres, nasilają ruchy dystoniczne,
natomiast odpoczynek, sen zmniejszają.
• Często obserwuje się ich krótkotrwałe ustępowanie
lub zmniejszanie po bodźcu dotykowym; np. dotknięcie
brody lub policzka może na chwilę zmniejszyć
nasilenie kręczu karku (tzw. „trick movements”).

Podział dystonii
• Dystonie pierwotne, w których ruchy dystoniczne są
podstawowym, często jedynym objawem. Niekiedy towarzyszy im
drżenie, podobne do spoczynkowego drżenia parkinsonowskiego.
• Dystonie plus, w których dystonia współistnieje z
parkinsonizmem (dopa-responsive dystonia) lub miokloniami.
• Dystonie wtórne, będące wynikiem urazów, zatruć, infekcji lub
występujące w przebiegu innych chorób zwyrodnieniowych (np.
pląsawica Huntingtona, choroba Wilsona). Do dystonii wtórnych
należą również: dystonia końca dawki w chorobie Parkinsona.
• Dystonie ogniskowe (np. kurcz powiek, kręcz karku, kurz
pisarski).
• Segmentalne (zespół Meige’a-kurcz powiek+dystonia czaszkowa).
• Wieloogniskowe.
• Uogólnione (DYT1).
• Połowicze (dystonia poudarowa).
• Dystonie idiopatyczne i objawowe (guz mózgu, udar, ch.
metaboliczne, zwyrodnieniowe, polekowe).

Dystonia Oppenheima (DYT1).
• Pierwotna, uogólniona dystonia torsyjna,
mutacja genu DYT 1
(TOR1A), kodującego białko - torsynę A, zlokalizowanego na
długim ramieniu chromosomu 9.
• Dziedziczenie AD.
• Pierwsze objawy około 12 roku życia.
• Początkowo jedna kończyna, (ramię lub kończynę dolną),
dystonia rozszerza się na szyję i krtań, a następnie ulega
uogólnieniu.
• Gdy pierwsze objawy dotyczą kończyny dolnej,
prawdopodobieństwo uogólnienia dystonii jest większe.
• Z postępem choroby dochodzi do powstania przymusowych,
utrwalonych pozycji ciała, ze zniekształceniem układu kostnego.
Dystonia reagująca na L-dopę (choroba
Segawy) (DYT5)
• DYT5 spowodowana jest mutacjami genu DYT5 w locus 14q22.
• 10% wszystkich dystonii wieku dziecięcego, dziedziczenie AD.
• Objawy parkinsonowskie, zmienność objawów w ciągu dnia,
poprawa rano, chód z tendencją do chodzenia na palcach.
• Poprawa po leczeniu małymi dawkami lewodopy lub lekami
antycholinergicznymi.

Pierwotna dystonia o początku
zachorowania
w wieku dorosłym.
• Częstość 30/100 000.
• Dziedziczenie AD, penetracja genu 10-15%.
• Obejmuje zwykle:
- szyję (dystonia szyjna, kręcz karku).
- twarz (kurcz powiek).
- usta i żuchwę (zespół Meige’a).
- struny głosowe (dysfonia spastyczna).
- ręce (kurs pisarski).
• Najczęściej: kręcz karku-20-60 rż
(kobiety>mężczyźni), blefarospazm.

Leczenie dystonii.
• W dystoniach ogniskowych i segmentalnych - toksyna
botulinowa, podawana miejscowo do mięśni
(czasowa, farmakologiczna denerwacja).
• Cholinolityki (np. triheksyfenidyl), benzodiazepiny
(klonazepam, diazepam), leki wypłukujące dopaminę
z zakończeń presynaptycznych (tetrabenazyna),
analogi GABA (baklofen), atypowe neuroleptyki
(klozapina, olanzapina).
• Przy braku reakcji na leczenie farmakologiczne
należy rozważyć możliwość leczenia chirurgicznego;
np. w kręczu karku - selektywne odnerwienie
dystonicznych mięśni, u niektórych chorych z
dystoniami uogólnionymi można uzyskać poprawę po
stereotaktycznych zabiegach neurochirurgicznych.
• Inwazyjny sposób leczenia dystonii - stymulacja jądra
niskowzgórzowego prądem o wysokiej częstotliwości.

Inne pozapiramidowe zaburzenia
ruchowe -balizm
• Balizm - charakteryzuje się mimowolnymi,
gwałtownymi, wyrzutowymi i obszernymi ruchami
kończyn.
• Występuje przeciwstronnie do uszkodzenia jądra
podwzgórzowego Luisa, spowodowanego
np. udarem lub guzem mózgu.
• Obustronny balizm jest zjawiskiem bardzo
rzadkim.
• W leczeniu stosuje się neuroleptyki i rezerpinę.
• W przypadku hemibalizmu o etiologii naczyniowej,
objawy ulegają samoistnemu zmniejszeniu lub też
wycofują się całkowicie w ciągu kilku tygodni
.

Mioklonie
• Są to mimowolne, krótkie, szybkie i nieregularne skurcze
pojedynczych mięśni lub grup mięśniowych, niekiedy z
efektem ruchowym określanym jako „zrywania mięśniowe”.
• Uszkodzenie kory mózgowej, móżdżku (jądra zębatego), jądra
czerwiennego, pnia mózgu, dolnej części oliwki, rdzenia
kręgowego, a także struktur obwodowych (splotów, korzeni
lub nerwów).
• W wyniku zatrucia, niedotlenienia mózgu, urazu
okołoporodowego.
• Mogą mieć charakter - ogniskowy, segmentalny i uogólniony.
• Wyróżnia się mioklonie spoczynkowe, zamiarowe
(prowokowane przez wykonywanie celowego ruchu
dowolnego) oraz odruchowe (wywoływane działaniem
zewnętrznych bodźców czuciowych).
• W leczeniu mioklonii stosowane są pochodne kwasu
walproinowego, klonazepam, primidon i piracetam.

Atetoza
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
Wyszukiwarka
Podobne podstrony:
Choroby rdzenia Gosia
Choroby układu pozapiramidowego 2
5. CHOROBY UKŁADU POZAPIRAMIDOWEGO, FIZJOTERAPIA, Jednostki chorobowe
Choroby nerwów czaszkowych Gosia
Szkol Choroby układu pozapiramidowego
Choroby układu pozapiramidowego
choroby ukladu pozapiramidowego, neurologia
choroby uk adu pozapiramidowegozaj8
CHOROBY UKŁADU POZAPIRAMIDOWEGO, NEUROLOGIA ( zxc )
Choroby układu pozapiramidowego
Choroby nerwów czaszkowych Gosia
choroby naczyn i serca(1)
ŻYWIENIE A CHOROBY 4b
Choroby układu nerwowego ppt
Produkty przeciwwskazane w chorobach jelit II
więcej podobnych podstron