
Przewodnik po patofizjologii
Tom I
Skrypt dla licencjackich studiów medycznych
Pod redakcją
dr hab. med. Danuty Rość
profesora nadzwyczajnego Akademii Medycznej w Bydgoszczy

Autorzy I tomu w porządku alfabetycznym
1.
dr n. med. Wanda Drewniak
2.
dr n. med. Iza Iwan- Ziętek
3.
prof. dr hab. med. Maria Kotschy
4.
dr hab. med. Danuta Rość
5.
dr. hab. med. Ewa Żekanowska
Opracowanie informatyczno- graficzne lek med. Przemysław Adamczyk

Spis treści:
Część pierwsza. Patofizjologia ogólna
Temat I. Patofizjologia. Choroba.
Temat II. Zapalenie.
Temat III. Transformacja nowotworowa.
Część druga. Patofizjologia gruczołów wydzielania wewnętrznego
Temat IV. Zaburzenia regulacji hormonalnej ustroju.
Temat V. Patologia podwzgórza i przysadki.
Temat VI. Patologia gruczołu tarczowego.
Temat VII. Patologia nadnerczy.
Temat VIII. Patologia przytarczyc.
Temat IX. Osteoporoza.
Temat X. Patologia gruczołów płciowych.
Temat XI. Patogeneza cukrzycy.
Część trzecia. Patofizjologia układu krwiotwórczego i hemostazy
Temat XII. Hematopoeza
Temat XIII. Patologia układu czerwonokrwinkowego.
Temat XIV. Patologia układu białokrwinkowego – zmiany ilościowe i jakościowe.
Temat XV. Białaczki.
Temat XVI. Proces hemostazy.
Temat XVII. Skazy krwotoczne.
Temat XVIII. Zakrzepica. Trombofilia.
Temat XIX. Rozsiane krzepnięcie śródnaczyniowe.

Część pierwsza. Patofizjologia ogólna
Temat I. Patofizjologia. Choroba.
Danuta Rość
1.Patofizjologia
Patofizjologia jest dziedziną nauk medycznych zajmującą się analizą oraz syntezą czynności
ustroju zaburzonych procesem chorobowym. Rozpatruje ona mechanizmy czynnościowe
prowadzące do rozwoju zmian chorobowych, ale także zmiany z choroby wynikające.
Zmienione w warunkach choroby funkcje organizmu oraz zaburzone regulacje i korelacje
tych funkcji patofizjologia ujmuje w sposób całościowy i odnosi do otaczającego organizm
środowiska człowieka.
W Polsce i w innych krajach europejskich dziedzina ta została wyodrębniona z
szerszej dziedziny medycyny tj. patologii. Nazwa patologia pochodzi z greckiego słowa
„pathos” – choroba, cierpienie i „logos” – nauka. Zatem patologia jest to nauka o chorobie.
Zainteresowania patologii są szerokie i obejmują cztery grupy zagadnień dotyczących
choroby: przyczyny powstawania choroby (etiologia), przebieg choroby (patogeneza),
zmiany strukturalne (patomorfologia) i zmiany czynnościowe (klinika). W Europie patologia
obejmuje dwie węższe dziedziny: patomorfologię – zajmującą się zmianami strukturalnymi i
ich diagnostyką makroskopową i mikroskopową oraz patofizjologię obejmującą pozostałe
aspekty choroby spośród czterech grup zagadnień wcześniej wymienionych.
Etiologia
Jest to nauka o przyczynach i uwarunkowaniach chorób. (greckie eitia- przyczyna, logos-
słowo, nauka). Czynniki chorobotwórcze zewnętrzne mogą być: biologiczne (np. bakterie,
wirusy, grzyby, pasożyty,), chemiczne (proste związki chemiczne np. kwasy i zasady),
fizyczne (uraz, zmiany temperatury, zmiany ciśnienia atmosferycznego, promieniowanie,
porażenie prądem) i społeczne (np. stres wynikający ze złych stosunków międzyludzkich).
Czynniki chorobotwórcze wewnętrzne to mechanizmy immunologiczne lub neurologiczne.
O chorobach, których przyczyn nie znamy, mówimy – idiopatyczne lub samoistne.
Choroba jatrogenna- powstała na skutek niezamierzonego działania lekarza np. chirurgiczne
usunięcie wola guzowatego tarczycy może spowodować objawy niedoczynności tego
narządu. Czynniki chorobotwórcze mogą mieć charakter złożony np. czynniki
meteorologiczne- na człowieka w określony sposób wpływa równocześnie temperatura
otoczenia, wilgotność powietrza, stężenie tlenu i dwutlenku węgla w powietrzu itp. Czynniki
ryzyka – to zewnątrzpochodne czynniki chorobotwórcze sprzyjające powstawaniu niektórych
chorób np. palenie tytoniu sprzyja istotnie powstawaniu raka płuca i chorób naczyniowych.
Patogeneza
Jest nauką o mechanizmach rozwoju choroby. Patogeneza choroby obejmuje poszczególne
ogniwa tzw. „łańcucha patogenetycznego”, który doprowadza do zaburzeń składających się
na pełny obraz choroby. Elementy łańcucha patogenetycznego zależą od czynnika
patogennego i od właściwości organizmu. Procesy patogenetyczne zachodzą na wielu
poziomach regulacji ustroju - od poziomu molekularnego, poprzez subkomórkowy,
komórkowy, tkanek, układów aż do ogólnoustrojowych procesów integracyjnych organizmu.
Choroba
jest
procesem
dynamicznym
o
niepowtarzalnym
przebiegu
charakterystycznym wyłącznie dla danego organizmu, który funkcjonuje w określonych
warunkach i podlega określonym wpływom.
Sanogeneza
Jest nauką o mechanizmach zjawisk, których celem jest eliminacja zmian chorobowych.
Opisuje procesy odpowiadające za stwierdzane objawy kliniczne w przebiegu różnych
chorób.

2.Choroba
Zdrowie i choroba pozostawały w sferze zainteresowań człowieka od dawna, ponieważ są
ważnym aspektem jego życia. Dawne pojmowanie zdrowia jest naiwne, zależne od nikłego
stopnia rozwoju nauk medycznych. Ojciec medycyny Hipokrates z Kos (460- 377 rok p.n.e)
uważał, że zdrowie to prawidłowy stosunek podstawowych płynów ustrojowych (krew, śluz,
żółć i czarna żółć), natomiast przewaga któregoś z nich powodowała chorobę. Asklepiades
(120- 56 r.p.n.e.) był zdania, że choroba jest skutkiem zmian w atomach i przestrzeniach
międzyatomowych. Według Galena przyczyn chorób należy upatrywać w zmianach
jakościowych i ilościowych płynów ustrojowych, zmianach budowy, liczby, rozmieszczenia
wielkości jednorodnych (tkanki) i niejednorodnych (narządy) – ich stałych składników oraz w
zaburzeniach pneumy- kierującej wszystkimi procesami ustrojowymi, decydującej o sile
życiowej.
Współczesna definicja Światowej Organizacji Zdrowia (1974) określa zdrowie jako
„stan pełnego dobrego samopoczucia (dobrostan) fizycznego, psychicznego i
społecznego, a nie tylko nieobecność choroby czy niedomagania”. Zdrowie to nadrzędna
wartość dla każdej jednostki. Dzięki zdrowiu może ona rozwijać się i realizować swoje
aspiracje i potrzeby oraz chronić i kształtować lokalne środowisko swojego życia i
działalności: pracy i uczenia się, zabawy i wypoczynku.
Według współczesnych poglądów „chorobę należy uważać za złożony łańcuch
wzajemnie ze sobą powiązanych zjawisk czynnościowych i morfologicznych, jako
następstw bezpośrednich uszkodzeń organizmu przez czynnik chorobotwórczy, a także
zjawisk odczynowych, obronnych i przystosowawczych oraz zaburzeń regulacji”
(Billewicz- Stankiewicz).
Organizm człowieka jest systemem otwartym. Znaczy to, że funkcjonuje on dzięki
wymianie gazów i biomasy ze środowiskiem go otaczającym. Do życia człowieka niezbędne
jest pobieranie tlenu i produktów pokarmowych z otoczenia a wydalanie dwutlenku węgla i
produktów przemiany do środowiska. Środowisko, w którym człowiek żyje ma na niego
znaczący wpływ korzystny lub niekorzystny, i w dużej mierze decyduje o jego zdrowiu lub
chorobie.
Pomiędzy organizmem człowieka a jego środowiskiem wykształciła się dynamiczna
równowaga określana jako ”homeostaza”. Polega ona na tym, że mimo zmian środowiska
człowieka liczne mechanizmy regulacyjne jego organizmu zapewniają funkcjonowanie
ustroju nie powodując dezintegracji systemu jakim jest człowiek. Jest to możliwe dzięki temu,
iż działania narządów i tkanek organizmu zapewniają jego funkcjonowanie w warunkach
„homeostazy wewnętrznej” ustroju człowieka.
Homeostaza ustroju człowieka to zdolność organizmu człowieka do utrzymania
w dynamicznej równowadze stałości środowiska wewnętrznego mimo niekorzystnych
zmian otaczającego go środowiska zewnętrznego. Środowisko wewnętrzne człowieka to
płyn zewnątrzkomórkowy obejmujący płyn śródmiąższowy i osocze krwi. W warunkach
zdrowia parametry określające to środowisko wewnętrzne pozostają względnie stałe. Wahania
są niewielkie – jest to równowaga homeostatyczna. Liczne narządy i układy zapewniają
utrzymanie stałości środowiska wewnętrznego ustroju człowieka.
W środowisku człowieka znajdują się przeróżne czynniki, które zmierzają do
zakłócenia homeostazy wewnętrznej ustroju. Są to czynniki chorobotwórcze zewnętrzne, a
mianowicie:
1. biologiczne: wirusy, priony, riketsje, bakterie, pasożyty, grzyby, substancje
białkowe o charakterze antygenów, toksyny pochodzenia bakteryjnego

2. fizyczne: różne formy promieniowania, prąd elektryczny, wysoka lub niska
temperatura, podwyższone lub obniżone ciśnienie atmosferyczne, uraz mechaniczny, fale
akustyczne, wibracje itp.
3. chemiczne: kwasy, ługi, szkodliwe chemikalia, toksyczne gazy, zanieczyszczenia
wody, powietrza i gleby, leki (efekty niepożądane, skutki przedawkowania), etanol, palenie
tytoniu,
4. społeczne: np. stres jako skutek złych stosunków międzyludzkich, niedożywienie,
niedobory witaminowe, skutki zaniedbań higieny osobistej.
Czynniki w/w wywołują reakcje organizmu w postaci odczynów regulacyjnych
biochemicznych i nerwowo- humoralnych, których celem jest zminimalizowanie zaburzeń i
zachowanie stałości środowiska wewnętrznego. Odczyny regulacyjne uruchamiane są na
wszystkich poziomach : molekularnym, subkomórkowym, komórkowym, na poziomie tkanek
i układów aż po integracyjną regulację humoralną i nerwową. Regulacja na poziomie molekuł
i komórek odbywa się w oparciu o reakcje chemiczne i biochemiczne, natomiast w regulacji
układowej zasadnicze znaczenie ma sprzężenie zwrotne. Jeżeli czynnik patogenny nie działa
nazbyt długo a jego nasilenie nie przekracza możliwości wyrównawczych ustroju – to
uruchomione mechanizmy doprowadzają do normalizacji zaburzonej funkcji.
Organizm człowieka może znaleźć się w szczególnie niekorzystnych warunkach.
Wówczas możliwe jest uruchomienie tzw. mechanizmów przystosowawczych. Zdolności
przystosowawcze ustroju są bardzo odmienne u poszczególnych osób, co jest uwarunkowane
genetycznie zarówno pod względem anatomicznym i strukturalnym. Przetrwanie organizmu
w zmiennych warunkach otoczenia zależy od zdolności przystosowania się (adaptacji). Jest
to ogół odczynów czynnościowych i strukturalnych zmierzających do zachowania
homeostazy ustroju czyli utrzymania stałego środowiska wewnętrznego i optymalnej
czynności ustroju w warunkach długotrwałego działania na ustrój niekorzystnych czynników
środowiskowych.
Procesy regulacyjne zaangażowane są w bieżącym utrzymaniu dynamicznej
równowagi homeostazy i są odpowiedzią organizmu na bodźce nagłe i krótkotrwałe,
natomiast mechanizmy przystosowawcze są wyzwalane przez bodźce działające dłużej i ze
zwiększoną mocą. Mechanizmy przystosowawcze związane są ze zwiększonym wydatkiem
energii a więc znaczniejszą eksploatacją organizmu.
Jeżeli ustrój znajduje się pod wpływem szczególnie silnego bodźca, który może go
uszkadzać lub wyzwalać szczególnie silne reakcje organizmu – staje się niemożliwe
utrzymanie homeostazy wewnętrznej ustroju a reakcje ustroju wychodzą znacznie poza zakres
odczynów regulacyjnych czy mechanizmów przystosowawczych. W takich warunkach
rozwija się choroba, kiedy odczyny regulacyjne są zaburzone i nieefektywne w zakresie
kontrolowania homeostazy wewnętrznej środowiska wewnętrznego człowieka. Po
przekroczeniu granicy możliwości utrzymania homeostazy wewnętrznej ustroju, procesy
regulacyjne zmierzające do wyrównania zaburzonej równowagi homeostatycznej stają się
niewystarczające – regulacja prawidłowa przechodzi w regulację patologiczną tj.
dysregulację.
Nozologia ogólna
Jest nauką o chorobie w ogólności. Wykrywa, opisuje cechy wspólne dla różnych grup
chorób, porządkuje je i bada prawa, jakim one podlegają.
Choroba organiczna- to taka, w której podłożem zmian czynnościowych są zmiany
strukturalne (anatomiczne). Uszkodzenia anatomiczne niełatwo cofają się. W tkankach lub
narządach pozostają zmiany strukturalne.

Choroba czynnościowa- nie stwierdza się uchwytnych zmian organicznych. Zmiany
patologiczne mają charakter czynnościowy; po ustąpieniu choroby nie ma pozostałości
morfologicznych.
Podstawowe pojęcia związane z chorobą
Symptomatologia- nauka o objawach choroby
Objawy podmiotowe- subiektywne,
Objawy przedmiotowe- obiektywne
Okres wylęgania (stadium incubationis)- okres pomiędzy zadziałaniem czynnika patogennego
a pojawieniem się objawów choroby
Okres utajenia – okres wylęgania w chorobach zakaźnych
Objawy prodromalne (prodroms, zwiastuny)- nieswoiste objawy subiektywne i obiektywne
zapowiadające wystąpienie pełnych i swoistych objawów choroby
Okres narastania objawów (stadium incrementi)- stopniowe pojawianie się objawów choroby
Okres pełnego rozwoju choroby (stadium manifestationis)- okres występowania wszystkich
objawów choroby
Okres największego nasilenia choroby , szczyt (stadium acmes)
Okres ustępowania objawów (stadium decrementi)
Ustępowanie choroby łagodne – per lysin, lysis
Ustępowanie choroby nagłe tzw. przełom - per crisin, crisis
Przebieg choroby ostry ( morbus acutus)- choroba o nagłym początku, znacznym nasileniu i
najczęściej o krótkim czasie trwania
Przebieg choroby przewlekły (morbus chronicus) – początek choroby powolny, czas trwania
długi, objawy nasilają się w czasie trwania choroby; taka choroba może prowadzić do
wyniszczenia organizmu.
Choroba podostra (morbus subacutus)- postać pośrednia
Zwolnienie choroby lub zaostrzenie (remissio lub exacerbatio) – w toku choroby okresy
mniejszego nasilenia objawów i powrotu z dużą intensywnością
Przepuszczanie (intermissio)- na pewien czas objawy choroby zupełnie znikają
Nawrót, wznowa (recidiva)- choroba pojawia się ponownie po okresie zupełnego ustąpienia
objawów
Powikłanie (complicatio)- nowa osobna choroba pojawiająca się wskutek zmian w wyniku
istniejącej lub przebytej choroby
Powrót do stanu nienaruszonego (restituto ad integrum)- wyzdrowienie zupełne
Trwałe następstwa choroby (restituto cum defectu)- pozostawienie trwałych strukturalnych
następstw choroby
Zejście śmiertelne (exitus letalis)- choroba kończy się śmiercią
Temat II. Zapalenie
Danuta Rość
Zapalenie jest to zespół zmian wstecznych, zaburzeń w krążeniu i zmian postępowych,
stanowiących miejscową odpowiedź żywej tkanki na działanie czynników szkodliwych
zewnątrzpochodnych lub wewnątrzpochodnych. Zapalenie jest odczynem obronnym, a
zespół mechanizmów składających się na proces zapalny zmierza do zlokalizowania i
wyeliminowania patogenu (czynnika szkodliwego), i następowego wygojenia i wyzdrowienia.

W przebiegu zapalenia, ze względu na intensywność objawów i czas trwania,
wyróżnia się fazę ostrą i przewlekłą. Fazę ostrą cechuje znaczna intensywność objawów i
czas trwania rzędu kilku sekund do kilku lub kilkunastu godzin, jest ona wczesną
odpowiedzią ustroju na czynnik uszkadzający. Fazę przewlekłą charakteryzuje mała
intensywność objawów, często brak klasycznych objawów zapalenia i czas trwania mierzony
w tygodniach i latach. Faza przewlekła może być kolejnym etapem zapalenia po fazie ostrej
lub postępować przewlekle od początku tj. od momentu zadziałania patogenu.
Patogeny zewnątrzpochodne obejmują m.in.:
1.czynniki
fizyczne:
uraz
mechaniczny,
wysoka
lub
niska
temperatura,
promieniowanie magnetyczne, elektryczne, jonizujące
2.czynniki chemiczne: proste związki chemiczne, kwasy, zasady, związki organiczne,
3.czynniki biologiczne: bakterie, wirusy, grzyby, pasożyty,
4.czynniki immunologiczne: antygeny, przeciwciała, kompleksy immunologiczne
Patogeny wewnątrzpochodne to mechanizmy immunologiczne np. odporność komórkowa i
reakcje neurologiczne.
Odpowiedzią żywej tkanki na czynnik uszkadzający jest pojawienie się w miejscu
zapalenia komórek zapalnych.
W procesie zapalnym biorą udział komórki uwalniające substancje zawarte w ich
ziarnistościach cytoplazmatycznych, tzw. mediatory zapalenia, które odpowiedzialne są za
mechanizmy i reakcje występujące w czasie procesu zapalnego. Mediatory zapalenia mogą
mieć także pochodzenie osoczowe, gdzie obecne są w formie białek prekursorowych, a ich
aktywacja odbywa się pod wpływem enzymów proteolitycznych.
Komórki zapalne
1.
Neutrofile
Są to główne komórki wczesnej fazy zapalenia. Po opuszczeniu szpiku przez około 6 godzin
przebywają we krwi, skąd w odpowiedzi na bodziec chemotaktyczny płynący z miejsca
powstałego uszkodzenia gromadzą się w tej okolicy tkanki. Neutrofile wyposażone są w
białka adhezyjne (integryny), które umożliwiają wędrówkę po powierzchni śródbłonka
naczyniowego i przybycie do miejsca uszkodzenia. Granulocyty obojętnochłonne rozpoznają
mikroorganizmy dzięki odpowiednim receptorom na powierzchni tych komórek po
uprzednim ich otoczeniu opsoninami – białkami dokonującymi zmian w błonie komórkowej
mikroorganizmów. Opsonizacja błon komórkowych drobnoustrojów umożliwia ich
przyleganie do błony komórkowej granulocytów i następowe wchłonięcie do wnętrza
komórki zapalnej.
Ziarnistości granulocytów zawierają liczne enzymy (mieloperoksydaza, kwaśne
hydrolazy, kolagenazy, fosfataza alkaliczna) umożliwiające zniszczenie pochłoniętych
drobnoustrojów czy obcego białka. Komórki te mają także zdolność do syntezy tlenku azotu
(NO). W procesie zapalnym NO funkcjonuje jako wolny rodnik i jest cytotoksyczny dla
pewnych drobnoustrojów, ale także dla komórek nowotworowych. Do układów
wytwarzających inne rodniki tlenowe należą układy enzymatyczne: dysmutazy
ponadtlenkowe, katalazy, peroksydaza glutationu. W wyniku ich działania powstają związki
silnie bakteriobójcze: rodnik wodorotlenowy, tlen singletowy (O-), kwas podchlorawy i
chloraminy.
2.
Mastocyty (komórki tuczne)
Dojrzałe mastocyty występują wyłącznie w tkankach. Często są pierwszymi komórkami
inicjującymi reakcję zapalną. Znajdują się w tkance łącznej wzdłuż naczyń i nerwów oraz w

miejscach kontaktu ustroju ze środowiskiem zewnętrznym (skóra, błona śluzowa jelit,
śluzówki dróg oddechowych).
Mastocyty uwalniają liczne mediatory jak: histamina, leukotrieny z grupy C, D i E,
prostaglandyna D2, ECF- A (eosynophilic chemotactic factor type A – czynnik
chemotaktyczny dla eozynofilów typu A), adenozyny a także enzymy: tryptaza i chymaza. Po
pobudzeniu wytwarzają i wydzielają cytokiny jak: Il–4, Il-5, Il-6, Il-8, Il-13 ale także GM-
CSF (patrz temat XII). Powodują aktywację i gromadzenie leukocytów w miejscu
uszkodzenia i w ten sposób mogą nasilać proces zapalny.
3.
Limfocyty
We krwi występują jako limfocyty B i T. Komórki te biorą udział w przewlekającym się
procesie zapalnym. Są mało ruchliwe, ale mogą wiązać rozpuszczalne antygeny. Limfocyty B
po kontakcie z obcym antygenowo białkiem ulegają przeobrażeniu w plazmocyty - komórki
zdolne do wytwarzania immunoglobulin. Są ważnym elementem odpowiedzi odpornościowej
humoralnej swoistej ze względu na antygen, z którym się kontaktowały. Limfocyty T biorą
udział w odpowiedzi komórkowej.
Limfocyty produkują liczne mediatory wpływające na stan immunologiczny ustroju.
Poprzez uwalnianie .mogą aktywować granulocyty obojętnochłonne i makrofagi (patrz
poniżej) nasilając ich właściwości fagocytowania obcego białka i drobnoustrojów oraz
czynniki hamujące ich zdolność poruszania się zatrzymując je w miejscu zapalenia.
Uwalniają interferon o właściwościach hamowania replikacji wirusów i limfotoksynę zdolną
do niszczenia obcych komórek. Wydzielają także czynniki chemotaktyczne gromadzące różne
komórki zapalne w miejscu uszkodzenia.
4.
Komórki plazmatyczne
Są komórkami powstałymi z limfocytów B po kontakcie z antygenem i mają zdolność
syntetyzowania immunoglobulin, skierowanych przeciw określonym obcym antygenom..
5.
Płytki krwi
Biorą udział w procesie zapalnym ostrym i przewlekłym. Zawierają mediatory powodujące
włączanie się do zapalenia mechanizmów inicjujących proces krzepnięcia krwi. Są to:
serotonina, adenozyna, tromboksan A2 a także histamina. Błona komórkowa płytek jest
bardzo podatna na działanie wielu bodźców (czynniki immunologiczne, kolagen, ADP,
trombina, trypsyna, serotonina, aminy katecholowe), co powoduje adhezję płytek krwi do
powierzchni ujemnie naładowanych (kolagen warstwy podśródbłonkowej), ich agregację i
reakcję uwalniania substancji zawartych w wewnątrzkomórkowych ziarnistościach płytek.
Fosfatydylcholina i fosfatydylseryna błon płytkowych (PF3) odpowiada za aktywację białek
osoczowego układu krzepnięcia krwi. Uwalniane z płytek czynniki chemotaktyczne i
wzrostowe decydują o interakcji płytek krwi z innymi komórkami zapalnymi jak: granulocyty
obojętnochłonne, monocyty/makrofagi, fibroblasty, a substancje naczynioruchowe i nasilające
przepuszczalność odpowiadają za napływ krwi do ogniska zapalnego.
6. Komórki śródbłonka naczyń
Na przebieg procesu zapalnego znaczący wpływ ma stan czynnościowy naczyń (skurcz,
rozkurcz, przepuszczalność) oraz metabolizm komórek śródbłonka i ich zdolność do
wytwarzania i wydzielania licznych biologicznie czynnych substancji. Komórki
śródbłonkowe posiadają właściwości antykoagulacyjne (antytrombina, siarczan heparanu,
układ białka C i S/trombomodulina), prokoagulacyjne (synteza niektórych czynników
krzepnięcia, ekspozycja kolagenu błony podstawnej dla działania płytek krwi i aktywacji
czynników krzepnięcia aktywacji drogą wewnątrzpochodną i zewnątrzpochodną) i
profibrynolityczne (synteza tkankowego aktywatora plazminogenu, hamujący wpływ na
inhibitor aktywatora plazminogenu tupu 1 - PAI-1 układu białko C i S / trombomodulina).
W początkowym okresie procesu zapalnego przeważają czynniki prokoagulacyjne.

Komórki śródbłonkowe ze względu na interakcje z komórkami krwi ( płytki , monocyty,
granulocyty) przy współudziale białek adhezyjnych oraz reagowanie na wiele mediatorów
odczynu zapalnego są ważnym elementem reakcji zapalnej choć o charakterze stacjonarnym

7. Makrofagi
Biorą udział przeważnie w zapaleniu przewlekłym. Powstają z przekształcenia krążących we
krwi monocytów. Podobnie jak one zawierają w ziarnistościach liczne enzymy proteolityczne
umożliwiające rozkład pochłoniętych drobnoustrojów. Monocyty, obok granulocytów
obojętnochłonnych, posiadają zdolność poruszania się i fagocytozy. Po aktywacji ich
metabolizm wybitnie się nasila; wydzielają wówczas cytokiny, (patrz temat XII), chemokiny,
enzymy i ich inhibitory, składniki dopełniacza, reaktywne związki tlenowe i azotowe,
fibronektynę (białko adhezyjne) i inne.
Czynność komórek zapalnych w miejscu zapalenia
1.
Migracja komórek
Jest to zdolność przechodzenia komórek zapalnych z krwi krążącej w świetle naczynia
poprzez śródbłonek wyściełający jego światło do warstwy podśródbłonkowej i do płynu
zewnątrzkomórkowego. Pod wpływem wazoaktywnych mediatorów zapalenia takich, jak
histamina czy bradykinina, dochodzi do gromadzenia się białek adhezyjnych na
powierzchchni śródbłonka i zwiększenia odstępów pomiędzy komórkami śródbłonkowymi do
0,1 –0,4
µ
m. W pierwszym etapie migracji neutrofile za pomocą białek adhezyjnych
przyczepiają się do śródbłonka naczyniowego, są aktywowane a następnie przechodzą przez
przestrzenie pomiędzy komórkami śródbłonkowymi do warstwy podśródbłonkowej.
Limfocyty T po pobudzeniu migrują do miejsc zapalnych, a limfocyty B osiadają w tkance
limfatycznej.
Komórki
zgromadzone
w
miejscu
zapalenia
uwalniają
czynniki
chemotaktyczne.
2. Chemotaksja
Jest to zjawisko poruszania się komórek zgodnie ze stężeniem danej substancji (przyciąganie
w kierunku wysokiego stężenia tej substancji- chemotaksja dodatnia, odpychanie –
chemotaksja ujemna). Czynniki chemotaktyczne mogą mieć pochodzenie egzogenne:
fragmenty rozpadłych komórek, toksyny bakteryjne i endogenne: składniki dopełniacza (patrz
poniżej), produkty utleniania lipidów oraz cytokiny (Il-8). Czynnik chemotaktyczny wiąże
się z receptorami błony komórkowej makrofaga lub granulocytu obojętnochłonnego, który
zmienia swój kształt i zaczyna się poruszać wysuwając pseudopodia w określonym kierunku i
pociągając resztę komórki.
3.
Fagocytoza
Granulocyty obojętnochłonne i makrofagi, które przybyły do obszaru zapalenia, pochłaniają
bakterie i trawią. Pierwszy etap fagocytozy to opsonizacja polegająca na zmianie błony
komórkowej pod wpływem opsonin umożliwiającej ich pochłonięcie, kolejny to pokrycie
bakterii IgG i białkiem układu dopełniacza oraz ich rozpoznanie i związanie, następnie
wchłonięcie przez komórkę obcego białka i w utworzonym w jej wnętrzu fagosomie –
strawienie przy pomocy obecnych tam enzymów lizosomalnych.
Mediatory zapalenia
Pochodzą z wymienionych już komórek , biorących udział w procesie zapalnym. Są
zgromadzone w ziarnistościach cytoplazmatycznych , a bodziec uszkadzający inicjujący
proces zapalny powoduje ich uwolnienie (np. histamina) lub syntezę de novo
(prostaglandyny). Drugą grupę mediatorów zapalnych stanowią mediatory osoczowe. Należą
do nich proteazy osoczowe a mianowicie białka układu dopełniacza, układu kininowego,
krzepnięcia i fibrynolizy. W osoczu występują one w formie prekursorowej i są aktywowane
przez enzymy lizosomalne uwalniane z komórek zapalnych.
Mediatory zapalne osoczowe
1.
Białka układu dopełniacza.

Układ dopełniacza obejmuje 28 białek współdziałających ze sobą w celu zniszczenia obcej
komórki. Aktywacja dopełniacza może odbywać się drogą klasyczną poprzez kompleksy
antygen- przeciwciało, antygeny powierzchniowe, białko C- reaktywne i enzymy
proteolityczne jak plazmina czy kalikreina i polega na odszczepieniu C3 składowej układu
dopełniacza przy związaniu C1 składowej z przeciwciałami.. Aktywatorami drogi
alternatywnej są polisacharydy ścian komórkowych bakterii, wirusy i zakażone przez nie
komórki, grzyby i pierwotniaki. W drodze alternatywnej uczestniczy properdyna i składnik
C3 dopełniacza.
Receptory dla poszczególnych składników układu dopełniacza, występujące na
komórkach układu immunologicznego i komórkach nabłonka, decydują o udziale tych
komórek w procesie fagocytozy i lizie komórek, ale także umożliwiają indukcję swoistej
odpowiedzi immunologicznej. Układ dopełniacza ma zasadnicze znaczenie w rozpoczęciu
procesu zapalnego, bowiem jego aktywacja doprowadza do uwolnienia kinin, histaminy, do
tworzenia czynników chemotaktycznych. Najważniejsze funkcje tego układu to: aktywacja
komórek posiadających zdolności do fagocytozy (granulocyty obojętnochłonne, makrofagi),
degranulacja komórek tucznych i bazofilów, opsonizacja i eliminacja komórek oraz działanie
cytotoksyczne w stosunku do komórek prowadzące do ich lizy.
2.
Układ kininowy
Kininy – kalidyna i bradykinina, posiadają zdolność pobudzenia receptorów bólowych,
zwiększają przepuszczalność ścian naczyniowych, rozszerzają naczynia krwionośne, mają
działanie chemotaktyczne i pobudzają limfocyty T.
Układ kininowy jest aktywowany przez czynnik Hagemana (XII) po jego zetknięciu z
ujemnie naładowaną powierzchnią np. ze ścianą naczynia pozbawioną śródbłonka
naczyniowego (kolagen jest aktywatorem czynnika XII). Powstająca w efekcie przemian w
układzie kinin - bradykinina odpowiedzialna jest za wymienione powyżej reakcje, a także
aktywuje neutrofile i makrofagi. Poprzez stymulację uwalniania chemokin przedłuża i
potęguje proces zapalny, co powoduje przejście ostrego zapalenia w proces przewlekły.
3.
Układ krzepnięcia
Uruchomienie procesu krzepnięcia krwi jest jednym z elementów odczynu zapalnego, a
zasadniczą rolę w nim odgrywają płytki krwi, komórki śródbłonka naczyniowego, białka
adhezyjne oraz białka osoczowego układu krzepnięcia krwi (patrz temat XVI). Inicjujący go
czynnik Hagemana równocześnie uruchamia proces fibrynolizy, układ kinin i ma wpływ na
układ dopełniacza. Czynnik Hagemana nasila przepuszczalność naczyń krwionośnych a
powstające pośrednie aktywne białka w procesie krzepnięcia mogą działać jako czynniki
chemotaktyczne (fibryna).
4.
Układ fibrynolizy
Znaczna ilość czynników wywołujących proces zapalny (uszkodzenia mechaniczne,
endotoksyny bakteryjne, niedotlenienie lub zakwaszenie tkanek) stymuluje syntezę i
następowe wydzielanie tkankowego aktywatora plazminogenu powodującego powstanie z
plazminogenu aktywnego proteolitycznie białka plazminy. Plazmina powoduje rozkład
fibryny (fibrynolizę) i aktywuje kininogenezę i granulocyty obojętnochłonne. Powstałe w
wyniku rozpadu fibryny produkty degradacji fibryny mają działają chemotaktyczne,
rozszerzają naczynia i zwiększają ich przepuszczalność.
Zapalenie ostre
W obrazie klinicznym cechuje go pięć głównych objawów miejscowych: zaczerwienienie
(rubor), ból (dolor), obrzęk (tumor), zwiększona ciepłota (calor ) i upośledzenie funkcji
(functio laesa).
Zaczerwnienie powstaje jako efekt rozszerzenia naczyń pod wpływem mediatorów
naczynioruchowych (serotonina, histamina, bradykinina). Podwyższona ciepłota jest

wynikiem nasilonego przepływu krwi w miejscu zapalenia. Ból powstaje na skutek
podrażnienia zakończeń nerwowych przez ucisk obrzękniętej tkanki i mediatiory zapalne
(kalidyna, bradykinina). Upośledzenie czynności narządu to wynik jego uszkodzenia przez
czynnik powodujący odczyn zapalny oraz uwalniane w procesie zapalnym mediatory
zapalenia zwłaszcza te o charakterze enzymów proteolitycznych.
Odczyny zapalne ogólne
Bardzo często organizm reaguje na zapalny czynnik uszkadzający uruchamiając odczyny
ogólne w postaci podwyższonej ciepłoty całego ciała, zwiększonej we krwi leukocytozy,
syntezy białek ostrej fazy a w przewlekającym się procesie zapalnym uruchamiając odczyny
immunologiczne. Podwyższona ciepłota ciała jest wynikiem pobudzania ośrodka
termoregulacyjnego przez cytokiny zapalne (Il-6) uwalniane przez komórki zapalne
(granulocyty obojętnochłonne, makrofagi). Podwyższona leukocytoza (zwiększenie liczby
granulocytów obojętnochłonnych we krwi powyżej 10000/
µ
l) jest spowodowana
stymulującym działaniem na szpik kostny cytokin (Il-3, GM- CSF) pochodzących z komórek
zapalnych zgromadzonych w miejscu uszkodzenia Reaguje on wzmożoną proliferacją
leukocytów, które są wyrzucane do krwi krążącej. Białka ostrej fazy są elementem
niespecyficznej reakcji układu immunologicznego w odpowiedzi na płynący z miejsca
zapalenia bodziec. Jest to grupa białek syntetyzowanych głównie w wątrobie w przebiegu
ostrych i przewlekłych stanów zapalnych, chorób zakaźnych, matrwicy (uraz, zawał mięśnia
sercowego, nerki czy płuca) oraz w chorobie nowotworowej. Makrofagi, które w tych
procesach uległy aktywacji, uwalniają interleukinę –1 (Il-1) i czynnik martwicy nowotworów
(TNF tumor necrosis factor). Mediatory te oddziaływują na komórki somatyczne (komórki
śródbłonka naczyń, fibroblasty), które wydzielają wówczas interleukinę –6 (Il-6). Jest to
główny induktor genów odpowiedzi zapalnej i immunologicznej organizmu. Cytokiny Il-1, Il-
6 i TNF aktywują w hepatocytach geny białek ostrej fazy. Zasadnicza część białek ostrej fazy
ma charakter inhibitorów proteaz (alfa1 inhibitor proteaz, białko C reaktywne, alfa 2
makroglobulina), a ich rola polega na zahamowaniu działania proteaz uwalnianych w procesie
zapalnym. Proteazy pochodzące z komórek zapalnych, obok niszczenia obcego białka
powodują destrukcję własnych tkanek, a powstałe białka ostrej fazy o cechach inhibitorów,
proces ten wytłumiają. Inna rola białek ostrej fazy to transport jonów lub innych białek np.
haptoglobina jest białkiem nośnikowym hemoglobiny pozakrwinkowej a ceruloplazmina
atomów miedzi.
Temat III. Transformacja nowotworowa
Danuta Rość
1.Definicja, przyczyny
Nowotwory stanowią w populacji ludzkiej drugą przyczynę śmierci po chorobach układu
krążenia. Z powodu choroby nowotworowej, co roku umiera około 6 milionów ludzi na
świecie i obserwuje się stały wzrost zachorowalności i śmiertelności z powodu nowotworów.
W Polsce średnia umieralność na nowotwory jest wyższa niż przeciętna europejska;
najczęstszym nowotworem wśród mężczyzn jest rak płuca (35 %) a wśród kobiet rak sutka
(14%). Przyczyną stałego wzrostu zachorowania na chorobę nowotworową w ludzkiej

populacji jest z jednej strony systematycznie wydłużające się życie ludzi, a z drugiej
postępujące zanieczyszczenie środowiska.
Istnienie choroby nowotworowej opisał już Hipokrates (V i IV wiek p.n.e) jako
wyniszczającą chorobę, uzewnętrzniającą się guzem i prowadzącą do śmierci.
Obecnie wiadomo, że nowotwór to choroba wielokomórkowego organizmu
charakteryzująca się:
-
nadmiernym, niekontrolowanym rozmnażaniem komórek danego organizmu
-
zaburzeniem różnicowania z pojawianiem się komórek o odmiennych cechach
morfologicznych i biologicznych niż komórki prawidłowe,
-
zdolnością do rozsiewu tych komórek przez naciekanie bądź tworzenie przerzutów do
odległych tkanek.
Uwzględniając zdobycze genetyki i biologii molekularnej w badaniach nad biologią
nowotworu można uznać nowotwór za chorobę genomu. Natomiast proces przekształcenia
prawidłowej komórki w komórkę nowotworową określa się mianem „transformacji
nowotworowej”. Istotą procesu transformacji nowotworowej są zmiany w obrębie genomu
komórki, które po utrwaleniu mogą być przenoszone na następne pokolenia komórek. Te
zmiany są przyczyną pojawiania się nowych cech komórki transformowanej np.
„nieśmiertelności” tych komórek w hodowlach „in vitro” oraz ich zdolności do wzrostu po
przeszczepieniu. Uważa się, że zmiany mutacyjne genów są w znacznym stopniu wywołane
mutagenami – czynnikami środowiskowymi. Podstawowym mechanizmem karcinogenezy są
niekorzystne interakcje pomiędzy genotypem danego organizmu a środowiskiem
zewnętrznym lub wewnętrznym człowieka.
Najczęstszymi mutagenami w populacji ludzkiej są: dym papierosowy i substancje
smoliste zawierające liczne substancje uważane za karcinogeny (uretan, formaldehyd, piren,
antarcenowe i nitrozaminowe pochodne benzenu, hydrazyna, chlorek winylu, związki arsenu,
niklu i kadmu, polon 210 i inne), wysokokaloryczna dieta zawierająca duże ilości
wysokonasyconych tłuszczów (zwłaszcza kwas linolenowy) z małą ilością substancji
resztkowych (błonnika), wirusy (hepatitis B virus, human papilloma viruses, Papova virus typ
B, adenowirusy, human herpes viruses, wirus ospy, retrowirusy B,C,D), zanieczyszczenia
wody, gleby i powietrza, promieniowanie jonizujące (promieniowanie ziemi i słońca,
promieniowanie związane z wykonywaniem zawodów- radiolodzy i górnicy rud uranowych,
promieniowanie stosowane w celach diagnostycznych i leczniczych, wybuchy bomb
atomowych, poligony nuklearne), czynniki karcinogenne związane z wykonywaniem
niektórych zawodów (benzen- chemicy, malarze i laboranci, nitrozaminy- konserwanty
produktów spożywczych, aminy aromatyczne – farbiarnie, związki arsenu, kadmu i chromu-
górnicy, chemicy i hutnicy, dioksyna- produkcja herbicydów, związki ołowiu- pracownicy
stacji benzynowych). Do szczególnie karcinogennych substancji należy benzen i jego
pochodne (węglowodory aromatyczne, pochodne antracenowe), metale ciężkie (kadm, chrom,
beryl, nikiel, kobalt, związki ołowiu), azbest, dioksyna, aflatoksyny wytwarzane przez grzyby
i roztocza, leki stosowane w leczeniu nowotworów (antymetabolity, antybiotyki).
2. Nowotwór jako choroba genomu
W organizmie wielokomórkowym rozrost tkanek znajduje się pod ścisłą kontrolą. Komórki
proliferują tylko w sytuacji zapotrzebowania na nie, w odpowiedzi na zewnętrzny bodziec.
Jest on sygnałem do podziałów określonych komórek. Wytworzone komórki w ilości
stosownej do aktualnych potrzeb ustroju, powodują włączenie się mechanizmu hamowania
ich proliferacji. W sytuacji prawidłowej proces tworzenia nowych komórek pozostaje w
ścisłej relacji z procesem apoptozy. Jest to zaprogramowana śmierć komórki, która po
wypełnieniu swojej funkcji ulega samoistnie destrukcji. Za namnażanie komórek oraz
hamowanie ich proliferacji oraz za proces apoptozy odpowiadają tzw. białka regulacyjne.

Wyłamanie się spod tej kontroli i niczym niepohamowana proliferacja jest istotą transformacji
nowotworowej. Komórka stransformowana dzieli się ciągle samoistnie bez stymulującego
wpływu czynników wzrostowych, kontaktów międzykomórkowych i określonych relacji z
podłożem. Transformacja nowotworowa jest wynikiem mutacji w genach kodujących
białka regulacyjne, które uczestniczą w kontroli wzrostu, różnicowania komórki i jej
gotowości do podziałów komórkowych. W przeważającej większości mutacje odpowiedzialne
za powstanie i rozwój nowotworu są mutacjami somatycznymi (mutacja zachodzi w
komórkach ciała, i nie jest przekazywana potomstwu- nie dotyczy komórek płciowych).
Proces karcinogenezy jest wieloetapowy. Pojawianie się kolejnych cech komórki
nowotworowej jak: niekontrolowany wzrost, zdolność do inwazji miejscowej, a następnie
odległej jest stopniowe określa się mianem progresji nowotworu. W procesie nowotworzenia
udział zasadniczy mają protoonkogeny i geny supresorowe. Znacząca jest rola genów
naprawy DNA (należących do grupy genów supresorowych) oraz genów kontrolujących
proces apoptozy, z których część ma charakter onkogenów, a większość jest zaliczana do
genów supresorowych.
Protoonkogeny kodują białka odpowiedzialne za proliferację komórkową. Wzrost i podział
komórki następuje po bodźcu, którym jest czynnik wzrostowy. Reaguje on z właściwym
receptorem znajdującym się na błonie komórkowej. Sygnał zostaje przekazany do wnętrza
cytoplazmy a następnie do jądra komórkowego. Protoonkogeny kodują następnie czynniki
transkrypcyjne odpowiedzialne za aktywację odpowiednich odcinków DNA, co doprowadza
do powstania białka regulującego wzrost i podziały komórki. W prawidłowo działającej
komórce produkcja białka regulacyjnego pozostaje pod ścisłą kontrolą.
Protoonkogeny zatem, zależnie od funkcji białek przez nie kodowanych w prawidłowej
komórce, można podzielić na następujące grupy – protoonkogeny kodujące:
1.
czynniki wzrostowe,
2.
swoiste receptory dla czynników wzrostowych,
3.
białka kaskady wewnątrzkomórkowej transdukcji sygnału,
4.
czynniki transkrypcyjne- białka wiążące się z DNA i regulujące ekspresję genów,
5.
białka regulujące podziały komórkowe.
Transformacja nowotworowa polegająca na mutacji genów odpowiedzialnych za
syntezę białka regulacyjnego może dokonać się na każdym etapie od pobudzenia komórki do
przekazywania sygnału wzrostu i proliferacji aż do wytworzenia nieprawidłowego białka.
Mutacje zmieniają protoonkogeny w onkogeny, a wytworzone nieprawidłowe białko
regulacyjne określa się nazwą onkoproteina.
Wyróżnia się następujące najczęstsze formy aktywacji genów
1.
amplifikacja – zwielokrotnienie kopii genu, co prowadzi do zwiększonej ilości
wytwarzanego białka- onkoproteiny
2.
nadekspresja- zwiększone wytwarzanie mRNA i wynikające stąd zwiększenie ilości
wytwarzanego białka
3.
mutacje- zmiany genu w jego nowy allel lub zmiany struktury chromosomów
Geny supresorowe
Są to geny, które kodują białka normalnie funkcjonujące w komórce jako inhibitory wzrostu
i podziału komórki. Są to zatem hamulce proliferacji. Sygnały hamujące, tak jak
pobudzające, przechodzą w komórce taką samą drogę od odebrania sygnału na powierzchni
komórki, poprzez jego transdukcję do jądra komórkowego, aktywację genów DNA i syntezę
białka regulującego proliferację komórek, w tym wypadku białka o charakterze inhibitora
podziałów komórkowych. Fizjologiczna rola genów supresorowych polega na regulacji
wzrostu organizmu.

Mutacja genu supresorowego prowadzi do braku syntezy białka hamującego wzrost i
podziały komórkowe. Dla procesu nowotworowego ma to znaczenie takie, iż nie ma w
komórce hamulca jej podziału mimo uszkodzonego DNA. Zmieniony DNA jest
przekazywany komórkom potomnym.
Ważnym genem supresorowym jest TP53, który jest włączany gdy w komórce pojawia się
uszkodzony DNA w wyniku działania promieniowania jonizującego lub mutagenów
chemicznych. Przy braku TP53 komórka z uszkodzonym DNA dzieli się bez przeszkód.
Powstaje klon uszkodzonych komórek, dzielących się łatwo i ulegających dalszym mutacjom
i tworzących nowotwór złośliwy.
Geny kontrolujące naprawę DNA
Działające w środowisku człowieka czynniki szkodliwe prowadzą do uszkodzeń DNA. Ich
naprawa jest warunkiem niezbędnym do utrzymania przekazywania niezmienionej informacji
genetycznej komórkom potomnym. Komórki posiadają enzymy umożliwiające im usuwanie
powstałych uszkodzeń DNA. Mutacje komórek płciowych w zakresie genów naprawy DNA
prowadzą do dziedzicznych zespołów nowotworowych albo są przyczyną zespołów
chorobowych, w których obok wielu zaburzeń występuje wzmożona predyspozycja do
nowotworów dziedzicznych.
Geny kontrolujące długość życia komórki
Apoptoza , tj. samobójcza zaprogramowana śmierć komórki, jest istotnym mechanizmem
kontroli nad proliferacją. Geny kodujące białka istotne dla rozpoczęcia i samego przebiegu
apoptozy są ważnymi elementami kancerogenezy.
Genami zapobiegającymi apoptozie są BCL2 i MDM2. Często w apoptozie biorą udział
geny supresorowe a głównie TP53. Zapobieganie nowotworowi polega na włączeniu
apoptozy w chwili pojawienia się uszkodzenia DNA, którego komórka nie jest w stanie
naprawić. Prowadzi to do śmierci komórki i nie jest możliwe przekazanie uszkodzonej
informacji genetycznej komórce potomnej.
Wieloetapowy rozwój nowotworu
Badania doświadczalne prowadzone w pierwszej połowie ubiegłego stulecia wykazały, że
karcinogeneza chemiczna (nowotworzenie pod wpływem jednego ze znanych karcinogenów
występujących w środowisku zewnętrznym człowieka) jest procesem wieloetapowym. Można
wyróżnić trzy główne jego etapy, choć czas ich trwania jest zmienny z okresami
przyśpieszania i zwalniania a niekiedy wręcz regresji procesu chorobowego. Są to:
1.
okres inicjacji- proces rozpoczyna się w następstwie ekspozycji organizmu na
karcinogen; można go wiązać z mutacją protonkogenu do onkogenu i powstaniem
onkoproteiny stymulującej podziały komórki,
2.
okres promocji – dochodzi do dodatkowego synergistycznego działania czynników
promocyjnych i mutacji genu supresorowego, spełniającego rolę hamulca nadmiernej
proliferacji,
3.
okres progresji – kolejne mutacje prowadzą do kumulacji błędów w DNA i selekcji
komórek nowotworowych. Mimo wielu błędów w zapisie DNA komórki
stransformowane dzielą się intensywnie. Następuje ich autononomizacja, nie
podlegają kontroli metabolicznej oraz układu odpornościowego i hormonalnego.
3. Biologia nowotworów
Transformacja nowotworowa zaburza lub uniemożliwia prawidłowe różnicowanie komórek a
nowotwór wyłamuje się spod mechanizmów kontroli i samoograniczania proliferacji.
Nowotwory złośliwe zbudowane są komórek niezróżnicowanych lub nisko zróżnicowanych a
więc niewyspecjalizowanych do pełnienia skomplikowanych funkcji ustrojowych. Rosną one

szybko naciekając tkanki sąsiednie a często dając przerzuty do tkanek odległych. Przerzuty
dokonują się drogą naczyń krwionośnych, naczyń chłonnych albo przez rozsiew do jam ciała.
Nowotwory łagodne zbudowane są z komórek wysoko zróżnicowanych. Im wyższe
jest zróżnicowanie morfologiczne komórek nowotworowych, tym funkcja tych komórek jest
zbliżona do komórek tkanki prawidłowej. Nowotwory pochodzące z tkanki gruczołów
wydzielania wewnętrznego mogą syntetyzować i uwalniać do krwi hormony, choć wówczas
ta produkcja nie podlega kontroli mechanizmu sprzężenia zwrotnego działającego w układzie
hormonalnym.
Guz nowotworowy nie jest zbudowany tylko i wyłącznie z jednego typu komórek,
ale stanowi strukturę składającą się z komórek stransformowanych i prawidłowych o różnej
aktywności biologicznej. W podścielisku guza znajdują się takie komórki jak: fibroblasty,
komórki śródbłonka naczyń, makrofagi i komórki napływowe, głównie krwinki białe. Wśród
komórek nowotworu można wyróżnić:
1.
komórki stransformowane- występują one w różnych fazach cyklu podziałowego,
komórki w fazie G o czasowo zahamowanych podziałach, oraz komórki niezdolne do
dalszych podziałów;
2.
komórki proliferujące - decydujące o głównej masie nowotworu; są to aktywne, łatwo
odradzające się komórki, które mogą tworzyć wiele subpopulacji o jednakowych lub
odmiennych cechach morfologicznych i biologicznych;
3.
hybrydy komórek – mogą one tworzyć się między komórkami nowotworowymi lub
między komórkami nowotworowymi a prawidłowymi.
Heterogenność komórek nowotworowych powoduje, że w chemioterapii stosuje się zestawy
leków działających na różne fazy cyklu komórkowego.
Środowisko zewnątrzkomórkowe nowotworów tworzą różne układy, a mianowicie;
zrąb łącznotkankowo- naczyniowy zawierający komórki: fibroblasty i komórki śródbłonka
oraz komórki napływające z krwi: limfocyty, makrofagi, leukocyty. Te komórki oraz cytokiny
przez nie wydzielane są wyrazem odczynu organizmu gospodarza na komórki guza i produkty
przezeń wydzielane.
Początkowo guz nie posiada unaczynienia, ale w miarę jego rozwoju w utkaniu
nowotworu dokonuje się neoangiogeneza (tworzenie naczyń krwionośnych de novo). Guz
wydziela czynniki pobudzające wzrost śródbłonka, który proliferuje z tkanki łącznej
gospodarza. Śródbłonek pączkuje, tworzy w utkaniu nowotworu sieć drobnych naczyń bez
unerwienia. Odżywianie guza następuje przez te naczynia. Tędy dostają się także leki
stosowane w terapii nowotworów.
W leczeniu nowotworów pierwsze zastosowania mają już leki hamujące angiogenezę.

Część druga. Patofizjologia gruczołów wydzielania
wewnętrznego.
Temat IV. Zaburzenia regulacji hormonalnej ustroju.
Maria Kotschy
Gruczoły dokrewne (wewnątrzwydzielnicze)
Układ wewnątrzwydzielniczy, czyli dokrewny należy do układów regulacyjnych człowieka,
podobnie jak układ nerwowy i immunologiczny. Gruczoły dokrewne są gruczołami, które nie
mają przewodów wprowadzających. Ich wydzielina zwana hormonami (od hormao –
pobudzam) przenika bezpośrednio do krwi w naczyniach krwionośnych, limfy w naczyniach
chłonnych, płynu mózgowo-rdzeniowego lub płynu międzykomórkowego.
Hormony są aktywnymi biologicznie substancjami wydzielanymi w niewielkich
ilościach (stężenie we krwi wynosi 10
-12
– 10
-7
nmol). Są przenośnikami informacji dla
komórek docelowych. Wspólna cechą wszystkich gruczołów dokrewnych jest ich bardzo
dobre unaczynienie, ponieważ naczynia krwionośne pełnią funkcję przewodów
wyprowadzających hormony.
Gruczoły dokrewne mają różne pochodzenie, budowę, położenie i zakres czynności.
Ze względu na pochodzenie ( tj. z jakiego listka zarodkowego się rozwijają) dzielimy jej na
3 grupy:
1. pochodzące z ektodermy (zewnętrznego listka zarodkowego): przysadka,
szyszynka, rdzeń nadnerczy,
2. pochodzące z mezodermy (środkowego listka zarodkowego): kora nadnerczy,
części wewnątrzwydzielnicze gruczołów płciowych,
3. pochodzące z endodermy (wewnętrznego listka zarodkowego): tarczyca,
przytarczyce, grasica, wyspy Langerhansa trzustki.
Ze względu na położenie i zakres czynności rozróżniamy:
1.
gruczoły pełniące wyłącznie funkcję wewnątrzwydzielniczą np. przysadka, tarczyca,
przytarczyce, nadnercza,
2.
gruczoły, które oprócz funkcji wewnątrzwydzielniczej pełnią czynność
zewnątrzwydzielniczą np. trzustka i gruczoły płciowe.
Pod względem czynnościowym odróżniamy następujące hormony:
1.
Hormony tzw. uwalniające (liberyny) wytwarzane i wydzielane w podwzgórzu,
regulujące syntezę i wydzielanie hormonów tropowych przysadki,
2.
Hormony tropowe wytwarzane i wydzielane przez przysadkę, regulujące
wytwarzanie i wydzielanie hormonów przez gruczoły dokrewne obwodowe,
3.
Hormony wytwarzane i wydzielane przez obwodowe gruczoły dokrewne działające
bezpośrednio na narząd docelowy; są to tak zwane hormony efektorowe.
Hormony podwzgórza, przysadki oraz gruczołów obwodowych stanowią jednolitą całość
czynnościową ze wspólnymi mechanizmami regulacyjnymi nerwowymi, hormonalnymi i
metabolicznymi.
Pod względem chemicznym hormony można podzielić na:
1.
Hormony białkowe i peptydowe (zbudowane z aminokwasów) – hormony
podwzgórza, przysadki, przytarczyc i trzustki,

2.
Hormony steroidowe (pochodne cholesterolu) – hormony kory nadnercza i hormony
płciowe,
3.
Hormony pochodne fenolu – noradrenalina, adrenalina, tyroksyna, trójjodotyronina.
Hormony działają za pośrednictwem wiążących je receptorów znajdujących się w różnych
częściach komórki.
1.
Hormony peptydowe i białkowe wydzielane do krwi jako pro- lub preprohormony
wiążą się z receptorami na błonie komórek docelowych.
2.
Hormony steroidowe wydzielane jako hormony aktywne działają na komórkę przez
wewnątrzkomórkowe receptory znajdujące się głównie w cytozolu, ale także w jądrze.
Ich synteza uwarunkowana jest prawidłową syntezą białek enzymatycznych
wchodzących w skład układów metabolicznych, które prowadzą do tworzenia
odpowiednich substancji czynnych, powodując zwiększenie transkrypcji właściwego
mRNA.
3.
Podobnie działają hormony gruczołu tarczowego i inne pochodne fenolu.
Zaburzenia czynnościowe wszystkich gruczołów układu dokrewnego polegają na:
1.
nadczynności lub
2.
niedoczynności wydzielniczej.
Zarówno nadczynność jak i niedoczynność mogą mieć charakter pierwotny lub wtórny.
Pierwotna nadczynność obwodowego gruczołu dokrewnego polega na zwiększeniu
wydzielania hormonów w następstwie jego procesu chorobowego (przerost i nowotwór).
Wtórna nadczynność obwodowego gruczołu dokrewnego polega na nadmiernym
pobudzeniu do wydzielania hormonów prawidłowego gruczołu obwodowego przez
zwiększone wydzielanie hormonów uwalniających podwzgórza lub tropowych przysadki
mózgowej.
Pierwotna niedoczynność obwodowego gruczołu dokrewnego polega na
niedostatecznym wydzielaniu hormonu w następstwie procesu chorobowego tego gruczołu.
Wtórna niedoczynność gruczołu dokrewnego spowodowana jest niedostatecznym
wydzielaniem hormonu przez gruczoł obwodowy w następstwie zmniejszonego wydzielania
lub braku hormonów uwalniającego podwzgórza lub tropowego przysadki.
Przyczyny zmian czynnościowych układu dokrewnego:
a. zaburzenia sprzężenia zwrotnego między podwzgórzem, przysadką i gruczołem
obwodowym,
b. zaburzenia katabolizmu (rozpadu) hormonów np. w niewydolności wątroby, w której
występuje m. in. obniżenie degradacji kortizolu lub estrogenów, powodujących u
mężczyzn ginekomastię, zanik jąder i zmiany owłosienia,
c. ektopowe wydzielanie hormonów w niektórych nowotworach (płuc, wątroby, trzustki,
grasicy i tarczycy),
d. genetycznie uwarunkowane defekty syntezy hormonów tzw. (endokrynopatie dziedziczne),
e. zaburzenia magazynowania, wydzielania i transportu hormonów np.:
-
nieprawidłowe magazynowanie i uwalnianie wazopresyny a także oksytocyny z
podwzgórza do przysadki,
-
uwalnianie insuliny z komórek β wysp trzustkowych w cukrzycy,
-
zmiany stężenia transkortyny (białka nośnikowego krwi dla kortizolu).
f. zaburzenie konwersji prohormonu w aktywny hormon np.:
-
proinsuliny w insulinę,
-
testosteronu w dihydrotestosteron (w narządach efektorowych brak 5-reduktazy),

g. zaburzenie odpowiedzi receptora komórkowego (np. dla insuliny somatotropiny,
parathormonu, testosteronu),
h/ niedobór wewnątrzkomórkowych nośników hormonalnych (np. w zespole feminizujących
jąder).
Temat V. Patologia podwzgórza i przysadki
Maria Kotschy
Podwzgórze
Podwzgórze jest częścią mózgu, znajdującą się w pobliżu 3 komory nad przysadką mózgową.
Struktura ta zespala czynności układu wegetatywnego i hormonalnego ustroju. W
podwzgórzu występuje szereg jąder. Neurony w jądrach nadwzrokowych oraz
przykomorowych syntetyzują wazopresynę i oksytocynę (neuropeptydy) uwalniane z
zakończeń nerwowych w tylnym płacie przysadki. W przedniej części podwzgórza
wytwarzane są czynniki lub hormony uwalniające (releasing factor RF lub RH) stymulujące
bądź hamujące wydzielanie odpowiednich hormonów przez przysadkę mózgową. Hormony te
są transportowane z podwzgórza do przysadki przez żylny układ wrotny. Znana jest już
budowa większości hormonów podwzgórza – są polipeptydami zawierającymi 10 – 41
aminokwasów. Niektóre z nich uzyskano już syntetycznie.
Hormony podwzgórza działające na przysadkę:
1.
kortikolibryna (CRF) peptyd – pobudza wydzielanie adrenokortikotropiny (ACTH),
uwalnia beta-endorfinę i inne substancje pochodne proopiomelanokortyny (POMC),
2.
tyreoliberyna (TRH) peptyd – pobudza wydzielanie i uwalnianie tyreotropiny (TSH),
3.
gonadoliberyna (GNRH) peptyd – u kobiet wydzielana cyklicznie, pobudza
uwalnianie folitropiny (FSH) oraz lutitropiny (LH),
4.
somatoliberyna (GRH) peptyd – pobudza wydzielanie hormonu wzrostu –
somatotropiny,
5.
somatostatyna (GH-IH) peptyd – hamuje wydzielanie hormonu wzrostu i niektórych
innych hormonów peptydowych np. insuliny i tyreoliberyny,
6.
prolaktoliberyna (PRH) peptyd – pobudza wydzielanie prolaktyny (PRL),
7.
prolaktostatyna (PIF), hamuje wydzielanie prolaktyny (PRL),
8.
melanoliberyna (MRF) uwalnia melanotropiny,
9.
wazopresyna (AVP) peptyd – kurczy naczynia i ma działanie antydiuretyczne,
10.
oksytocyna (OT) – kurczy mięśnie gładkie naczyń i macicy,
11.
dopamina (PIH) hamuje uwalnianie prolaktyny.
Uwalnianie hormonów z podwzgórza jest regulowane przez ujemne sprzężenie zwrotne
między podwzgórzem, przysadką oraz obwodowym gruczołem dokrewnym. Zarówno
hormony przysadki jak i hormony gruczołów dokrewnych obwodowych hamują wydzielanie
hormonów uwalniających podwzgórza.
Ryc.1 Regulacja czynności układu podwzgórze – część gruczołowa przysadki – dokrewny
gruczoł obwodowy (Patofizjologia red. S. Maśliński, J. Ryżewski, PZWL, W-a 2002)
Przedstawiono zasadę ujemnego sprzężenia zwrotnego; + pobudzenie, - hamowanie,

hamowania neuronów wydzielniczych podwzgórza (typowego dla zasady ujemnego sprzężenia
zwrotnego) na rycinie zaznaczono również możliwość pobudzenia tych neuronów przez
hormony obwodowe.
Choroby podwzgórza.
Choroby podwzgórza takie jak np. nowotwory, zapalenia, procesy degeneracyjne i zaburzenia
rozwojowe, objawiają się zaburzeniem funkcji endokrynnych przysadki mózgowej. Mogą
pojawiać się też zaburzenia hormonalne powstałe na podłożu problemów psychologicznych
np. w stresie czy w anorexia nervosa. Wyrazem zaburzeń czynności podwzgórza jest tzw.
akromegalia pochodzenia podwzgórzowego polegająca na zwiększonym wydzielaniu
somatoliberyny lub zmniejszonym wydzielaniu somatostatyny. Zmieniona czynność układu
dopaminergicznego podwzgórza może powodować hiperprolaktynemię.
Przysadka
Przysadka jest gruczołem nieparzystym. Leży u podstawy mózgu w zagłębieniu siodła
tureckiego. Z podstawą mózgu łączy się za pośrednictwem lejka. Jest kształtu owalnego o
średnicy 1cm i masie 0,5 grama. W przysadce rozróżniamy dwa płaty: przedni i tylny.
Embriologicznie płat przedni rozwija się z endodermy cewy pokarmowej i dzięki temu ma
budowę gruczołową.
Hormony tylnego płata przysadki.
Płat tylny wywodzi się z pnia mózgu, jest częścią nerwową przysadki, zbudowaną z
elementów neuroglejowych. Hormonami syntetyzowanymi w podwzgórzu ale uwalnianymi
przez płat tylny przysadki jest wazopresyna zwana też hormonem antydiuretycznym (ADH) i
oksytocyna produkowana w jądrach nadwzrokowym i przykomorowym podwzgórza.
Wazopresyna - AVP (hormon antydiuretyczny ADH).
Hormon ten działa na dystalne kanaliki nerkowe zwiększając ich przepuszczalność dla
wody i poprzez zwiększoną resorbcję zwrotną wody doprowadza do zagęszczenia moczu.
Wazopresyna, powoduje u człowieka także skurcz mięśni gładkich ściany naczyń powodując
ich obkurczenie a w następstwie nadciśnienie tętnicze. Działa także synergistycznie z
hormonem kortikoliberyną (CRH) podwzgórza, zwiększając uwalnianie hormonu
adrenokortikotropowego (ACTH) przysadki.
Niedobór wazopresyny powoduje chorobę o nazwie moczówka prosta, która polega
na zaburzeniu zagęszczania moczu. Chory oddaje duże ilości moczu (poliuria) o niskim
ciężarze właściwym a więc o niskiej osmolarności. Utrata wody z moczem powoduje
ogromne odwodnienie organizmu i pragnienie. Upośledzenie wydzielania tego hormonu
występuje przede wszystkim w guzach mózgu, po urazach OUN, zabiegach
neurochirurgicznych, w procesach zapalnych a także po uszkodzeniu przysadki.
Istnieje też „obwodowa moczówka prosta” spowodowana brakiem lub uszkodzeniem
receptorów dla ADH w kanalikach dystalnych nerek. ADH jest obecny w organizmie, ale nie
może zadziałać.
Nadmiar wazopresyny prowadzi do tzw. Zespołu Schwartza-Barttera objawiającego
się zatrzymaniem przez nerki wody, rozcieńczeniem krwi, zmniejszeniem diurezy i
wydalaniem moczu o zwiększonej osmolarności. Najczęściej nadmierne wydzielanie
wazopresyny występuje w owsianokomórkowym raku płuca.
Oksytocyna
Wspomaga zapłodnienie komórki jajowej, odgrywa rolę podczas porodu i bierze udział w
wydzielaniu mleka podczas laktacji działając obkurczająco na ciężarną macicę i pęcherzyki

gruczołów sutkowych. Te właściwości oksytocyny wykorzystuje się w położnictwie.
Wazopresyna i oksytocyna mają cechy neuromediatorów i lub neuromodulatorów w
ośrodkowym układzie nerwowym. Wazopresyna usprawnia pamięć i ułatwia zapamiętywanie.
Oksytocyna jest czynnikiem sprzyjającym amnezji.
Zmniejszone wydzielanie oksytocyny występuje pod wpływem niektórych rodzajów
stresów (np. działania obniżonej temperatury otoczenia) oraz niektórych leków.
Zwiększone uwalnianie oksytocyny powodują: poród, odruch wytrysku mleka, bodźce
seksualne, niektóre bodźce stresowe.
Hormony przedniego płata przysadki.
Przedni płat przysadki syntetyzuje co najmniej 6 hormonów, pełniących ważną rolę w
regulacji procesów metabolicznych ustroju. Hormony te regulują czynność innych
obwodowych gruczołów dokrewnych, dlatego nazywane są hormonami tropowymi.
Rozróżniamy następujące hormony przedniego płata przysadki:
1.
hormon adrenokortikotropowy – kortikotropina (ACTH) (peptyd) działający na
nadnercza,
2.
hormon tyreotropowy – tyreotropina (TSH) (glikoproteina) pobudza wydzielanie
hormonów tarczycy,
3.
hormon wzrostu – somatotropina (GH), wpływa na wzrost człowieka,
4.
hormon folikulotropowy – folitropina (FSH) (glikoproteina) wpływa na dojrzewanie
pęcherzyków Graafa w jajniku u kobiet i spermatogenezę u mężczyzn,
5.
hormon luteinizujący - lutitropina (LH) (glikoproteina) wpływa na syntezę
progesteronu u kobiet i testosteronu u mężczyzn,
6.
prolaktyna (PRL) (polipeptyd), wpływa na wydzielanie mleka,
7.
beta-lipoproteina (LPH),
8.
melanotropiny alfa, beta, gamma.
Działanie hormonów tropowych.
Hormony tropowe przysadki wpływają na wydzielanie podległych im obwodowych
gruczołów dokrewnych. Kortikotropina (ACTH) pobudza wydzielanie hormonów kory
nadnerczy, tyreotropina (TSH) wpływa na czynność tarczycy, folitropina (FSH) i lutitropina
(LH) pobudzają pęcherzyki jajnikowe Graafa. Z kolei hormony wydzielane przez obwodowe
gruczoły dokrewne hamują wydzielanie hormonów tropowych przysadki i uwalniajacych
(liberyn) podwzgórza, tworząc pętlę ujemnych sprzężeń zwrotnych. Również hormony
przysadki ACTH, TSH, FSH i LH hamują wydzielanie hormonów uwalniających
podwzgórza.
Hormon wzrostu (GH) zwany też somatotropiną, powoduje wzrost wszystkich tkanek, które
mają możliwość wzrastania, poprzez wpływ na przemianę białek, weglowodanów, tłuszczów
i soli mineralnych. GH mając działanie anaboliczne wpływa również na masę mięśniową, a
współzależnie z dopaminą, bierze udział w regulacji stanu psychicznego człowieka.
Wydzielanie tego hormonu jest największe w pierwszych godzinach snu, zwiększa się także
pod wpływem bólu, zimna, wysiłku fizycznego, głodu i obniżenia zawartości glukozy we
krwi. Hormon wzrostu powoduje również uwolnienie z wątroby a także z trzustki
somatomedyn (czynników wzrostowych) współdziałających z nim synergistycznie. Znane jest
współdziałanie GH z intermedyną C jako insulinowo-podobnego czynnika wzrostu (IGF-1).
Przypuszcza się, że somatotropina działając bezpośrednio na tkankę chrzęstną sprawia, że
chondrocyty stają się wrażliwe na IGF-1. Somatotropina jest swoista dla człowieka, gdyż
preparaty pochodzenia bydlęcego nie działają na karłowatość przysadkową. Somatotropina

ma działanie antyinsulinowe, upośledza zatem tolerancję glukozy. Schemat działania
hormonu wzrostu przedstawia ryc. 2.
Nadmiar hormonu wzrostu (GH).
Zwiększone wydzielanie GH związane jest najczęściej z powstaniem gruczolaka przysadki,
zbudowanego przede wszystkim z komórek kwasochłonnych. Może być jednak także
następstwem zwiększonego wydzielania somatoliberyny podwzgórza.
Następstwa jego działania zależą od wieku. Nadmiar GH może powodować u dzieci przed
okresem skostnienia nasad kości długich proporcjonalny wzrost olbrzymi (gigantysmus).
Natomiast po okresie skostnienia nasad GH wywołuje wybiórcze powiększenie obwodowych
części ciała (dłoni, stóp, nosa, żuchwy), pogrubienie skóry i powiększenie narządów
wewnętrznych (serca, wątroby). Zespół tych objawów nazywamy akromegalią.
Niedobór hormonu wzrostu.
Zmniejszenie wydzielania GH w okresie wzrostu człowieka prowadzi do karłowatości
przysadkowej. U dzieci niedobór somatotropiny powoduje upośledzenie wzrostu i często
zwiększa wrażliwość na insulinę. Wzrost karłów przysadkowych ulega zahamowaniu od 2 – 3
roku. Ich stan ogólny jest dobry i rozwój umysłowy prawidłowy. Często upośledzenie
wydzielania hormonu wzrostu współistnieje z zaburzeniami wydzielania gonadotropin. Dlatego
ci chorzy nie osiągają dojrzałości płciowej.
Prolaktyna (PRL).
Prolaktyna jest polipeptydem, o budowie podobnej do somatotropiny i wywiera podobne
działanie anaboliczne. Występuje u mężczyzn i kobiet. We krwi kobiet waha się zgodnie ze
stężeniem estrogenów i progesteronu w cyklu miesiączkowym i ciąży. Wzrasta w 1. fazie
cyklu, maleje w drugiej, zwiększa się znacznie w ciąży, maleje podczas porodu i połogu
wpływając wraz z oksytocyną na wydzielanie mleka przez gruczoły mleczne kobiet.
Nadmierne wydzielanie prolaktyny (hiperprolaktynemia) może być spowodowane
obecnością gruczolaka przysadki lub obniżonym wydzielaniem dopaminy przez podwzgórze.
Prolactinoma – guzy syntetyzujące prolaktynę są najczęstszymi nowotworami
przysadki. Namierne wydzielanie prolaktyny u kobiet powoduje mlekotok i brak miesiączki, u
mężczyzn nie występuje mlekotok, ale stwierdza się bóle głowy, zaburzenia widzenia,
impotencję. Objawy te wynikają przede wszystkim z ucisku guza na przysadkę i jej okolice.
Leczenie hiperprolaktynemii polega na usunięciu chirurgicznym guza lub terapii
bromokryptyną (antagonistą dopaminy). Nadmierne wydzielanie prolaktyny może być nieraz
spowodowane przez nowotwory różnych narządów np. płuc, trzustki i grasicy.
Hormony gonadotropowe
Hipogonadyzm hipogonadotropowy jest wtórną niedoczynnością gruczołów płciowych
spowodowaną:
a. niedoborem hormonów gonadotropowych przysadki,
b. lub brakiem gonadoliberyny podwzgórza.
Głównym objawem tego zespołu jest brak dojrzewania płciowego u osób z prawidłowym lub
nadmiernym wzrostem, niedorozwój drugorzedowych cech płciowych i brak trzeciorzędnych
cech płciowych. Leczenie androgenami przynosi poprawę.
Uszkodzenie części gruczołowej przysadki.
Część gruczołowa przysadki może być powoli niszczona przez rosnący w tej okolicy guz.
Objawy wówczas występujące określa się zespołem Glińskiego- Simmondsa.
Inną postacią niedoczynności przysadki jest zespół Sheehana (poporodowa martwica
przysadki), który powstaje nagle po porodzie powikłanym krwotokiem, jak skutek
wykrzepiania wewnątrz naczyń odżywiających przysadkę. Manifestuje się on brakiem
miesiączki i niepłodnością.
U mężczyzn wtórny niedobór androgenów w następstwie ustania czynności jąder
powoduje duże zaburzenia w sferze płciowej.
Z powodu braku ACTH występuje niewydolność kory nadnerczy a braku TSH –
niedoczynność gruczołu tarczowego.
Tabela 1 przedstawia wykaz hormonów syntetyzowanych w obwodowych gruczołach
dokrewnych. (Patofizjologia red. S. Maśliński i J. Ryżewski PZWL W-a 2002)
Miejsce powstania
Hormon
Struktura
Kora nadberczy (warstwa
pasmowata oraz siatkowata
Kortyzol
Kortykosteron
Dehydroepiandrosteron
Androstendion
Steroid
Steroid
Steroid
Steroid
Kora adnerczy (warstwa
kłębkowata)
Aldosteron
Steroid
Rdzeń nadnerczy
Adrenalina (A)
Noradrenalina (NA)
Enkefalina metioninowa
Enkefalina leucynowa
Amina katecholowa
Amina katecholowa
Peptyd
Peptyd
Gruczoł tarczowy
Tyroksyna (T
4
)
Trijodotyronina (T
3
)
Kalcytonina (CT)
Tetrajodotyronina
Peptyd
Gruczoły przytarczyczne
Parathormon (PTH)
Peptyd
Wyspy trzustkowe
(Langerhansa):
Komórki A
Komórki B
Komórki D
Komórki F (PP)
Glukagon
Insulina
somatostatyna (SS)
polipeptyd trzustkowy
(PP), pancreatic
polypeptide)
Peptyd
Peptyd
Peptyd
Peptyd
Jajnik (warstwa ziarnista
pęcherzyków jajnikowych)
Estradiol
Estron
estriol
inhibiny
aktywiny
Steroid
Steroid
Steroid
Peptydy
Peptydy
Jajnik (ciałko żółte)
Progesteron
Relaksyna
Steroid
Peptyd
Łożysko
Ludzka gonadotropina
łożyskowa (HCG* - human
chorionic gonadotropin)
Ludzka somatomam-
motropina kosmówkowa
(HCS); synonimy: ludzki
laktogen łożyskowy (HPL),
chorionic growth hormone-
prolactin (CGP)
Relaksyna
Glikoproteina: HCG- (m.cz.
18 000)
HCG- (m.cz. 28 000)
peptyd zbliżony do hGH

estrogeny
progesteron
Peptyd
Steroidy
Steroid
Jądro (komórki śródmiąższowe
Leydiga)
Testosteron
Steroid
Jądro (komórki Sertolego)
Inhibiny
Aktywiny
Peptydy
Peptyd
Grasica
Tymozyna
Tymozyna
tymopoetyna I i II
tymulina (TSF-grasiczy
czynnik surowiczy)
grasiczy czynnik
humoralny (THF-thymic
humoral factor)
Peptyd
Peptyd
Peptydy
Peptyd
Peptyd
Szyszynka
Melatonina
N-acetylo-5-
metoksytryptamina
Ryc. 2.
Regulacja osi podwzgórze-hormon wzrostu-somatomedyna wg E. Małeckiej-Tandery
(Patofizjologia Kliniczna, Volumed, Wrocław 2001)
SS –somatostatyna
GH – hormon wzrostu
GHRH – czynnik uwalniający hormon wzrostu
IGF – somatomedyna (insulinopodobny czynnik wzrostu)
Rozdział VI. Patologia gruczołu tarczowego
Iza Iwan- Ziętek
1. Tarczyca, hormony tarczycy
Tarczyca zbudowana jest z dwóch płatów połączonych cieśnią. Płaty składają się z
licznych pęcherzyków, wewnątrz których odbywa się produkcja hormonów: tyroksyny ( T4),
trójjodotyroniny (T3) i reverse trójjodotyroniny (rT3). Synteza ich zależy od podaży jodu
dostarczanego z pokarmem i powietrzem oddechowym. We krwi większość T3 i T4 wiąże się
z białkami nośnikowymi (globuliny, prealbuminy, albuminy). Tylko niewielka ilość
tyroksyny i trójjodotyroniny występuje w stanie wolnym, ale tylko w tej formie hormon jest
biologicznie aktywny. Hormony tarczycy zwiększają produkcję ciepła i zużycie tlenu.
Uczestniczą w regulacji metabolizmu białek tłuszczów i węglowodanów. Wzmagają działanie
katecholamin, pobudzają filtrację kłębuszkową, biorą udział w regulacji gospodarki
wapniowo-fosforanowej. Odgrywają również bardzo istotną rolę w rozwoju ośrodkowego i
obwodowego układu nerwowego. Poza w/w hormonami w okołopęcherzykowych
komórkach C tarczycy odbywa się synteza kalcytoniny, hormonu biorącego udział w regulacji
gospodarki wapniowo-fosforanowej.
2. Wole obojętne

Definicja
Wole obojętne jest to powiększenie tarczycy, przebiegające ze stanem eutyreozy, bez
objawów zapalenia i zezłośliwienia.
Częstość występowania
Jest to najczęstsze schorzenie układu endokrynologicznego, zaliczane do chorób społecznych.
W przypadku, gdy wole obojętne występuje w mniejszym odsetku niż 5% populacji dzieci w
wieku 6-14 lat mówimy o wolu sporadycznym, przy wyższym odsetku określane jest jako
wole endemiczne.
Etiologia
Wole obojętne rozwija się najczęściej w warunkach niedoboru jodu. Dlatego w rejonach, w
których ilość tego pierwiastka w wodzie i glebie jest niska, zaleca się profilaktykę jodową
polegającą na jodowaniu soli, paszy dla zwierząt, nawozów itp. Innymi czynnikami
przyczyniającymi się do powstania wola są: stresy fizjologiczne (okres dojrzewania,
przekwitania, ciąża), defekty enzymatyczne syntezy hormonów tarczycy, procesy
immunologiczne, nadmierne spożywanie związków wolotwórczych zawartych w roślinach
jadalnych np. kalarepie, kalafiorze, kapuście, rzepie, rzodkwi, orzeszkach ziemnych oraz duże
zanieczyszczenia przemysłowe. Również niektóre leki jak: barbiturany, diuretyki, PAS,
węglan litu, salicylany, jod (podawany w dużych dawkach) mogą zaburzać syntezę
hormonów tarczycy.
Patogeneza
Wole obojętne powstaje głównie w wyniku stymulacji przez TSH komórek tarczycy, co
doprowadza do ich przerostu.TSH wydzielany jest w nadmiarze, ponieważ istniejący
niedobór jodu przyczynia się do spadku syntezy hormonów tarczycy oraz
aktywacji wewnątrztarczycowych czynników wzrostowych m. in. EGF ( Epidermal Growth
Factor), IGF-1 (Insulin –like Growth Factor 1), które powodują zwiększenie liczby
tyreocytów
Początkowo wole obojętne ma charakter wola miąższowego koloidowego. W okresie
tym nie stwierdza się zaburzeń hormonalnych tarczycy. W późniejszym okresie wole to może
przekształcić się w wole guzowate, powstające w wyniku namnażania się komórek
funkcjonujących autonomicznie (niezależnych od TSH), co może doprowadzić do utajonej
lub jawnej nadczynności gruczołu.
Obraz kliniczny
Obraz kliniczny wola zależy od jego wielkości. Zwykle widoczne jest gołym okiem lub
wyczuwalne przy badaniu palpacyjnym. Gdy jego rozmiary są małe, nie daje żadnych
dolegliwości i stanowi tylko problem kosmetyczny. Pacjenci z większym wolem skarżą się na
uczucie duszności spowodowanej uciskiem mechanicznym na tchawicę, trudności przy
połykaniu (dysfagia) uwarunkowane przemieszczeniem przełyku, bóle w śródpiersiu,
spowodowane wolem zamostkowym.
Badania dodatkowe
1. Ocena wielkości tarczycy ( badanie palpacyjne i ultrasonograficznie),
2. oznaczenie stężenia jodu w rannej próbce moczu (niskie wartości),
3. oznaczenie stężenia TSH, T3, T4 – w granicach normy (z wyjątkiem ciężkich
przypadków niedoboru jodu),
4. stosunek molarny T3/T4 podwyższony, gdyż ustrój wytwarza więcej biologicznie
aktywniejszego hormonu i zawierającego mniej atomów jodu,

5. oznaczanie stężenia tyreoglobuliny (wzrost stężenia w przypadku niedoboru jodu),
6. scyntygrafia tarczycy , badanie rtg.
Leczenie
Leczenie wola obojętnego może być zachowawcze (podawanie jodu, hormonów tarczycy lub
jodu + hormonów tarczycy), operacyjne bądź jodem promieniotwórczym.
3.Nadczynność tarczycy - hipertyreoza
Definicja
Nadczynność tarczycy to zespół objawów klinicznych uwarunkowany nadmiarem krążących
hormonów tarczycy.
Częstość występowania
Nadczynność tarczycy występuje u około 2% populacji dorosłych. Dzieci chorują ponad 10
razy rzadziej niż dorośli.
Postaci i przyczyny nadczynności tarczycy:
1.
nadczynność pochodzenia immunologicznego-Choroba Gravesa-Basedowa - stanowi
ona 60 % przypadków nadczynności, występuje pięciokrotnie cześciej u kobiet niż u
mężczyzn,
2.
nadczynność spowodowana autonomią czynnościową - Wole guzowate nadczynne
-
choroba Plummera (mnogie guzki autonomiczne),
-
choroba Goetscha (pojedynczy guzek tarczycy),
-
rozproszona autonomia w formie zmian mikroguzowatych w całej tarczycy;
3. niekiedy choroba Gravesa-Basedowa współitnieje z nadczynnym guzkiem tarczycy
(zespół Marine-Lenharta).
Rzadko do nadczynności tarczycy dochodzi na tle guza przysadki produkujacego TSH
(wtórna nadczynność tarczycy), ektopowego wydzielania hormonów przez nowotwory,
nadmiaru HCG (gonadotropina kosmówkowa) w ciąży, stanów zapalnych tarczycy i
stosowania leków np. związków jodu, amidaronu.
3a. Choroba Graves-Basedowa
Etiologia i patogeneza
Jest to schorzenie autoimmunologiczne, w którym dochodzi do wytwarzania autoprzeciwciał
TSH-R (stim)Ab. Przeciwciała te reagując z receptorami TSH, zlokalizowanymi w błonie
komórkowej tyreocytów, wybiórczo pobudzają aktywność tarczycy, co prowadzi do wzrostu
wydzielania T3 i T4. Poza przeciwciałami przeciwreceptorowymi u chorych mogą
występować przeciwciała antyperoksydazowe (anty-TPO) i przeciwciała
antytyreoglobulinowe (anty-Tg). Pojawienie się tych przeciwciał związane jest z wrodzonym
defektem supresorowych limfocytów T. Również czynniki środowiskowe takie jak: zakażenia
bakteryjne i wirusowe, stres, palenie papierosów, uraz, starzenie się ustroju, ciąża mogą
bezpośrednio powodować defekt w limfocytach T-supresorowych.
Genetyczne uwarunkowanie choroby związane jest z antygenami zgodności tkankowej HLA:
B8 i DR3

3b. Wole guzowate nadczynne
Etiopatogeneza
Wole guzowate nadczynne rozwija się głównie na podłożu wola spowodowanego niedoborem
jodu. Wykazano, że w prawidłowo funkcjonującej tarczycy znajdują się tyreocyty nie
podlegające regulacji ze strony układu podwzgórzowo- przysadkowego tworzące
fizjologiczną autonomię tarczycy.
Niedobór jodu sprzyja namnażaniu tych komórek, co w efekcie może prowadzić do
powstania guzków autonomicznych, które często przez wiele lat nie doprowadzają do
powstania klinicznych objawów nadczynności. Rozwojowi nadczynności tarczycy u chorych
z wolem guzowatym sprzyja dodatkowa podaż jodu.Tkanka autonomiczna, w odróżnieniu od
zdrowej, nie blokuje bowiem wbudowywania jodu w przypadku jego wzrostu w surowicy, co
prowadzi do nadmiernego tworzenia hormonów. Dlatego większość endokrynologów nie
zaleca stosowania preparatów zawierających jod u chorych z wolem guzowatym w stanie
eutyreozy.
W pojedynczych przypadkach przyczyną autonomicznej nadczynności tarczycy są
mutacje genu receptora TSH.
Obraz kliniczny nadczynności tarczycy
Niezależnie od przyczyny nadczynności tarczycy obraz kliniczny jest charakterystyczny i
zbliżony we wszystkich jej postaciach, ponieważ jest on wynikiem patologicznie
zwiększonego stężenia hormonów tarczycy. Nasilają one przemiany metaboliczne oraz
podwyższają wrażliwość tkanek na katecholaminy (dzięki zwiększonej ilości błonowych
receptorów adrenergicznych).
Do objawów będących konsekwencją zwiększonej przemiany materii należy:
zmniejszenie masy ciała, wzmożony apetyt, niekiedy hiperglikemia, nietolerancja ciepła,
aksamitna skóra, nadmierna potliwość. Pobudzenie układu nerwowego powoduje u chorych
nerwowość, bezsenność, drobnofaliste drżenie rąk, miopatię (osłabienie mięśni i ich zanik)
oraz objawy oczne łagodne - przejaw uogólnionej sympatykotonii. Uzewnętrzniają się one
pod postacią objawu: Graefego (nienadążanie górnej powieki za gałką oczną podczas jej
ruchu w dół), Kochera (nienadążanie dolnej powieki przy ruchu gałki ocznej do góry),
Dalrymple’a (nadmierne rozszerzenie szpary powiekowej), Stellwaga (rzadkie mruganie),
Joffroya (niemożność zmarszczenia czoła przy patrzeniu do góry).
W przebiegu nadczynności tarczycy dochodzi do toksycznego wpływu hormonów tarczycy na
układ sercowo-naczyniowy w postaci: tachykardii, migotania przedsionków , nadciśnienia ze
zwiększoną amplitudą ciśnienia krwi. W przypadkach braku leczenia może rozwinąć się
kardiomiopatia i niewydolność krążenia. Zwykle w nadczynności stwierdza się również:
wole, biegunki, zaburzenia miesiączkowania u kobiet, spadek libido, łamliwość paznokci,
wypadanie włosów, osteoporozę.
Rozpoznanie choroby Gravesa-Basedowa ułatwia stwierdzenie „triady merseburskiej”
(wytrzeszcz, wole, tachykardia). Tarczyca jest wówczas zwykle równomiernie powiększona i
słyszalny jest nad nią szmer naczyniowy.
U kilku procent pacjentów z chorobą Gravesa-Basedowa zmiany oczne nie są łagodne
lecz przybierają postać bardziej złośliwą. Proces ten nazywamy oftalmopatią postępującą
czyli wytrzeszczem naciekowo-obrzękowym. Powstaje on w wyniku reakcji
autoimmunologicznej w tkance łącznej pozagałkowej, komórkach mięśni okołoruchowych
oraz w powiekach. Istotną rolę w tym schorzeniu odgrywają przeciwciała skierowane przeciw
antygenom fibroblastów oraz limfocyty cytotoksyczne Uczulone limfocyty uwalniają
cytokiny powodujące odczyn zapalny i powstanie nacieków złożonych z limfocytów i
mukopolisacharydów, a w późniejszym etapie włóknienie tkanek. Opisany proces powoduje u

chorych wysunięcie gałek ocznych do przodu, wysychanie i owrzodzenie rogówki,
ograniczenie ruchomości gałki ocznej oraz utrudnione patrzenie zbieżne. Chorzy zgłaszają
uczucie pieczenia spojówek, łzawienia, niekiedy skarżą się na podwójne widzenie
(upośledzenie czynności mięśni pozagałkowych), pogorszenie ostrości wzroku (ucisk na nerw
wzrokowy).
U 5% chorych występuje obrzęk przedgoleniowy również pochodzenia
autoimmunologicznego polegający na odkładaniu mukopolisacharydów w tkance podskórnej
okolicy przedgoleniowej.
Trudności diagnostyczne występują głównie u osób starszych z wolem guzkowym ,
gdyż wówczas objawy są mniej nasilone i często pacjenci mają tylko zaburzenia rytmu serca
lub niewydolność krążenia „oporną” na preparaty naparstnicowe.
Ponadto u chorych z nadczynnością (głównie z chorobą Gravesa- Basedowa) pod
wpływem operacji, urazu psychicznego, zakażenia, porodu, odstawienia leków
tyreostatycznych może rozwinąć się przełom tarczycowy hipermetaboliczny. Wynika on z
nagłego uwolnienia do krwi z gruczołu bardzo dużych ilości hormonów tarczycy. Jest to stan
zagrażający życiu, wymagający natychmiastowej interwencji, manifestujący się: wysoką
temperaturą, zlewnymi potami, biegunką, wymiotami prowadzącymi do odwodnienia,
niewydolnością krążenia, niewydolnością wątroby, znacznym pobudzeniem, psychozą. W
miarę rozwoju choroby może rozwinąć się śpiączka. .
Badania dodatkowe
1. Badania hormonalne :
-obniżony poziom TSH i podwyższony FT4 i FT3 (FT3 i FT4 - wolne hormony tzn. nie
związane z białkami)
-
podwyższony poziom TSH, FT3 i FT4 - u chorych z guzem przysadki
produkującym TSH
-
zmniejszenie stężenie TSH, prawidłowe stężenie FT3 i FT4- utajona
nadczynność tarczycy
2. Badania immunologiczne:
-przeciwciała przeciw receptorowi TSH, antytyreoperoksydazowe, antytyreoglobulinowe
3. Inne:
- TSH, scyntygrafia
- biopsja aspiracyjna cienkoigłowa tarczycy (BAC) - gdy występują zmiany guzkowe.
Leczenie
Leczenie nadczynności tarczycy może być zachowawcze tyreostatykami (pochodne
tiouracylu i imidazolu), jodem promieniotwórczym (131 I ) bądź operacyjne
4. Niedoczynność tarczycy – Hipotyreoza
Definicja
Niedoczynność tarczycy jest to stan kliniczny spowodowany niedoborem lub brakiem
tyroksyny a niekiedy osłabioną reaktywnością tkanek na hormony tarczycy. Niedoczynność
tarczycy u dzieci powoduje kretynizm, a u dorosłych przebiega jako obrzęk śluzowaty.
Częstość występowania
Niedoczynność tarczycy występuje 5 razy częściej u kobiet niż u mężczyzn.
Wrodzona niedoczynność tarczycy jest najczęstszą wrodzoną endokrynopatią i występuje
1:4000 noworodków.

Etiopatogeneza
Niedoczynność wynikająca z niedoboru TSH nazywana jest niedoczynnością wtórną zaś z
niedoboru TRH niedoczynnością trzeciorzędową. Niedoczynność pierwotna spowodowana
jest uszkodzeniem samego gruczołu tarczowego.
Wrodzona niedoczynność tarczycy najczęściej spowodowana jest zaburzeniami
rozwojowymi wyrażającymi się całkowitym brakiem lub niedorozwojem tarczycy bądź
nieprawidłowym jej położeniem, co jest wynikiem procesów immunologicznych oraz
zaburzeń genetyczych.
Inną grupą zaburzeń, które mogą doprowadzić do wrodzonej niedoczynności są rodzinne
dyshormonogenezy (nieprawidłowości receptorowe lub enzymatyczne w syntezie
hormonów). Wrodzona niedoczynność podwzgórzowo-przysadkowa występuje bardzo
rzadko, a jej przebieg kliniczny jest łagodniejszy niż pierwotnej niedoczynności tarczycy.
Nabyta niedoczynność tarczycy najczęściej powstaje wskutek przewlekłego
limfocytarnego zapalenia tarczycy typu Hashimoto (autoimmunologiczne zapalenie tarczycy).
Rzadziej spotykaną postacią jest jatrogenna niedoczynność, np. po podaniu jodu
radioaktywnego, zabiegu chirurgicznym, radioterapii okolicy szyjnej bądź po stosowaniu
tyreostatyków czy litu. Również uszkodzenie podwzgórza i przysadki mogą wywołać
niedoczynność (wtórną i trzeciorzędową).
Obraz kliniczny wrodzonej niedoczynności tarczycy
Objawy wrodzonej hipotyreozy zależą od etiologii, ilości hormonów tarczycy i czasu trwania
choroby. Zwykle dzieci bezpośrednio po urodzeniu nie mają żadnych objawów, a rozpoznanie
ustala się na podstawie przesiewowych oznaczeń TSH wykonywanych w pierwszych dniach
życia. Jeśli u noworodka zostanie wykryty nieprawidłowy poziomu TSH natychmiast
wdrażane jest leczenie substytucyjne, co zapobiega rozwinięciu się objawów chorobowych.
Włączenie leczenia dopiero w 4 miesiącu życia dziecka powoduje trwałe uszkodzenie OUN.
W przypadku bardzo dużego niedoboru hormonów tarczycy, w okresie życia
płodowego, u noworodka mogą pojawić się cechy hipotyreozy. Są one wyrażone :
przedłużającą się żółtaczką fizjologiczną, apatią, zaparciami, hipotermią, suchą,
marmurkowatą skórą, otwartym ciemieniem tylnym, hipotonią mięśni, przepukliną pępkową,
słabym łaknieniem. Jeśli natychmiast nie zostanie wdrożone leczenie dołączają się stopniowo
następne objawy somatyczne: zaburzenia wzrostu, nieprawidłowe proporcje ciała (duża
głowa, długi tułów, krótkie kończyny), opóźnione wyżynanie zębów, osłabienie odruchów
ścięgnistych, płaski nos, szeroko rozstawione oczy, ochrypły płacz, kruche włosy, pogrubiałe
brzegi powiek, obrzęki, bradykardia, niedokrwistość, opóźnione lub przedwczesne
wystąpienie dojrzewania płciowego.
Obraz kliniczny nabytej niedoczynności tarczycy
W przypadku wystąpienia niedoczynności tarczycy u dziecka po 2 r. ż. nie stwierdza
się niedorozwoju umysłowy, ale dzieci te osiągają słabsze wyniki w nauce, są niższe, mają
osłabioną siłę mięśniową i zaburzenia dojrzewania płciowego.
Niedoczynność tarczycy u dorosłych
Objawy niedoczynności tarczycy u dorosłych są mniej nasilone i po wprowadzeniu terapii
substytucyjnej odwracalne. W pierwszym (utajonym) okresie hipotyreozy brak jest typowych
symptomów a zaburzenia dotyczą głównie gospodarki lipidowej (wzrost stężenia cholesterolu
i jego frakcji LDL). Pełnoobjawowa niedoczynność tarczycy charakteryzuje się zaś:
wzrostem masy ciała (obrzęk śluzowaty), zwiększeniem wrażliwości na zimno, suchą,
chłodną w dotyku skórą, obrzękiem twarzy, kończyn, spowolnieniem sprawności fizycznej i
psychicznej, sennością, brakiem zainteresowania otoczeniem, wypadaniem włosów,

skłonnością do zaparć , kamicą żółciową, bradykardią, często nadciśnieniem tętniczym
rozkurczowym, płynem w worku osierdziowym i otrzewnowym (wynik przechodzenia
albumin oraz wody do przestrzeni pozałożyskowej). Obrzęk w tkance podskórnej
spowodowany jest odkładaniem hydrofilnych mukopolisacharydów, U starszych osób może
wystąpić jednak niedoczynność monosymptomatyczna charakteryzująca się tylko stanami
depresyjnymi, osłabieniem pamięci czy stałym uczuciem zimna. W skrajnych przypadkach u
chorych może rozwinąć się śpiączka hipometaboliczna charakteryzująca się dużą
śmiertelnością. Typowymi jej objawami są: zamroczenie lub śpiączka, hipowentylacja z
hiperkapnią , hipotermia, hipotensja.
Badania dodatkowe:
1.
wzrost stężenia cholesterolu, frakcji LDL cholesterolu, trójglicerydów
2.
obniżenie stężenia FT4 i podwyższenie stężenia TSH ( pierwotna niedoczynność
tarczycy)
3.
obniżenie stężenia FT4 i prawidłowe lub obniżone stężenie TSH (niedoczynność
wtórna)
4.
USG, BAC, scyntygrafia, stężenie przeciwciał antyperoksydazowych i
antytyreoglobulinowych
Leczenie
Długotrwała substytucja syntetyczną solą sodową lewoskrętnego izomeru tyroksyny
(lewotyroksyna).
Temat VII . Patologia nadnerczy
Iza Iwan- Ziętek
Nadnercza, hormony nadnerczy
Nadnercza leżą na górnych biegunach nerek, są narządem parzystym. W każdym nadnerczu
znajdują się dwa narządy endokrynne: rdzeń i kora. Rdzeń jest pochodzenia ektodermalnego a
kora mezodermalnego.
Kora nadnerczy składa się z trzech warstw. Najbardziej zewnętrznie leży
warstwa kłębkowata (zona glomerulosa) produkująca mineralokortykosteroidy ( aldosteron,
dezoksykortykosteron –DOC); w środku, najszersza, warstwa pasmowata (zona fasciculata)
produkująca głównie glikokortykosteroidy (kortyzol, kortyzon, kortykosteron)
i najbliżej rdzenia warstwa siatkowata (zona reticularis) produkująca głównie androgeny
(dehydroepiandrosteron -DHEA, androstendion, 11beta-hydroksyandrostendion).
Wydzielanie hormonów przez warstwę pasmowatą i siatkowatą
zależy od przysadkowego hormonu adrenokortykotropowego (ACTH), który powstaje z
prekursora proopiomelanokortyny (POMC). W procesie proteolizy z POMC powstaje
również melanotropina (MSH), lipotropina (LPH) i endorfiny. Wydzielanie ACTH
pobudzane jest przez hormon podwzgórza kortykoliberynę (CRH). ACTH wywiera
niewielki wpływ na syntezę i uwalnianie mineralokortykosteroidów. Główną rolę w regulacji
wydzielania tych hormonów odgrywa układ reninowo-angiotensynowy. Rdzeń nadnerczy
produkuje katecholaminy: adrenalinę, noradrenalinę i dopaminę. Hormony rdzenia nadnerczy
łącznie z glikokortykosteroidami i mineralokortykosteroidami są niezbędne do przetrwania
sytuacji stresowych.
Kortyzol (lipoka)

Choroby kory nadnerczy
1. Zespół Cushinga
Definicja
Jest to zespół objawów klinicznych wywołany nadmiarem glikokortykosteroidów, któremu
często towarzyszy również wzmożone wydzielanie androgenów.
Etiologia i podział
Jeżeli pierwotna przyczyna zaburzeń zlokalizowana jest w podwzgórzu lub przysadce
mówimy o chorobie Cushinga czyli
ośrodkowym zespole Cushinga. Pozostałe przypadki
kwalifikujemy do zespołu, który może mieć podłoże:
-jatrogenne (najczęstszy),
-nadnerczowe: gruczolak, rak, barwnikowy rozrost mikroguzkowy i makroguzkowy,
-ektopowe (wydzielanie ACTH przez guzy pozaprzysadkowe np. rak trzustki, rdzeniasty
tarczycy, owsianokomórkowy płuc, grasiczak),
-indukowane alkoholem.
Patogeneza
Obraz kliniczny choroby wynika z nadmiernego działania steroidów na tkanki i narządy.
Powodują one wzrost glukoneogenezy z następową hiperglikemią, nasilenie katabolizmu
białkowego, zaburzenie syntezy kolagenu, spadek wchłaniania wapnia z przewodu
pokarmowego i zwiększone wydalanie go z moczem; zatrzymywanie sodu i wody przez nerki
i zwiększone wydalanie potasu z moczem; zwiększenie wrażliwości mięśniówki gładkiej
naczyń na katecholaminy i angiotensynę.
Obraz kliniczny
W przypadku przewlekłego działania steroidów rozwija się typowy obraz chorobowy, na
który składają się:-zmiana wyglądu twarzy i sylwetki ciała - twarz o kształcie księżyca w
pełni, „bawoli” kark, duży brzuch (nagromadzenie tkanki tłuszczowej), szczupłe kończyny
(zanik mięśni) -osłabienie siły mięśniowej (katabolizm białek),-osteoporoza (zaburzenia
gospodarki wapniowej),-nadciśnienie tętnicze (zwiększona wrażliwość presyjna naczyń,
nasilona wątrobowa synteza, angiotensynogenu),-hirsutyzm, zaburzenia miesiączkowania,
trądzik (w przypadku nadmiaru androgenów),-u mężczyzn zanik popędu płciowego i
potencji,-zaburzenia psychiczne (euforia, depresja), -zmniejszona tolerancja glukozy bądź
cukrzyca steroidowa (antagonistyczne działanie kortyzolu w stosunku do insuliny),-
ścieńczenie tkanki podskórnej predysponujące do wybroczyn i wylewów podskórnych
(katabolizm tkanki łącznej), -czerwonofioletowe rozstępy skórne,-zmiany w składzie krwi:
poliglobulia, leukocytoza, eozynopenia, monocytopenia, trombocytoza, -zmniejszenie
odporności (działanie kataboliczne na białka), -utrata zdolności do reakcji anafilaktycznych, -
zahamowanie wzrostu u dzieci (działanie kataboliczne na białka, zaburzenia gospodarki
wapniowej).
Badania dodatkowe
I . Rozpoznanie hiperkortyzolizmu: podwyższony poziom kortyzolu we krwi, zniesienie
dobowego rytmu stężenia kortyzolu w surowicy, zwiększone wydalanie 17-
hydroksykortykoidów w moczu (17-KS-metabolit kortyzolu).
II. Ustalenie etiologii hiperkortyzolizmu
1.Poziom ACTH

- guzy kory nadnerczy - poziom ACTH niski;
- choroba Cushinga – poziom ACTH w normie lub podwyższony;
- ektopowe wydzielanie ACTH – poziom ACTH bardzo wysoki.
2.Test hamowania dużą dawką deksametazonu (silny syntetyczny steryd)
-
guzy kory nadnerczy – brak spadku stężenia kortyzolu;
-
choroba Cushinga – spadek stężenia kortyzolu;
-
ektopowe wydzielanie ACTH – brak spadku stężenia kortyzolu.
3.Badania lokalizacyjne-tomografia komputerowa, rezonans magnetyczny
Leczenie
W przypadku stwierdzenia nowotworu usuwany jest on operacyjnie.
2. Niewydolność kory nadnerczy
Definicja
Jest to zespół objawów klinicznych spowodowanych niedoborem hormonów kory nadnerczy.
Podział
Niedoczynność nadnerczy może być pierwotna-wynikła z uszkodzenia samych nadnerczy
(choroba Addisona) i wtórna, u podstaw której leży niedobór ACTH (uszkodzenie
podwzgórza bądź przysadki).
2a. Choroba Addisona (pierwotna niedoczynność kory nadnerczy)
Etiologia :
Uszkodzenie kory nadnerczy przez: proces autoimmunologiczny (75%), przerzuty
nowotworowe, gruźlica, sarkoidoza, grzybice, u chorych na AIDS w wyniku zakażeń.
Patogeneza
Objawy są spowodowane brakiem hormonów wytwarzanych przez trzy warstwy części
korowej nadnerczy.
Okresy choroby:
-bezobjawowa niewydolność kory nadnerczy,
-klinicznie jawna niewydolność kory nadnerczy (gdy zniszczeniu ulegnie ponad 90% kory
nadnerczy),
-przełom nadnerczowy,
-śpiączka nadnerczowa.
Obraz kliniczny choroby
Do typowych objawów choroby należy: osłabienie mięśniowe , chudnięcie i odwodnienie (
brak łaknienia, nudności, wymioty, bóle brzucha), wzmożone spożywanie soli (nadmierna
utrata sodu z moczem-brak mineralokortykosteroidów, zawroty głowy, mroczki przed
oczami, drżenie, uczucie głodu (niskie ciśnienie, hipoglikemia), hiperpigmentacja skóry i
błon śluzowych (przy niedoborze kortyzolu wzrasta, na drodze ujemnego sprzężenia
zwrotnego poziom POMC z którego powstaje ACTH i MSH), utrata owłosienia łonowego i
pachowego u kobiet (spadek androgenów), niewydolność krążenia ( osłabienie mięśnia
sercowego, odwodnienie), obniżona odporność na zakażenia, stresy, urazy,-zaburzenia
psychiczne: spowolnienie, ostre psychozy,

Badania dodatkowe
1. zmiany w ekg,
2. zmiany w składzie krwi: lekka niedokrwistość, liczba krwinek. białych na granicy normy,
limfocytoza, eozynofilia,
3. zwiększenie gamma-globulin
4. hiperkaliemia (zaburzenia akcji serca), hiponatremia (brak mineralokortykosteroidów).
2b. Wtórna niedoczynność nadnerczy
Przyczyny:
Najczęstsze przyczyny to: przewlekła kortykoterapia , guzy okolicy przysadki lub
podwzgórza, martwica poporodowa przysadki, urazy, infekcje, napromieniowanie przysadki,
operacje przysadki
Patogeneza
U chorych obniża się wówczas uwalnianie kortyzolu i androgenów, wydzielanie
aldosteronu pozostaje jednak prawidłowe.
Obraz kliniczny
Przebieg wtórnej niedoczynności jest lżejszy niż choroby Addisona a głównym
objawem różnicującym jest brak przebarwień (we wtórnej niedoczynności) z obecnością
odbarwień ( w wyniku niedoboru ACTH i melanotropiny).
Badania dodatkowe wykonywane w pierwotnej i wtórnej niedoczynnosci
W chorobie Addisona występuje niski poziom kortyzolu w surowicy , wysoki ACTH , niski
17 -OHCS w moczu oraz hiperkaliemia, hiponatremia i hipoglikemia na czczo.
W teście rezerwy nadnerczowej ( po stymulacji syntetycznym preparatem ACTH ocenia się
stężenie 17-OHCS w moczu) w chorobie Addisona występuje brak istotnego wzrostu 17-
OHCS w moczu (zniszczone nadnercza).
We wtórnej niewydolności nadnerczy stężenie kortyzolu, ACTH w surowicy jest
niskie. Niskie jest również stężenie 17-OHCS w moczu.
Po stymulacji preparatem ACTH poziom 17-OHCS stopniowo wzrasta.
Leczenie
Leczenie niewydolności nadnerczy polega na substytucji brakujących hormonów.
2c. Przełom nadnerczowy (ostra niewydolność nadnerczy)
Definicja
Jest to zespół objawów klinicznych wywołanych nagłym dużym niedoborem kortyzolu.
Etiologia i patogeneza
Przyczyną tego schorzenia jest nagła destrukcja nadnerczy powstała w wyniku zatoru lub
wylewu do gruczołu. Zmiany te najczęściej powstają w przebiegu posocznicy
meningokokowej (zespół Waterhouse-Friderichsena), po urazach, wylewach oraz przy
niedostatecznej substytucji w sytuacjach stresowych u chorych z przewlekłą niewydolnością
nadnerczy. Jest to poważny stan często prowadzący do śmierci.
Obraz kliniczny

U chorych występują bóle brzucha , nudności, wymioty (utrata wody, sodu chlorków)
podwyższona temperatura, odwodnienie, sucha skóra, niewydolność krążenia. Pacjenci
początkowo są pobudzeni, potem wpadają w śpiączkę.
Badania dodatkowe:
hipoglikemia , hiperkaliemia, hiponatremia oraz kwasica metaboliczna
3. Zespół nadnerczowo-płciowy
Definicja
Zespół nadnerczowo-płciowy jest to wrodzone zaburzenie syntezy kortyzolu, dziedziczone
autosomalnie recesywnie.
Patogeneza
Niedobór kortyzolu powoduje zwiększone wydzielanie CRH i ACTH, co doprowadza
do przerostu kory nadnerczy, która wobec niemożności produkcji kortyzolu syntetyzuje
głównie androgeny oraz produkty steroidogenezy wytwarzane w nadmiarze przed miejscem
bloku enzymatycznego. Androgeny działają anabolicznie na białka.
Objawy kliniczne
W przypadku nadmiaru androgenów dzieci początkowo szybciej rosną, ale również
szybciej zachodzi u nich kostnienie, co w efekcie powoduje iż osoby te w wieku dojrzałym są
niskie. Gdy nadmiar tych hormonów występuje u dziewczynek w życiu płodowym powoduje
maskulinizację narządów płciowych zewnętrznych . W późniejszym okresie doprowadza do
rozwoju męskiego typu budowy (szeroka obręcz barkowa, wąska miedniczna, słabo
wykształcone gruczoły piersiowe) oraz hirsutyzmu (owłosienie typu męskiego). Takie cechy
określamy mianem wirylizmu. Nadmiar androgenów u chłopców odpowiada za nadmierny i
przedwczesny rozwój narządów płciowych zewnętrznych. Hormony te na drodze sprzężenia
zwrotnego ujemnego hamują wydzielanie gonadotropin z przysadki, co powoduje
niepłodność u obu płci..
Najczęstsze bloki enzymatyczne prowadzące do rozwoju zespołu nadnerczowo-płciowego
to:
- niedobór 21-hydroksylazy, w którym progesteron i 17- hydroksyprogesteron nie ulegają
przekształceniu do 11-dezoksykortykosteronu i 11-dezoksykortyzolu oraz
- niedobór 11B-hydroksylazy , w którym 11-dezoksykortykosteron i 11-dezoksykortyzol nie
ulegają przekształceniu do kortykosteronu i kortyzolu
Niedobór 21-hydroksylazy powoduje niedobór kortyzolu i aldosteronu.
Zależnie od niedoboru enzymu wyróżniamy:
1.Prostą wirylizację „simple-virilizing”- postać łagodna –95%. Wśród objawów
stwierdzane są tylko te, które wynikają z niedoboru kortyzolu i nadmiaru androgenów
(maskulinizacja noworodków żeńskich i przedwczesne dojrzewanie u chłopców)
2. Postać z utratą soli „salt-wasting”- ciężka – dochodzi w niej nie tylko do dużego
niedoboru kortyzolu, ale również do znacznego zmniejszenia syntezy aldosteronu. Dlatego
objawy chorobowe występują już w drugim tygodniu życia noworodka w postaci: zaburzeń
gospodarki wodno-elektrolitowej, utraty łaknienia, wymiotów, biegunek oraz wstrząsu
hipowolemicznego. U dziewczynek maskulinizacja narządów płciowych występuje już w
życiu płodowym, u chłopców narządy płciowe są prawidłowo wykształcone. W przypadku
braku leczenia dziecko umiera.
Badania dodatkowe
Podwyższony poziomu 17-OH- progesteronu w surowicy.

Niedobór 11B-hydroksylazy
Patogeneza
Niedobór kortyzolu i aldosteronu (dużo prekursora 11- dezoksykortykosteronu
zwiększającego ciśnienie)
Objawy kliniczne: wirylizacja, nadciśnienie tętnicze.
4. Hiperaldosteronizm
Aldosteron odpowiedzialny jest za zatrzymywanie sodu w ustroju oraz wydalanie potasu i
wodoru. Wyróżnia się: hiperaldosteronizm pierwotny (zespół Conna) i wtórny.
4a. Hiperaldosteronizm pierwotny
Definicja
Jest to schorzenie spowodowane gruczolakiem wydzielającym aldosteron, rakiem lub
przerostem warstwy kłębkowatej kory nadnerczy.
Patogeneza
Nadmiar produkowanego aldosteronu powoduje zatrzymanie sodu i wody w ustroju co
zwiększa objętość osocza. Aktywacja mechanoreceptorów plamki gęstej i przepływ jonów
sodu hamuje produkcję reniny.
Obraz kliniczny:
Do typowych objawów zespołu należy: nadciśnienie tętnicze (zatrzymanie sodu i wody) i
jego powikłania narządowe, hipokalemia i wynikające z niej zaburzenia(osłabienie
mięśniowe, zaparcia, zmiany zapisu EKG), wielomocz (zwyrodnienie wodniczkowe cewek
nerkowych), zwiększone pragnienie, zasadowica metaboliczna (parestezje, niekiedy objawy
tężyczki).
Badania dodatkowe:
1. niski poziom potasu,
2. zwiększone wydalanie potasu z moczem,
3. zwiększone stężenie aldosteronu w osoczu,
4. poziom sodu prawidłowy lub trochę podwyższony,
5. niska aktywność reninowa osocza (skutek nadmiernego wydzielania aldosteronu).
4b. Hiperaldosteronizm wtórny
Definicja
Hiperaldosteronizm wtórny wynika z nadmiernej produkcji aldosteronu spowodowanej
czynnikami pozanadnerczowymi. W zaburzeniu tym dochodzi do zwiększonej produkcji
reniny przez aparat przykłębuszkowy nerek.
Etiologia i patogeneza
Hiperaldosteronizm wtórny rozwija się w przypadku zmniejszonej przemiany aldosteronu
(niewydolność nerek, krążenia, marskość wątroby), guzów produkujących reninę,
niedokrwieniu nerek, spadku objętości wewnątrznaczyniowej (zespół nerczycowy, diuretyki),

Leczenie
W przypadku guzów stosuje się zabieg operacyjny. Przerost kory stanowi wskazanie do
stosowania leków przeciwnadciśnieniowych i antagonistów aldosteronu (spironolakton).
Choroby rdzenia nadnerczy
1.Guz chromochłonny nadnerczy (Pheochromocytoma)
Definicja
Jest to nowotwór wywodzący się z utkania chromochłonnego produkujący katecholaminy. W
80% guz znajduje się w rdzeniu nadnerczy, rzadziej w okolicy aorty i tętnic biodrowych
wspólnych. Guzy pozanadnerczowe znajdujące się powyżej przepony wydzielają tylko
noradrenalinę.
Patogeneza
Wydzielane w nadmiarze katecholaminy oddziałują presyjnie na układ krążenia, mają wpływ
na gospodarkę węglowodanową i tłuszczową.
Obraz kliniczny choroby
Głównym objawem guza chromochłonnego jest nadciśnienie. Gdy guz produkuje głównie
adrenalinę u chorych występuje nadciśnienie napadowe, gdy zaś noradrenalinę nadciśnienie
utrwalone (na które nakładają się przełomy nadciśnieniowe). W przypadku przełomu
wzrostowi ciśnienia towarzyszy przyspieszona akcja serca, ból głowy, wymioty, bladość
powłok, drżenie rąk, poty, lęk, rozszerzone źrenice. Napad często poprzedzony jest uciskiem
na jamę brzuszną np. przy wysiłku fizycznym, defekacji, kucaniu, a nawet podczas badania
lekarskiego. We krwi chorych wzrasta poziom glukozy i kwasów tłuszczowych. W
przypadku, gdy guz produkuje adrenalinę u chorych dochodzi często do ortostatycznych
spadków ciśnienia.
Badania dodatkowe:
1.oznaczenie stężenia katecholamin we krwi oraz w moczu (znacznie podwyższone ich
stężenie występuje bezpośrednio po napadzie),
2.oznaczanie stężenia metabolitów katecholamin w moczu (wzrost stężenia kwasu
wanilinomigdałowego, metoksyadrenaliny, metoksynoradrenaliny),
3.- badania pomocne w ustaleniu umiejscowienia guza ( TK, USG, MRI, scyntygrafia,
arterografia).
Leczenie
Leczenie polega na operacyjnym usunięciu guza.
Niekiedy u chorych współistnieją zaburzenia ze strony wielu gruczołów dokrewnych. Mogą
one przebiegać z ich niedoczynnością ( zespoły autoimmunologicznego uszkodzenia wielu
narządów dokrewnych) lub nadczynnością (gruczolokowatośc wewnątrzwydzielnicza).
Zespoły autoimmunologicznego uszkodzenia wielu gruczołów wydzielania wewnętrznego
Typ I (zespół Blizzarda): objawy występują u dzieci
- choroba Addisona
- niedoczynność przytarczyc
- kandydoza skóry i błon śluzowych

- upośledzenie czynności limfocytów
Typ II (zespół Carpentera)
- choroba Addisona
- cukrzyca typ I
- choroba Hashimoto
Zespół Schmidta = ch. Addisona + ch. Hashimoto
Typ III
-autoimmunologiczna choroba tarczycy
-cukrzyca typ I
- zanikowe zap. Błony śluzowej żołądka lub niedokrwistość
złośliwa
Gruczolakowatość wewnątrzwydzielnicza ( adenomatosis multiglandularis), MEN
(multiple endocrine neoplasia)
1.Zespół MEN I (Wermera) gruczolak przytarczyc, guz trzustki (gastrinoma lub insulinoma),
gruczolak przysadki
2.Zespół MEN II (Sipple`a)
-podtyp II a rak rdzeniasty, gruczolak przytarczyc (może wtórny do wzrostu kalcytoniny),
pheochromocytoma
- podtyp II b zamiast gruczolaka przytarczyc neurozwojaki i nerwiaki i inne zaburzenia
somatyczne
Temat VIII. Patologia przytarczyc
Iza Iwan- Ziętek
1. Regulacja gospodarki wapniowo- fosforanowej
U dorosłego człowieka 99% wapnia znajduje się w kościach i zębach. Pozostała część tego
pierwiastka występuje w tkankach miękkich oraz we krwi. Prawidłowy poziom wapnia w
osoczu krwi wynosi 2,25-2,75 mmol/l z czego około 50 % stanowi wapń zjonizowany, 40%
związany z białkami i 10% tzw. wapń kompleksowy (związany z jonami cytrynianowym,
fosforanowym, wodorowęglanowym ). Wiązanie białka z albuminami zależy od pH.
Zwiększenie zasadowości krwi obniża poziom wapnia zjonizowanego, w kwasicy zaś
dochodzi do dysocjacji kompleksów wapnia z albuminą, co powoduje wzrost wapnia
zjonizowanego w surowicy. Stwierdzono, że tylko wapń zjonizowany (wolny) jest formą
aktywną biologicznie, która odgrywa istotną rolę w procesie krzepnięcia krwi, stabilizacji
potencjału błonowego, skurczu mięśni. Poziom wapnia w surowicy regulowany jest przez
układ dokrewny, głównie przez parathormon (PTH), kalcytoninę oraz aktywną postać
witaminy D3 - 1,25-dwuhydroksycholekalcyferol (1,25(OH)2D3). Wymienione hormony
wpływają również na stężenie fosforanów, które stanowią ważną składową hydroksyapatytów
znajdujących się w kościach. Ponadto fosfor tworzy jeden z układów buforowych krwi,
wchodzi w skład kwasów nukleinowych, nukleotydów, związków wysokoenergetycznych
(ATP) oraz błon komórkowych..
Parathormon (PTH) jest hormonem peptydowym syntetyzowanym w komórkach
głównych przytarczyc. Podstawowy bodziec do wydzielania PTH stanowi obniżenie stężenia
wapnia zjonizowanego w surowicy. Najważniejszymi narządami docelowymi tego hormonu

są nerki i kości. Na przewód pokarmowy działa pośrednio przy współudziale witaminy D3. W
nerkach parathormon zwiększa wydalanie fosforanów. Powstała hipofosfatemia pobudza
nerkową hydroksylazę do syntezy 1,25 dihydroksycholekalcyferolu, który nasila wchłanianie
wapnia z przewodu pokarmowego. PTH nasila również wchłanianie zwrotne wapnia w
nerkach jednak w pierwotnej nadczynności przytarczyc, gdy poziom parathormonu jest
wysoki , podwyższony ładunek filtrowanego Ca powoduje w konsekwencji zwiększone
wydalanie wapnia w moczu. W układzie kostnym PTH w stężeniach fizjologicznych stymuluje
w równym stopniu osteoblasty i osteoklasty. Jednak w przypadku dużych stężeń tego
hormonu i obecności wit. D dochodzi do zwiększonej resorpcji kości, co powoduje
uwolnienie znacznych ilości wapnia zjonizowanego do krwi. Działanie PTH doprowadza w
efekcie do hiperkalcemii.
Prowitamina D3 powstaje w skórze z 7-dehydrocholesterolu pod wpływem
promieniowania ultrafioletowego albo pochodzi ze spożytego pokarmu.Ulega hydroksylacji
najpierw w wątrobie przy węglu 25, a potem w nerkach przy węglu 1. Powstający w ten
sposób bardzo aktywny metabolit 1, 25 (OH)2-D3 nazywamy jest kalcytriolem. W jelicie
cienkim powoduje on zwiększone wchłanianie wapnia, w przytarczycach zmniejszone
wydzielanie PTH, zaś w kośćcu nasila wbudowywanie wapnia (w stężeniach fizjologicznych).
W wysokich stężeniach nasila aktywność osteoklastów, a tym samym zwiększają resorpcję
Ca i P.
Hormonem obniżającym poziom wapnia w surowicy jest kalcytonina., wydzielana
przez komórki C tarczycy. Hamuje ona resorpcję Ca i P z tkanki kostnej.
2. Pierwotna nadczynność przytarczyc
Definicja
Pierwotna nadczynność przytarczyc jest to zespół zaburzeń gospodarki wapniowo-
fosforanowej i metabolizmu kostnego wywołany zwiększoną produkcją parathormonu (PTH)
oraz brakiem sprzężenia zwrotnego pomiędzy poziomem wapnia w surowicy a sekrecją PTH
Częstość występowania
Badania epidemiologiczne wskazują na występowanie 25 zachorowań na 100000 osób w
ciągu roku.
Etiologia
Najczęściej przyczyną pierwotnej nadczynności przytarczyc jest pojedynczy gruczolak (80%),
rzadziej mnogie gruczolaki, przerost lub rak gruczołów przytarczycznych, które wytwarzają
nadmierne ilości PTH.
Patogeneza
W przypadku, gdy stężenie PTH w ustroju człowieka przekracza wartości fizjologiczne
dochodzi do zwiększonej osteoklastycznej resorpcji tkanki kostnej, zwiększonego
wchłaniania wapnia w jelicie cienkim oraz nasilonego wydalania fosforanów z moczem i
zwiększonego zwrotnego wchłaniania wapnia w cewkach nerkowych.
Obraz kliniczny
W początkowym okresie przebieg schorzenia jest w większości przypadków bezobjawowy, a
jedyną manifestacją choroby są okresowo występujące odchylenia od normy w poziomie Ca.
Również rozpoznanie schorzenia w późniejszym okresie może sprawiać trudności, gdyż u
pacjentów często występują izolowane objawy np. tylko kamica nerkowa, czy choroba
wrzodowa bądź też mało charakterystyczne dolegliwości.

Do typowych objawów pierwotnej nadczynności przytarczyc jako skutku nadmiernej
ilości PTH należą:
- zmiany w układzie kostnym, które mogą prowadzić do złamań w wyniku uogólnionej lub
miejscowej osteoporozy, ogniskowych ubytków w postaci osteitis fibrosa cystica; dla
pierwotnej nadczynności przytarczyc charakterystyczna jest resorpcja podokostnowa; chorzy
często skarzą się na bóle kostne, stawowe;
- zmiany w nerkach w postaci kamicy, tubulopatii lub wapnicy, co sprzyja zakażeniom i
może prowadzić do mocznicy; objawem charakterystycznym jest wielomocz
wazopresynooporny;
- zmiany w przewodzie pokarmowym powodują zwolnienie motoryki jelit, co powoduje
nudności, wymioty, zaparcia; u 10% chorych występują wrzody żołądka, dwunastnicy
(podwyższony poziom Ca zwiększa wydzielanie gastryny i kwasu solnego), a niekiedy
również zapalenie trzustki (wapń aktywuje trypsynę);
- zmiany w układzie nerwowo-mięśniowym w postaci osłabienia i zaników mięśniowych;
- ponadto mogą występować objawy neuropsychiatryczne
Badania dodatkowe
1.
hiperkalcemia
2.
wzrost aktywności fosfatazy zasadowej we krwi
3.
hipofosfatemia (50%)
4.
podwyższony poziom PTH we krwi
5.
USG, TK, scyntygrafia – w celu zlokalizowania patologicznej tkanki przytarczyc
Leczenie
Leczenie polega na chirurgicznym usunięciu zmiany chorobowej tj. tkanki wydzielającej
nadmierną ilość PTH.
3. Wtórna nadczynność przytarczyc
Definicja
Jest to schorzenie, w którym zwiększone wydzielanie PTH jest odpowiedzią na hipokalcemię.
Etiologia
Do przyczyn powodujących wtórną nadczynność przytarczyc należy:
1.niewydolność nerek, w której dochodzi do spadku syntezy aktywnego kalcytriolu,
wzrostu stężenia fosforanów i spadku poziomu wapnia zjonizowanego w surowicy krwi
(hipokalcemia pobudza przytarczyce),
2.
niedostateczne wchłanianie wapnia lub witaminy D z przewodu pokarmowego,
3.
niedostateczna podaż wapnia w diecie,
4.
zwiększona utrata wapnia z moczem,
5.
nadmiar kalcytoniny (zwiększone odkładanie wapnia w kościach),
6.
marskość wątroby (zaburzona hydroksylacja witaminy D3),
Objawy kliniczne
Objawy wynikające ze wzrostu PTH są podobne do tych, które występują w pierwotnej
nadczynności przytarczyc; wykazują one różne nasilenie zależnie od choroby podstawowej.
Badania dodatkowe:
1. u chorych z niewydolnością nerek występuje: podwyższony poziom fosforanów, mocznika
kreatyniny oraz PTH w surowicy, poziom wapnia jest obniżony lub na dolnej granicy normy;

2. u chorych z prawidłową czynnością nerek występuje: wysoki poziom PTH, prawidłowe
stężenie fosforanów, małe stężenie wapnia w surowicy.
4. Niedoczynność przytarczyc
Definicja
Jest to zespół objawów klinicznych spowodowany brakiem przytarczyc lub niedoborem
parathormonu.
Etiologia
Najczęstszą przyczyną jest nie zamierzone wycięcie przytarczyc w czasie operacji tarczycy.
Rzadziej powstaje na skutek uszkodzenia tych gruczołów jodem promieniotwórczym czy
promieniami rtg, w wyniku procesów autoimmunologicznych, urazów mechanicznych szyi,
zapaleń tarczycy oraz zaburzeń wrodzonych.
Patogeneza
Powyższe przyczyny doprowadzają do niedoboru parathormonu, co przyczynia się do spadku
poziomu wapnia, wzrostu fosforanów w surowicy oraz zahamowania syntezy 1,25(OH)2D3.
Hipokalcemia nasilając przepuszczalność błon komórkowych, zwiększa pobudliwość struktur
nerwowo-mięśniowych powodując napady tężyczki. Zmiana stosunku stężeń wapnia do
fosforu w płynie zewnątrzkomórkowym sprzyja natomiast wytrącaniu się kryształów
fosforanów wapnia w tkankach miękkich np. w OUN.
Obraz kliniczny
U pacjentów z niedoczynnością przytarczyc może wystąpić:
1. tężyczka jawna – napad tężyczkowy charakteryzuje się drgawkami przy zachowanej
świadomości, kurcze mięśni są symetryczne i bolesne; obejmują one początkowo mięśnie rąk
(ręka położnika), potem kolejno przedramiona, ramiona, twarz (skurcz mięśni powiek, „usta
karpia”), klatki piersiowej i kończyn dolnych (ustawienie końsko-szpotawe);
2. tężyczka utajona – typowe dla niej są dodatnie objawy Chvostka (po uderzeniu nerwu
twarzowego w okolicy policzka występuje podciągnięcie kącika ust) i Trousseau (po
jednominutowym ucisku ramienia mankietem manometru dłoń układa się w kształcie „ręki
położnika”); u pacjentów łatwo wywołać napad przez hiperwentylację;
3. równoważniki tężyczkowe – kurcz mięśni powiek, głośni, oskrzeli, skurcz naczyń
wieńcowych czy mózgowych;
4. zmiany troficzne narządowe – zaćma (postępująca bardzo szybko), zwapnienia jąder
podstawy mózgu, nieprawidłowości wzrostu włosów i paznokci.
W pierwotnej niedoczynności przytarczyc może wystąpić nadpobudliwość , bezsenność,
depresja, objawy pozapiramidowe. Gdy schorzenie wystąpi u dzieci, pacjenci osiągają niski
wzrost oraz często mają niedorozwój umysłowy.
Badania dodatkowe
1.
niski poziom wapnia we krwi i moczu
3.
podwyższony poziom fosforanów w surowicy (zwykle)
4.
śladowe stężenie PTH oraz 1,25(OH)2D3 w surowicy
5.
rtg, TK – zwapnienia w OUN
Leczenie
Przy objawach tężyczki podaje się dożylnie roztwory soli wapnia. W celu normalizacji
poziomu wapnia podaje się przewlekle sole wapnia i aktywne metabolity wit. D.

Temat IX. Osteoporoza
Iza Iwan- Ziętek
Definica
Osteoporoza jest układową chorobą kośćca przebiegającą ze zmniejszeniem masy kostnej i
zaburzeniami mikroarchitektury tkanki kostnej, charakteryzującą się zwiększonym ryzykiem
złamań.
Częstość występowania
Zaliczana jest do chorób społecznych tzn. charakteryzuje ją wysoka zapadalność,
występowanie groźnych dla życia powikłań powodujących obciążenie ekonomiczne chorych i
społeczeństwa.Wśród czynników etiologicznych ważna jest grupa czynników związanych ze
stylem życia.
Patogeneza
Zwykle nie daje objawów do chwili złamania kości (najczęściej nadgarstka lub trzonu
kręgowego), występujących po niewielkim urazie lub bez uchwytnej przyczyny. W ciągu
całego życia człowieka zachodzi proces remodelowania kości, czyli jej „odmładzania”,
przeciętnie od 2 do 10 % masy kostnej rocznie. Prawidłowo u młodych organizmów
aktywność kościotwórcza przewyższa aktywność kościogubną. Około 30 roku życia człowiek
osiąga szczytową masę kostną, po 40 r. ż. resorpcja przewyższa syntezę kości a tempo ubytku
masy kostnej jest indywidualne.
Do czynników ryzyka osteoporozy zaliczamy: wiek ( spadek masy kostnej wraz z
wiekiem), płeć (80% kobiety), czynniki genetyczne (rasa biała i żółta), niska masa kostna,
defekt syntezy kolagenu, czynniki związane ze stylem życia: niedobór w diecie Ca, P, białka ,
witaminy C i D, brak nasłonecznienia, niska aktywność fizyczna, palenie tytoniu,
nadużywanie alkoholu i kofeiny, czynniki hormonalne: niedobór estrogenów, androgenów,
nadczynność tarczycy, przytarczyc, hiperkortyzolemia a także choroby przewodu
pokarmowego, nerek, wątroby.
Podział
Osteoporoza może być uogólniona lub miejscowa (zawsze proces wtórny wywołany jest
znanym czynnikiem patogennym). Osteoporoza uogólniona jest najczęściej procesem
pierwotnym. Do pierwotnej osteoporozy zalicza się się: osteoporozę występującą u młodych
ludzi (bardzo rzadka postać) oraz osteoporozę pomenopauzalną (typ II) i osteoporozę starczą
(typ I).Osteoporoza typu II występuje u kobiet po 50 r. ż. na skutek niedoboru estrogenów i
dotyczy głównie kości beleczkowej. Typ I osteoporozy występuje u osób po 70 r ż. ( u kobiet
i mężczyzn) w wyniku niedoboru wapnia, witaminy D, braku ruchu oraz zaawansowanego
wieku a zmiany dotyczą zarówno części gąbczastej, jak i zbitej kości..
Obraz kliniczny
U chorych występują bóle kostne, złamania, zmniejszenie wzrostu, zmiany postawy ciała
(okrągłe plecy).

Badania dodatkowe
1. Badania laboratoryjne: morfologia, liczba leukocytów ze składem odsetkowym , OB.,
analiza dobowej zbiórki moczu ( Ca, kreatynina – wskaźniki resorpcji kości), poziom wapnia
fosforu, fosfatazy zasadowej (wskaźnik tworzenia kości), azotu mocznikowego , kreatyniny w
surowicy (ocena czynności nerek)p;
2.
W przypadku osteoporozy wtórnej: T3, T4, FT4 (w celu wykluczenia nadczynności
tarczycy) , elektroforeza białek (w celu wykluczenia szpiczaka mnogiego, w którym
stwierdza się obecność białka monoklonalnego), stężenie C-końcowych segmentów
immunoreaktywnego PTH;
3.
Pomiar gęstości kości;
Najczęściej stosuje się absorpcjometrię promieniowania rtg o pojedynczej energii ( do
badania kości położonych bezpośrednio pod skórą np. kość piętowa, przedramienia) oraz o
podwójnej energii (do badania kości kręgosłupa i bliższego odcinka kości udowej).
Leczenie
U kobiet stosuje się terapię hormonalną. Ponadto chorych można leczyć witaminą D3,
preparatami wapnia, kalcytoniną, bifosfonianami.
Szersze omówienie problematyki osteoporozy zostanie zamieszczone w tomie II. .
Temat X. Patologia gruczołów płciowych
Iza Iwan-Ziętek
Z uwagi na obszerny zakres zagadnień dotyczących patologii gruczołów płciowych w
niniejszym opracowaniu zasygnalizowano tylko podstawowe zagadnienia a ich rozszerzenie
będzie przedmiotem studiów na wyższych latach lub samokształcenia.
1. Różnicowanie płciowe
Gonady są narządem parzystym.Występują one u kobiet w postaci jajników, natomiast u
mężczyzn jako jądra. Płeć człowieka determinowana jest przez wiele genów, znajdujących się
na chromosomach płciowych i autosomalnych. Główną rolę odgrywa jednak chromosom Y,
w którym obecny jest gen SRY (sex – determining region Y), odpowiedzialny za
wykształcenie się jąder. W przypadku jego braku rozwija się zarodek żeński. U płci męskiej z
przewodów sródnerczowych (przewody Wolffa) pod wpływem testosteronu wykształcają się
najądrza, nasieniowody i kanaliki nasienne. U płci żeńskiej, wskutek braku testosteronu,
przewody te zanikają, a jajowody, macica i górna część pochwy powstają z przewodów
okołośródnerczowych Müllera..
Różnicowanie zewnętrznych narządów płciowych męskich zależy od obecności
dihyrotestosteronu (powstaje z testosteronu pod wpływem 5 alfa reduktazy). W przypadku
jego braku powstają narządy płciowe zewnętrzne żeńskie. Pełne zróżnicowanie obu płci
dokonuje się w okresie pokwitania, które powinno rozpocząć się u dziewcząt przed 13 rokiem
życia, a u chłopców około 14 roku. . W populacji polskiej pierwsza miesiączka przypada
średnio na 13 r. ż. Jeśli pokwitanie wystąpi przed 8 r. ż. u dziewcząt i przed 9 r. ż. u
chłopców, mówimy o przedwczesnym dojrzewaniu.

2. Cykl miesiączkowy
U kobiet błona śluzowa macicy ulega cyklicznym zmianom przebiegającym przeciętnie w 28
dniowych odstępach czasowych. Wahania cyklu mogą się jednak mieścić w granicach 24-32
dni. Utrata krwi w czasie 3-5 dniowej miesiączki wynosi przeciętnie od 20 do 40 ml. Cykl
miesiączkowy składa się z dwóch faz rozdzielonych owulacją.
W pierwszej, folikularnej fazie, początkowo występuje krwawienie miesięczne, a
następnie pod wpływem FSH dochodzi do dojrzewania pęcherzyka Graafa oraz odnowy
błony śluzowej macicy. Owulacja, czyli pękniecie pęcherzka Graafa i uwolnienie komórki
jajowej, spowodowana jest wzrostem stężenia estradiolu, który na zasadzie sprzężenia
zwrotnego dodatniego powoduje gwałtowny wyrzut LH (wyrzut owulacyjny) Pozostała część
pęcherzyka Graafa (po uwolnieniu komórki jajowej) zwana ciałkiem żółtym, zaczyna
produkować progesteron.
W drugiej fazie cyklu, lutealnej dochodzi do spadku gonadatropin pod wpływem
wysokiego poziomu progesteronu i estradiolu. Równocześnie następuje wzrost i wydzielanie
śluzu przez rozrastające się gruczoły błony śluzowej macicy. Spadek LH i FSH powoduje
przekształcenie się ciałka żółtego w nieczynne hormonalnie ciałko białawe, zmniejszanie się
poziomu progesteronu i estrdiolu, co umożliwia wydzielanie gonadotropin i prowadzi do
złuszczania części czynnej błony śluzowej macicy.
3.Niepłodność
Pojęcie
Niepłodność ma miejsce wówczas, gdy po roku regularnego współżycia bez zabezpieczeń nie
dochodzi do poczęcia. W myśl Światowej Organizacji Zdrowia niepłodność jest chorobą
społeczną.
Częstość występowania i przyczyny
Dotyczy ona 20% małżeństw. Rozróżniamy niepłodność pierwotną i wtórną. Pierwotna
dotyczy par, w których kobieta nigdy nie była w ciąży, natomiast wtórna par, gdy kobieta
była kiedyś w ciąży. Uważa się, że w około 30% przypadków przyczyna niepłodności leży po
stronie mężczyzny, w 30%-po stronie kobiety, w 30% przypadków u obojga małżonków,
natomiast w 10% przyczyna jest nieznana (niepłodność idiopatyczna). Badanie niepłodności
należy prowadzić równocześnie u obojga partnerów. Częstą przyczyną niepłodności są
zaburzenia hormonalne.
4. Zaburzenia cyklu miesiączkowego
4a. Pierwotny brak miesiączki to jej brak u dziewczyny do 17 roku życia. Może to być
następstwem:
-
braku, niedorozwoju (zespół Turnera) i zaburzeń czynności jajników,
-
zespołu nadnerczowo-płciowego,
-
niedoczynności tarczycy, przysadki lub podwzgórza,
-
nieprawidłowej budowy narządów płciowych np. braku macicy lub zarośnięcia błony
dziewiczej.
Pierwotny brak miesiączki występuje również u osobników z żeńskim fenotypem ale męskim
kariotypem, w tzw. zespole feminizujących jąder ( zespół Morrisa). U tych chorych wskutek
niewrażliwości ustroju na androgeny mimo obecności jąder w ustroju rozwija się sylwetka
typu kobiecego (żeńskie zewnętrzne narządy płciowe, rozwinięte gruczoły sutkowe, ślepo

zakończona pochwa). Jeżeli jądra znajdują się w jamie brzusznej należy je usunąć, gdyż ze
względu na panującą tam temperaturę mogą ulec zezłośliwieniu.
4b.Wtórny brak miesiączki
Oznacza jej brak przez okres sześciu miesięcy u kobiety wcześniej miesiączkującej w wieku
prokreacyjnym.
Przyczyny
Wtórny brak miesiączki może być skutkiem procesów patologicznych jajników (guzy,
przedwczesne przekwitanie), nadnerczy (zespół Cushinga, guzy produkujące androgeny),
tarczycy, przysadki bądź podwzgórza. Najczęstszą jednak przyczyną wtórnego braku
miesiączki jest ciąża.(stan fizjologiczny) i zespół policystycznych jajników.
Zespół policystycznych jajników (PCO)
Częstość występowania
Zespół policystycznych jajników występuje u około 5% kobiet w wieku reprodukcyjnym.
Patogeneza
U chorych występuje zwiększony poziom androgenów, który jest prawdopodobnie
wynikiem zwiększonej stymulacji komórek osłonki pęcherzka Graafa przez podwyższony
poziom LH.
Objawy kliniczne
Chore są często otyłe i wykazują cechy hiperandrogenizmu (hirsutyzm,, trądzik,
łysienie androgenowe). Miesiączki u tych kobiet występują u nich nieregularnie, cykle są
nieowulacyjne, co uniemożliwia im zajście w ciążę. Występuje powiększenie jajników z
pogrubiałą osłonką białawą (utrudniającą dodatkowo owulację) oraz liczne drobne torbielki.
Badania dodatkowe
1. badania hormonalne - stwierdza się podwyższony poziom LH, zaburzenie proporcji
LH/FSH (najczęściej powyżej 2:1, podczas gdy prawidłowy ich stosunek wynosi około 1:1),
2. hiperinsulinemia z insulinoopornością,
3. podwyższone stężenie androgenów (testosteron, androstendion).
4c. Inne zaburzenia cyklu miesiączkowego
Mogą polegać na częstym miesiączkowaniu (cykl krótszy niż 21-24 dni), rzadkim (cykl
dłuższy niż 32 dni), obfitym (krwawienie trwające 5-7 dni, z dużą utratą krwi), skąpym (1-
2dniowe, mało obfite) lub bolesnym miesiączkowaniu.
5. Hipogonadyzm – niedoczynność gonad
Hipogonadyzm dzielimy na pierwotny (uszkodzone jądra lub jajniki) i wtórny (uszkodzenie
przysadki lub podwzgórza). W hipogonadyzmie pierwotnym występuje podwyższone stężenie
gonadotropin i niski poziom hormonów gonadalnych, zaś w hipogonadyzmie wtórnym
zarówno poziom gonadotropin jak i hormonów gonadalnych jest niski.
5a. Hipogonadyzm u kobiet
Hipogonadyzm pierwotny
Dysgenezja gonad tj. obecność szczątkowych jajników pozbawionych gonocytów ,
charakteryzuje się żeńskim fenotypem, pierwotnym brakiem miesiączki, niedorozwojem II i

III rzędowych cech płciowych. Najczęstszą postacią dysgenezji gonad jest zespół Turnera
(45XO).
Objawy kliniczne
Dzieci z tym zespołem mają niską wagę urodzeniową, małą długość ciała, luźny fałd skóry na
szyi oraz obrzęknięte dłonie i stopy. Często również występuje u nich ubytek przegrody
międzykomorowej serca, zwężenie cieśni aorty oraz wady układu moczowego. Ze względu na
brak gonad dziewczęta nigdy nie osiągają dojrzałości płciowej, nie miesiączkują, są
niepłodne, mają niedorozwój zewnętrznych narządów płciowych oraz gruczołów sutkowych.
Osiągają niski wzrost, mają zaburzenia proporcji ciała, koślawość kolan i łokci, płetwistość
szyi, nisko schodzące owłosienie na karku, nieprawidłowości uzębienia oraz anomalie kostne.
Badania dodatkowe
U osób z zespołem Turnera występuje niski poziom estrogenów, wysoki gonadotropin,
ujemna chromatyna płciowa (70%).
-Przedwczesne wygaśnięcie jajników u kobiet uprzednio regularnie miesiączkujących,
rozwija się przede wszystkim w wyniku procesu autoimmunologicznego. Poza utratą
miesiączki dochodzi również u nich do zaniku jajników, II i III rzędowych cech płciowych
-Zespół jajników opornych na gonadotropiny jest to niewydolnośc jajników powstała
na skutek ich oporności na sygnały pochodzące z wyższych pięter układu dokrewnego.
Hipogonadyzm wtórny
Przyczyny:
Mogą to być: choroby podwzgórza, uogólniona niedomoga przysadki, odosobniony niedobór
gonadotropin wrodzony lub nabyty (po operacjach, po urazach), hiperprolaktynemia,
jadłowstręt psychiczny.
Zaburzenia ze strony gonad mogą rozwinąć się również u chorych z zaburzeniami
tarczycy i nadnerczy.
5b. Hipogonadyzm u mężczyzn
Patogeneza
Objawy występujące w hipogonadyzmie są następstwem niedoboru androgenów.
Objawy kliniczne
W przypadku, gdy proces patologiczny rozpocznie się przed okresem dojrzewania, u chorego
pojawiają się następujące objawy: niedorozwój narządów płciowych zewnętrznych,
niepłodność, nikły rozwój mięśni, brak mutacji, zarostu na twarzy, szczątkowe owłosienie
pachowe i łonowe, budowa eunuchoidalna (wysoki wzrost z długimi kończynami górnymi) i
odkładanie się tkanki tłuszczowej na biodrach i sutkach.
Jeżeli proces chorobowy rozpocznie się dopiero w wieku dojrzałym występuje:
niepłodność, impotencja, spadek libido, zanik zarostu, a niekiedy nawet ginekomasta i
mlekotok.
Hipogonadyzm pierwotny
Przyczyny:
Mogą to być:

a.
wrodzony brak jąder (anorchizm); stan ten należy różnicować z wnętrostwem (nie
zstąpienie jąder): prawidłowo jądra zstępują do moszny w 8 miesiącu życia
płodowego lub tuż po urodzeniu;
b.
zespół Del Castillo (zespół samych komórek Sertolego); u chorych występuje
prawidłowa morfologia i czynność komórek Leydiga produkujących testosteron a brak
komórek plemnikotwórczych; patogeneza nie jest znana, podejrzewa się uraz,
zaburzenia autoimmunologiczne, badź spaczoną wędrówkę gonocytów do zawiązków
pierwotnej gonady; ze względu na to że II i III-rzędowe cechy płciowe są prawidłowo
rozwinięte zespół jest trudno rozpoznawalny;
c.
zespół Klinefeltera spowodowany jest nieprawidłowościami chromosomów płciowych
(kariotyp 47 XXY, mozaicyzm XXY/XY); zwykle jest rozpoznawany dopiero w
wieku dojrzałym, kiedy pacjent zgłasza się do lekarza z powodu bezpłodności; u
chorych występuje zeszkliwienie kanalików nasiennych, słabo rozwinięte wtórne
cechy płciowe, osteoporoza, rozedma płuc, ginekomasia (u 60%);
d.
wrodzone defekty biosyntezy testosteronu;
e. kastracja;
f.
choroby w przebiegu których dochodzi do zapalenia jąder (np. świnka, wirusy
Coxackie lub Herpes);
g.
przedwczesne przekwitanie męskie.
Czynność jąder może być również uszkodzona u mężczyzn w wyniku chorób
wenerycznych, poprzez żylaki powrózka nasiennego, wodniaki jąder, wnętrostwem, po
urazach jąder, chemioterapii, promieniowaniu rtg, jak również przy przegrzewaniu jąder
(zawodowi kierowcy, osoby noszące obcisłe ubrania z tworzyw sztucznych) czy stosowaniu
niektórych leków ( przeciwdepresyjne, uspakajające, nasercowe).
Hipogonadyzm wtórny
Przyczyny:
Hipogonadyzm wtórny może być spowodowany:
a. niedoborem gonadoliberyn - zespół Kallmana (hipogonadyzm skojarzony z utratą
węchu),
b.
izolowanym niedoborem gonadotropin,
c.
niedoborem LH (zespół płodnego eunucha),
d.
nowotworami podwzgórza, przysadki ( np. prolactinoma) i innymi ich
uszkodzeniami.
Objawy hipogonadyzmu występują również u mężczyzn z chorobami układowymi np.
marskością wątroby, mocznicą, cukrzycą oraz innymi chorobami endokrynologicznymi
(tarczycy, nadnerczy).
Badania dodatkowe:
1. badanie nasienia,
2. badania hormonalne:
3. poziom testosteronu, FSH, LH, prolaktyny,
4. testy dynamiczne z GnRH, hCG, kloimifenem (blokuje hamujący wpływ estrogenów na
wydzielanie LH),
5. badania chromosomalne,
6. biopsja jąder.

Powyżej wymieniono najczęściej wykonywane badania w przypadku hipogonadyzmu
u mężczyzn . Uzyskane w nich wyniki są różne, zależnie od przyczyny która go wywołała.
6. Klimakterium
Definicja, okresy
Jest to długotrwały i stopniowy proces zanikania czynności jajników wskutek braku
oddziaływania hormonalnego: podwzgórze – przysadka - jajnik, sterowanego przez centralny
układ nerwowy. Jest to okres między zdolnością prokreacyjną a starością występujący od 45
do 55 roku życia kobiet. W klimakterium można wyróżnić następujące podokresy:
a.
premenopauzalny (wstępny) występujący około 40 roku życia, bez wyraźnych
zaburzeń hormonalnych, jednak z podwyższonym poziomem gonadotropin
przysadkowych,
b.
perimenopauzalny (objawowy) występujący po 45 roku życia, charakteryzujący się
spadkiem liczby pęcherzyków Graafa w jajnikach, zanikiem ich wrażliwości na
stymulację przysadkową, obniżeniem poziomu estrogenów i progesteronu oraz
cyklami często bezowulacyjnymi. Pojawiają się wary (uderzenia gorąca),
przedłużające się obfite krwawienia oraz krótsze cykle miesięczne.
c.
menopauzy właściwej, czyli ostatniego krwawienia miesięcznego; w populacji
polskiej przypada ono przeciętnie między 48-50 rokiem życia.
d.
zanik miesiączki przed 45 rokiem określa się jako menopauzę przedwczesną, a
trwanie krwawienia po 55 roku życia menopauzą opóźnioną.
e.
pomenopauzalny (bez krwawień)- z obniżonym poziomem estrogenów, progesteronu i
podwyższonym stężeniem gonadotropin.
Objawy kliniczne
Okres klimakterium, a w nim właściwej menopauzy, wywołuje dolegliwości fizyczne,
psychiczne związane z obawą kobiet przed utratą pełnowartościowego życia i początkiem
starości. Występują w nim nocne poty, kołatanie serca, wary, bezsenność, pogarszanie
pamięci, lęki, utrata libido, zaburzenia uwagi, trudności w decydowaniu, spadek zapału do
działania, płaczliwość, wzrost zachorowań na chorobę wieńcową, osteoporoza, utrata
elastyczności skóry, przerzedzenie włosów, męskie owłosienie twarzy, zanik narządów
rodnych, stany zapalne pęcherza, wypadanie macicy, nie trzymanie moczu oraz zanikanie
gruczołów sutkowych.
Leczenie
Większości objawów można zapobiegać stosując wczesną hormonalną terapię zastępczą.
7. Andropauza
Jest to okres u mężczyzn objawiający się niedoborem testosteronu. Badania wykazały, że od
40 roku życia stężenie testosteronu zmniejsza się o 1% rocznie. Andropauza przebiega
odmiennie niż menopauza u kobiet, u których wygaśnięcie jajników następuje około 50 roku
życia. U mężczyzn nie ma tak gwałtownych i nieodwracalnych zmian w funkcji komórek
Leydiga, a zatem nie ma też przyczyn do ustania czynności hormonalnej jąder i funkcji
rozrodczych. Zmniejszanie się stężenia testosteronu powoduje większą zapadalność na
choroby układu krążenia, otyłość, zaburzenia gospodarki węglowodanowej i lipidowej,
osteoporozę, depresję, zmniejszenie poczucia własnej wartości, zaburzenia erekcji
zmniejszenie libido, zaburzenia snu.
Leczenie hormonalne

Temat XI. Patogeneza cukrzycy
Wanda Drewniak
1. Homeostaza glukozy
Utrzymanie równowagi między dopływem glukozy do krwi a jej zużyciem jest możliwe w
ustroju dzięki współdziałaniu wielu złożonych mechanizmów regulacyjnych. Prawidłowa
glikemia zależy od stopnia absorbcji glukozy w jelitach, zużycia oraz produkcji w wątrobie i
nerkach, a także jej wykorzystania przez tkanki obwodowe.
Do krwi glukoza jest dostarczana przez przewód pokarmowy wraz z pożywieniem.
Drugim źródłem glukozy jest wytwarzanie jej w wątrobie i w nerkach w procesie
glikoneogenezy. Procesem przeciwstawnym jest glikoliza, która stanowi główny szlak
katabolizmu glukozy. Zużycie glukozy przebiega we wszystkich komórkach ustroju i może
odbywać się zarówno w warunkach tlenowych jak i beztlenowych.
Decydujące znaczenie w utrzymaniu prawidłowego poziomu glikemii we krwi
/normoglikemii/ mają dwa hormony trzustkowe insulina i glukagon. Synteza tych hormonów
odbywa się w endokrynnej części trzustki, którą stanowią wyspy Langerhansa. Insulina jest
syntetyzowana w komórkach
β
stanowiących około 80% wszystkich komórek wysp i
zlokalizowanych w ich części centralnej. W komórkach
α
położonych obwodowo jest
syntetyzowany glukagon .
Cząsteczka insuliny zawiera 51 aminokwasów i składa się z dwóch łańcuchów A i B
połączonych wiązaniami dwusiarczkowymi. Wydzielana jest z trzustki do układu żyły
wrotnej w sposób ciągły zapewniając stałe podstawowe stężenie hormonu. Glukoza we krwi
dopływającej do trzustki jest najważniejszym i najsilniejszym czynnikiem zwiększającym
uwalnianie insuliny.
Charakterystyczną cechą odpowiedzi komórek
β
na bodziec glukozowy jest
dwufazowość wydzielania insuliny. Profil jej sekrecji składa się z fazy początkowej szybkiej
– tj. szybkiego wyrzutu insuliny i następującej po niej fazy drugiej powolnej – stopniowego
narastania wydzielania hormonu. Pierwsza faza odpowiada wyrzutowi insuliny zdeponowanej
w ziarnistościach, druga sekrecji insuliny syntetyzowanej de novo w komórkach
β
.
Utrzymanie prawidłowego poziomu glikemii we krwi /normoglikemii/ wymaga
ścisłego współdziałania w ustroju trzech głównych zjawisk w przemianie węglowodanów
zależnych od insuliny:
1.
pobudzania wydzielania insuliny w komórkach
β
trzustki,
2.
pobudzania przez insulinę wychwytu glukozy przez tkanki obwodowe głównie przez
mięśnie,
3.
hamowania przez insulinę wytwarzania glukozy endogennej czyli nowotworzenia glukozy
z aminokwasów w tkankach, głównie w wątrobie /glukoneogeneza/.
Działanie biologiczne insuliny obejmuje wpływ na przemianę węglowodanową, tłuszczową i
białkową.
Wpływ insuliny na metabolizm węglowodanów.
1. Insulina umożliwia transport glukozy do tkanek insulinozależnych /tkanka tłuszczowa,
mięśnie szkieletowe, mięsień sercowy, fibroblasty/. Transport glukozy do wnętrza komórek
tych tkanek następuje przy pomocy swoistych przenośników białkowych glukozy /receptorów/
głównie GLUT- 4 /Glukose Transporter/ obecnych w błonie komórkowej i cytoplazmie.
Wyróżnia się również receptory GLUT-1, GLUT - 2, GLUT-3, GLUT-5 niezależne lub w

niewielkim stopniu zależne od insuliny. Szczególnie ważną grupę stanowią przenośniki
GLUT-4.
2. Insulina zwiększa wewnątrzkomórkowe zużycie glukozy w kilku szlakach metabolicznych:
-
wzmaga się glikoliza - główny szlak katabolizmu glukozy, w toku którego glukoza ulega
przemianie do pirogronianu,
-
w cyklu kwasów trikarboksylowych /cykl Krebsa/ zwiększa się wytwarzanie acetylo –CoA
na drodze pobudzania oksydacyjnej dekarboksylacji pirogranianu,
-
w cyklu kwasu cytrynowego acetylo – CoA ulega wzmożonemu utlenieniu,
-
w cyklu pentozofosforanowym /pentozowym/ glukoza ulega nasilonemu utlenianiu do
dwutlenku węgla.
3.
Hamuje nowotworzenie glukozy z aminokwasów /glukoneogeneza /. Insulina jest silnym
i szybko działającym inhibitorem produkcji glukozy. Poprzez ograniczenie
lipolizy/hydrolizy tłuszczów /i proteolizy /rozpadu białka/ insulina redukuje ilość
substratu do syntezy glukozy.
4.
Zwiększa wytwarzanie glikogenu /zapasowego materiału energetycznego/.
W zakresie metabolizmu tłuszczów insulina powoduje:
1. zwiększenie syntezy kwasów tłuszczowych w adipocytach /komórki tłuszczowe/ z acetylo
– CoA,
2. nasilenie estryfikacji kwasów tłuszczowych do trójglicerydów,
3. zmniejszenie poziomu wolnych kwasów tłuszczowych we krwi na drodze hamowania
lipolizy wewnątrzkomórkowej poprzez zmniejszenie aktywności lipazy lipoproteinowej w
tkance tłuszczowej. Głównym zadaniem insuliny w tkance tłuszczowej jest szybkie
przekształcenie glukozy w tłuszcz i utrzymanie jego zapasu.
W zakresie gospodarki białkowej, insulina jest hormonem anabolicznym, zwiększającym
syntezę i hamującym rozpad białka, ponieważ:
1.
zwiększa syntezę białka w wyniku nasilenia transportu aminokwasów do wnętrza komórki,
2.
zmniejsza rozpad białka poprzez hamowanie nowotworzenia glukozy z aminokwasów.
Drugim bardzo ważnym hormonem utrzymującym homeostazę glukozy jest glukagon.
Metaboliczny wpływ glukagonu obejmuje przemianę węglowodanów, tłuszczów i białek.
Przemiana węglowodanów
Glukagon powoduje rozpad glikogenu w wątrobie i zwiększa oddawanie glukozy do krwi, co
prowadzi do hiperglikemii.
Przemiana tłuszczów
Glukagon działa lipolitycznie w tkance tłuszczowej i w wątrobie. Poprzez aktywację lipazy
trójglicerydowej powoduje uwalnianie kwasów tłuszczowych i ich utlenienie.
Przemiana białek
Glukagon nasila glukoneogenezę (tworzenie glukozy z białek), co potęguje hiperglikemię
spowodowaną rozpadem glikogenu w wątrobie. Działanie tego hormonu jest ukierunkowane
na zapewnienie zaopatrzenia energetycznego tkanek poprzez stały dopływ glukozy do mózgu
i innych tkanek glukozależnych.
2. Definicja.

Terminem cukrzyca (diabetes mellitus) określa się grupę chorób metabolicznych, które
charakteryzują się podwyższonym stężeniem glukozy /hiperglikemią/ we krwi. Jest to wynik
nieprawidłowego wydzielania insuliny przez komórki
β
trzustki co prowadzi do :
a.
niedoboru insuliny różnego stopnia aż do całkowitego jej braku,
b.
i/lub nieprawidłowego działania tego hormonu.
3. Etiologiczna klasyfikacja cukrzycy.
W ciągu ostatnich czterdziestu lat klasyfikacja cukrzycy zmieniała się kilkakrotnie zależnie
od przyjmowanych kryteriów podziału.
Aktualna klasyfikacja została opracowana i opublikowana w 1999 r. w Genewie przez
Komitet Ekspertów WHO z wykorzystaniem wcześniejszych z 1997 r. propozycji
Amerykańskiego Towarzystwa Diabetologicznego /American Diabetes Association – ADA/
Zasadnicze postacie uwzględnione w tym podziale to:
1.
Cukrzyca typu 1.
Cukrzyca insulinozależna /Insulin – Dependent Diabetes Mellitus – IDDM/
2.
Cukrzyca typu 2.
Cukrzyca insulinoniezależna /Non Insulin – Dependent Diabetes Mellitus – NIDDM/
3.
Cukrzyca ciężarnych.
4.
Inne specyficzne typy cukrzycy.
4. Etiopatogeneza cukrzycy typu 1.
Właściwe leczenie cukrzycy jest oparte na zrozumieniu jej patogenezy.
Zatem konieczne jest przybliżenie mechanizmów odpowiedzialnych za upośledzenie
homeostazy glukozy, które są odrębne w cukrzycy typu 1 i typu 2.
Cukrzyca typu 1 jest następstwem przewlekłego autoimmunologicznego niszczenia
komórek
β
wysp trzustki w wyniku predyspozycji genetycznej i działania czynników
środowiskowych. Predyspozycja genetyczna jest zakodowana w obrębie kompleksu genów
układu HLA zlokalizowanego na ramieniu krótszym chromosomu 6p 21.
W procesie niszczenia komórek B biorą udział elementy odporności komórkowej,
makrofagi, limfocyty T cytotoksyczne/ supresorowe, limfocyty T pomocnicze oraz
wydzielane przez nie mediatory: cytokiny i wolne rodniki. W trzustkach chorych, którzy
umierają w krótkim czasie po ujawnieniu się cukrzycy stwierdza się naciek wysp
trzustkowych przez limfocyty T i B oraz makrofagi /insulinitis/ ze zniszczeniem większości
komórek
β
wydzielających insulinę, przy zachowaniu komórek
α
syntetyzujących glukagon.
W procesie tym dochodzi również do aktywacji immunologicznej odpowiedzi
humoralnej z wytworzeniem szeregu przeciwciał. W obrębie komórek
β
znajduje się szereg
antygenów, które mogą stymulować powstawanie przeciwciał. Autoprzeciwciała są
wskaźnikami humoralnymi destrukcji komórek
β
.
Największe znaczenie mają stwierdzane we krwi chorych przeciwciała:
-
ICA /Islet Cell Anitibody/- przeciwwyspowe,
-
IAA/Insulin Auto- Antibody/ - przeciwinsulinowe,
-
anty-GAD /anti- Glutamic Acid Decarboxylase/- przeciw dekarboksylazie kwasu
glutaminowego,
-
anty –IA-2 /antibodies/- przeciw fosfatazie tyrozyny.
W historii naturalnej cukrzycy typu 1 można wyróżnić kilka faz rozwoju. Pierwszą
fazę stanowi okres predyspozycji genetycznej. W drugiej fazie zaczyna się toczyć proces
destrukcji komórek
β
na skutek przerwania tolerancji immunologicznej wobec własnych
antygenów. Proces ten zostaje uruchomiony w wyniku zadziałania różnych czynników m.in.
infekcyjnych /głównie wirusy/, składników pokarmowych, substancji toksycznych. W tym
okresie destrukcja komórek
β
pozostaje jeszcze bez wpływu na równowagę biochemiczną,

ponieważ zdolność trzustki do sekrecji insuliny nadal znacznie przewyższa zapotrzebowanie
organizmu. W sytuacji, gdy destrukcja komórek
β
przekracza 80%, rozpoczyna się obniżona
sekrecja insuliny.
W dalszym etapie postępu destrukcji komórek
β
pojawia się nieprawidłowa
odpowiedź na doustną stymulację glukozą i łatwo dochodzi do trwałego zaburzenia
homeostazy glukozy. Stan ten na krótko wyprzedza kliniczne ujawnienie się cukrzycy, która
rozwija się przy zniszczeniu około 90% komórek
β
. Destrukcja może przebiegać w różnym
tempie, dlatego obserwuje się okresy remisji choroby. Proces może trwać kilka, kilkanaście
tygodni czasem miesięcy. Najczęściej mija nie dłużej niż dwa lata od ujawnienia się choroby
do całkowitego zniszczenia komórek
β
.
5. Etiopatogeneza cukrzycy typu 2.
Cukrzyca typu 2 jest zróżnicowanym zespołem, w którym hiperglikemia jest wywołana w
dwojaki sposób:
1. poprzez spadek uwalniania insuliny z komórek
β
trzustki w odpowiedzi na bodziec
glukozowy,
2. wskutek oporności tkanek obwodowych na działanie insuliny /insulinooporność/.
W cukrzycy typu 2 najczęściej od początku choroby stwierdza się upośledzenie syntezy i
wydzielania insuliny.
Zaburzenie wydzielania insuliny polega na obniżeniu szczytu wydzielania tego
hormonu w pierwszej fazie i opóźnionym wydzielaniu w drugiej fazie po bodźcu
glukozowym /posiłek /.
Równocześnie współistnieje insulinooporność t.j. oporność tkanek obwodowych na
działanie insuliny pobudzająca wychwyt glukozy przez komórki insulinozależne /głównie
mięśnie/ co prowadzi do hiperglikemii.
Podwyższony poziom glukozy stymuluje komórki
ββββ
trzustki do dalszego
kompensacyjnego zwiększenia wydzielania insuliny, co prowadzi do hiperinsulinemii.
Z czasem dochodzi do wyczerpania zdolności sekrecyjnej trzustki i obniżenia poziomu
insuliny we krwi.
Zmniejszenie wrażliwości tkanek na insulinę (insulinooporność) może rozwinąć się w
następstwie wielu czynników:
Czynniki genetyczne
Przyjmuje się, że defekty genetyczne stanowią czynnik predysponujący do cukrzycy typu 2.
Istnieje szereg mutacji genowych prowadzących do insulinooporności:
1. mutacja genu insuliny - w jej wyniku powstaje nieprawidłowa cząsteczka tego hormonu i
rozwja się hiperinsulinemia - mechanizm przedreceptorowy,
2. mutacja w obrębie genu receptora insuliny – mechanizm receptorowy,
3. mutacja genów transporterów glukozy, prowadząca do nieprawidłowej struktury i funkcji
transporterów glukozy – mechanizm poreceptorowy.
Czynniki immunologiczne
ze szczególną rolą interleukiny 1 i 6, a także tkankowego czynnika martwicy guza /TNF -
α
/.
Czynniki środowiskowe
głównie nieprawidłowe nawyki żywieniowe i mała aktywność fizyczna .
Skutki insulinooporności zależnie od rodzaju tkanki są następujące:

- komórki mięśni i tkanki tłuszczowej posiadają mniejsze zdolności zużycia glukozy, co prowadzi do
hiperglikemii,
- w komórkach wątroby /hepatocytach/ zostaje zniesione hamujące działanie insuliny na
produkcję wątrobową glukozy /glukoneogenezę /; zachowana zostaje jednak wrażliwość na
glukagon; w tym mechanizmie nasilona glukogeneza potęguje hiperglikemię wywołaną
zmniejszonym zużyciem.
- w komórkach
ββββ
wysp trzustki insulinooporność powoduje zniesienie hamującego działania
insuliny na jej sekrecję, skutkiem czego jest wystąpienie hiperinsulinemii.
Insulinooporność jest podstawową przyczyną hiperglikemii w cukrzycy typu 2.
W cukrzycy typu 2 hiperglikemia współistnieje z hiperinsulinemią.
Hiperglikemia powoduje :
-
toksyczne uszkodzenie komórek
ββββ
trzustki i wtórne zmniejszenie sekrecji insuliny,
-
obniżenie wrażliwości komórek
ββββ
na bodziec glukozowy,
-
pogłębienie insulinooporności tkanek obwodowych.
Wytwarza się błędne koło przyczyn i skutków. Upośledzenie wydzielania insuliny i
zmniejszenie zużycia glukozy w tkankach obwodowych powoduje narastanie hiperglikemii.
Hiperglikemia zaś poprzez toksyczne działanie na komórki
ββββ
wysp trzustki, wywołuje
zmniejszone wydzielanie insuliny przez te komórki. Toksyczne działanie glukozy na tkanki
obwodowe powoduje narastanie insulinooporności.
6. Kryteria laboratoryjne rozpoznania cukrzycy.
a. Kryteria rozpoznawania zaburzeń tolerancji glukozy.
Nowe kryteria rozpoznawania cukrzycy opracował i ogłosił w 1999r Komitet Ekspertów
Światowej Organizacji Zdrowia przy udziale Amerykańskiego Towarzystwa
Diabetologicznego /ADA /.
Rozróżnia się trzy kategorie zaburzeń gospodarki węglowodanowej przebiegającej z
hiperglikemią. Do nich należą
-
cukrzyca,
-
upośledzona tolerancja glukozy - impaired glucose tolerance /IGT/
-
upośledzona glikemia na czczo - impaired fasting glucose concentration /IFG/
Badanie glikemii w określonych warunkach umożliwia rozpoznawanie i zróżnicowanie
stanów hiperglikemii oraz zaliczenia ich do jednej z wymienionych kategorii.
Warunki badania glikemii:
- na czczo,
- badanie glikemii przygodnej /w czasie niezależnym od posiłku/,
- w dwugodzinnym teście doustnego obciążenia 75,0 g glukozy /OGTT/.
Test doustnego obciążenia 75,0 g glukozy /OGTT).
Badanie wykonuje się rano po 8-12 godzinach głodu. Badany w ciągu 5min. wypija 75
g glukozy rozpuszczonej w 300 ml płynu z dodatkiem substancji smakowej. W czasie testu
nie może wykonywać wysiłków ani palić tytoniu. Krew na badanie pobiera się w 0 i 120 min.
testu. Glukozę oznacza się w osoczu lub surowicy.
Wartości dla osocza krwi żylnej:
♦
glikemia na czczo:
-
prawidłowa glikemia na czczo: < 110 mg /< 6,1 mmol/l/

-
podwyższenie glikemii na czczo:
≥
110 mg /
≥
6,1 mmol/l/
ale <126 mg /< 7,0 mmol/l
- cukrzyca dwukrotne stwierdzenie glikemii na czczo:
≥
126 mg /
≥
7,0 mmol /l/
♦
glikemia w dwie godziny po doustnym podaniu 75.0 g glukozy.
- prawidłowa tolerancja glukozy: < 140 mg /< 7,8 mmol/l/
- upośledzenie tolerancji glukozy;
≥
140 mg /
≥
7,8 mmol/
ale < 200 mg / <11,1mmol/l/
- cukrzyca - dwukrotne stwierdzenie glikemii
≥
200 mg /
≥
11,1mmol/l/
♦
glikemia przygodna:
- prawidłowa glikemia przygodna: < 110 mg /< 6,1 mmol/l/
- podejrzenie nieprawidłowej glikemii przygodnej:
≥
110 mg /
≥
6,1 mmol/l/
ale < 200 mg /11,1mmol/l
- cukrzyca - dwukrotne stwierdzenie glikemii
≥
200 mg /
≥
11,1mmol/l/
W sytuacji wystąpienia typowych dolegliwości i objawów klinicznych cukrzycy rozpoznanie
może być ustalone przy /nawet pojedynczym /uzyskaniu wyniku glikemii
≥
200 mg - /
≥
11,1mmol/l / w osoczu krwi żylnej .
7. Objawy kliniczne cukrzycy
Kliniczne objawy cukrzycy zależą od patofizjologicznego działania hiperglikemii .
Hiperglikemia o niewielkim stopniu nasilenia może nie powodować typowych objawów
klinicznych, ale może być przyczyną nasilenia się cukrzycy i wystąpienia ciężkich
przewlekłych powikłań w postaci angiopatii i neuropatii cukrzycowej, patologii ciąży i
obniżenia odporności przeciwbakteryjnej.
Znacznie podwyższona glikemia doprowadza do:
-
glukozurii /cukromoczu/,
-
diurezy osmotycznej,
-
zaburzeń elektrolitowych,
-
ketonemii i ketonurii.
Cukromocz stwierdza się gdy, poziom glukozy we krwi przekracza próg nerkowy tj. przewyższa
maksymalną zdolność reabsorcji zwrotnej glukozy w nerkach. Przekroczenie stężenia glukozy we
krwi powyżej 180 mg /dl powoduje pojawienie się glukozy w moczu. Wydalanie glukozy z
moczem może odbywać się jedynie w odpowiedniej ilości wody.
Diureza osmotyczna pojawia się w przypadku, kiedy podwyższenie glikemii powoduje
hipermolalność płynu pozakomórkowego. Sprzyja to przemieszczaniu się wody z przestrzeni
wewnątrzkomórkowej do pozakomórkowej w celu utrzymania równowagi osmotycznej. Nasila to
wydalanie glukozy i wody przez nerki i w stanach szczególnej intensywności prowadzi do
odwodnienia ustroju.
Zaburzenia elektrolitów polegają na pojawieniu się hiponatremii /niedobór jonu sodowego/
hiperkalemii /podwyższony poziom potasu/ we krwi obok komórkowego niedoboru potasu.
Niedobór sodu i potasu zależy od utraty tych jonów wraz z ciałami ketonowymi, niedobór
potasu dodatkowo od wzmożonego katabolizmu białek i lipidów.
Ketonemia rozwija się w wyniku gromadzenia się związków ketonowych we krwi. Przy braku
insuliny narasta stężenie związków ketonowych tj. acetooctanu i
β
-hydroksymaślanu

produkowanych przez wątrobę oraz acetonu, który tworzy się spontanicznie z acetooctanu.
Pojawienie się związków ketonowych w moczu nosi nazwę ketonurii.
Cukrzyca typu 1 jest stanem ostrej hiperglikemii
Główne objawy kliniczne narastają szybko tj .w ciągu kilku dni i mają określony charakter,
dlatego rozpoznanie cukrzycy typu 1 na ogół nie sprawia trudności. Niekiedy jednak początek
bywa powolny, objawy narastają w ciągu dłuższego czasu. Cukrzyca typu 1 dotyczy głównie
dzieci i osób młodych. Zwykle procesy destrukcji komórek B trzustki przebiegają dość
trzustki gwałtownie i prowadzą do niedoboru insuliny w ustroju.
Objawy cukrzycy typu 1 /typowe/:
-
nagły początek,
-
wzmożone pragnienie – polidypsja /picie dużej ilości płynów obojętnych/,
-
wielomocz – poliuria,
-
chudnięcie,
-
spadek sił,
-
kwasica towarzysząca hiperglikemii i zaburzenia gospodarki elektrolitowej i wodnej
manifestujące się cechami odwodnienia skóry i śluzówek.
Cukrzyca typu 2 jest stanem przewlekłej hiperglikemii.
Często bywa rozpoznawana po wielu latach jej trwania, dopiero w chwili wystąpienia
przewlekłych powikłań naczyniowych. Często dotyczy osób z otyłością. Nieuchwytny
początek stwierdza się u ponad 85% chorych. Ocenia się, że w przybliżeniu jedynie 50%
przypadków cukrzycy typu 2 jest zdiagnozowanych i leczonych. Bywa, że cukrzycę
rozpoznaje się przy stwierdzeniu uporczywej czyraczności, infekcji grzybiczych, zgorzeli
stopy lub postępujących zaburzeń wzroku. Hiperglikemia rozwija się stopniowo z powodu
narastającej insulinooporności.
Ryzyko wystąpienia choroby wzrasta:
-
z wiekiem,
-
przy braku aktywności fizycznej,
-
u osób z otyłością,
-
u osób z nadciśnieniem tętniczym lub zaburzeniami lipidowymi,
-
u osób z wywiadem rodzinnym występowania cukrzycy.
8. Powikłania cukrzycy
8a. Ostre powikłania cukrzycy.
W cukrzycy może dojść do rozwoju głębokich zaburzeń metabolicznych, które powodują
powstanie ostrych i ciężkich zespołów klinicznych przebiegających z zaburzeniami
świadomości a nawet utratą przytomności. Zaburzenia metaboliczne mogą rozwinąć się w
wyniku ostrego względnego lub bezwzględnego niedoboru insuliny a co za tym idzie szybko
narastającej hiperglikemii bądź odwrotnie przy niedoborze glukozy dochodzi do gwałtownie
nasilającej się hipoglikemii. Z powodu narastających zaburzeń metabolicznych mogą
rozwinąć się stany zagrażające życiu.
Do najczęściej występujących należą:
1.
Kwasica i śpiączka ketonowa.
2.
Ostra hipoglikemia polekowa i śpiączka neuroglikopeniczna.

Kwasica ketonowa jest kompleksowym zaburzeniem metabolizmu węglowodanów,
białek i tłuszczów z powodu braku działania biologicznego insuliny.
Głęboki deficyt insuliny powoduje wystąpienie znacznej hiperglikemii. W tych warunkach
jest ona wywołana zmniejszonym zużytkowaniem glukozy w tkankach z powodu zahamowania
transportu glukozy do wnętrza komórek w tkance mięśniowej, tłuszczowej i łącznej. Nasileniu
ulega również glikogenaliza /rozpad glikogenu/ w wątrobie i mięśniach i zwiększone oddawanie
glukozy do krwi. Znaczna hiperglikemia we krwi powoduje wzmożoną diurezę osmotyczną,
skutkiem której organizm organizm traci duże ilości energii, wody i elektrolitów (głównie sodu i
potasu). Rozwijające się znacznego stopnia odwodnienie ustroju może doprowadzić do spadku
ciśnienia tętniczego, a nawet do wstrząsu oligowolemicznego. Następstwem tego jest
zmniejszenie przepływu krwi przez ważne dla życia narządy takie jak mózg i nerki. Klinicznym
tego skutkiem są zaburzenia świadomości i skąpomocz.
Jednocześnie niedobór insuliny powoduje w wątrobie nasilony rozpad białek i zwiększone
przetworzanie uwolnionych aminokwasów w glukozę. Przy niedoborze insuliny bezpośrednio
pobudzające działanie na glukoneogenezę i ketogenezę wywiera glukagon.
W zakresie metabolizmu tłuszczów warunkach całkowitego niedoboru insuliny następuje
zahamowanie syntezy triglicerydów i zwiększenie lipolizy. W wyniku tego duże ilości
kwasów tłuszczowych trafiają z komórek tkanki tłuszczowej do wątroby, gdzie są
przekształcane poprzez acetylo-CoA do acetooctanu i powstający z niego
β
– hydroksy maślan
.Wytwarzanie acetooctanu przez wątrobę jest znacznie nasilone, dochodzi więc do
nagromadzania się we krwi acetaoctanu jak również
β
-hydroksymaślanu. Aniony tych obu
związków są silnymi kwasami, wiążą dużę ilości zasad i prowadzą do rozwoju kwasicy
metabolicznej. Zwiększone stężenie związków ketonowych powoduje wydalanie ich z
moczem łącznie z jonami sodowymi i potasowymi, potęgując niedobory jonów
spowodowanie diurezą osmotyczną. Znaczna część acetooctanu ulega samoistnej
dekarboksylacji do acetanu, który jest wydalany z moczem i wydychanym powietrzem.
Śpiączka ketonowa jest skutkiem narastania kwasicy metabolicznej i obniżenia pH we krwi i
tkankach. W wyniku czego dochodzi do upośledzenia funkcji wyższych ośrodków mózgu i
wystąpienia śpiączki. Ta postać śpiączki cukrzycowej występuje najczęściej u chorych na
cukrzycę typu 1, u których doszło do znacznego i nagłego niedoboru insuliny.
Śpiączka hipoglikemiczna jest uwarunkowana zmniejszeniem dowozu glukozy do mózgu
przy krytycznym obniżeniu stężenia glukozy we krwi poniżej 50 mg/dl.
Hipoglikemia u chorych leczonych insuliną jest spowodowana najczęściej
przekroczeniem dawki w stosunku do aktualnego zapotrzebowania. Dominujące objawy
kliniczne ujawniają się jako zaburzenia ośrodkowego układu nerwowego /neuroglikopenia/ i
są wyrażone w różnym stopniu w zależności od szybkości i stopnia narastania hipoglikemii.
Zależnie od nasilenia objawów należy wyróżnić stany lekkie, ciężkie i śpiączkę
neuroglikopeniczną. Stany niedocukrzenia przebiegające z utratą przytomności są bardzo
poważnym powikłaniem insulinoterapii, a przedłużająca się śpiączka może doprowadzić do
nieodwracalnego uszkodzenia struktur mózgowych.
8b. Przewlekłe powikłania cukrzycy.
Niezależnie od etiologii każde zaburzenie tolerancji glukozy, któremu towarzyszy
hiperglikemia, prowadzi do powikłań cukrzycy o charakterze angiopatii i neuropatii.
Te przewlekłe powikłania mogą rozwijać się zarówno w przebiegu cukrzycy typu 1 ,jak i
cukrzycy typu 2 a także towarzyszą innym postaciom cukrzycy.

Hiperglikemia odgrywa zasadniczą rolę w występowaniu tych powikłań. Wśród wielu
mechanizmów za pośrednictwem których hiperglikemia prowadzi do uszkodzenia funkcji i
struktury śródbłonka naczyń podkreśla się glikację białek,czyli nieenzymatycze wiązanie
glukozy z wolnymi grupami aminowych białek. W patogenezie rozwoju przewlekłych
powikłań cukrzycy, obok hiperglikemii bierze udział wiele czynników środowiskowych oraz
czynniki genetyczne.
Zmiany w dużych naczyniach /makroangiopatia/ szczególnie często występują w
cukrzycy typu 2 i są odpowiedzialne za ok. 60% zgonów w tej grupie chorych. Cukrzyca w
sposób zasadniczy zwiększa ryzyko przyspieszenia rozwoju miażdżycy naczyń, szczególnie
wieńcowych, mózgowych oraz kończyn dolnych.
Zmiany cukrzycowe w obrębie drobnych naczyń /mikroangiopatia/ występują
zarówno w cukrzycy typu 1 jak i typu 2. Mikroangiopatia cukrzycowa manifestuje się w
postaci charakterystycznych zmian głównie w obrębie narządu wzroku, nerek, nerwów
obwodowych oraz stóp.
Wśród przewlekłych powikłań cukrzycy należy wymienić:
♦
Zespoły makroangiopatii:
- chorobę niedokrwienną serca,
- udar mózgu,
- miażdżycę zarostową tętnic kończyn dolnych,
- nadciśnienie tętnicze.
♦
Zespoły mikroangiopatii pod postacią:
-
retinopatii-zespołu swoistych dla cukrzycy zmian czynnościowych i morfologicznych
naczyń włosowatych siatkówki, które w stadium znacznego ich zawansowania mogą
prowadzić do upośledzenia lub nawet utraty wzroku,
-
nefropatii-zespołu klinicznego, którego podłożem są swoiste zmiany w naczyniach
kłebuszków nerkowych, prowadzące początkowo do białkomoczu z czasem występuje
zespół nerczycowy i przewlekła niewydolność nerek,
-
zespół stopy cukrzycowej obejmującego zmiany w tkankach miękkich, czasem
dotyczą także kości; podłożem są zmiany w obrębie układu nerwowego i/lub zmiany
naczyniowe, jak i podatność na infekcje bakteryjne.
♦
Neuropatię:
-
obwodową symetryczną czuciowo-ruchową i jedno- lub wieloogniskową,
-
układu autonomicznego.
9. Leczenie cukrzycy.
Obecnie jednym z najważniejszych celów leczenia cukrzycy jest uzyskanie maksymalnego
wyrównania metabolicznego,czyli uzyskania normoglikemii, co sprzyja zapobieganiu
angiopatii cukrzycowej. Właściwe leczenie cukrzycy wymaga indywidualnego podejścia w
każdym przypadku.
Na to składa się:
- dostosowanie stylu życia do przebiegu choroby,
- podjęcie diety cukrzycowej, jako integralnej składowej leczenia,
- stosowanie wysiłku fizycznego, który w istotny sposób wpływa na procesy metaboliczne i
odgrywa szczególnie ważną rolę w utrzymaniu homeostazy glukozy,
- ocena i monitorowanie wyrównania metabolicznego poprzez aktywne włączenie się chorego
i wykonywanie oznaczeń glikemii, /w przypadkach koniecznych nawet kilka razy na dobę/,
- stosowanie leczenia farmakologicznego:
a/ w cukrzycy typu 1 wyłącznie insuliny,

b/ w cukrzycy typu 2 podstawą leczenia są leki doustne z odpowiednio dobranej grupy; w
niektórych przypadkach cukrzycy typu 2 konieczne jest leczenie skojarzone t.j. lekami
doustnymi i insuliną lub wyłącznie insuliną.
- edukacja chorych, która jest istotnym elementem leczenia; ma ona na celu poszerzenie
wiedzy chorego na temat cukrzycy, zmianę postawy wobec niej; umożliwia to lepsze
wyrównanie metaboliczne, zapobieganie ostrym i przewlekłym powikłaniom choroby.

Część trzecia. Patofizjologia układu krwiotwórczego i hemostazy
Temat XII. Hematopoeza (krwiotworzenie)
Danuta Rość
Hematopoeza lub hemopoeza jest to proces wytwarzania komórek krwi, który polega na
stałym odnawianiu i uzupełnianiu komórek krwi, niszczonych fizjologicznie w śledzionie
po spełnieniu ich funkcji. Komórki krwi tzw. elementy morfotyczne krwi są wysoce
wyspecjalizowanymi komórkami spełniającymi określone funkcje w ustroju, a należą do
nich: erytrocyty, granulocyty obojętno-, kwaso- i zasadochłonne, monocyty, płytki krwi i
limfocyty.
Dla celów dydaktycznych wyróżnić można tzw. mielopoezę, zachodzącą głównie w
szpiku kostnym i polegającą na tworzeniu wszystkich wymienionych wyżej komórek z
wyjątkiem limfocytów i limfopoezę tj. tworzenie i przemiany limfocytów, która tylko w
około 10% ma miejsce w szpiku, a odbywa się głównie w układzie chłonnym ustroju tj. w
grasicy, węzłach chłonnych, migdałkach podniebiennych i kępkach Peyera.
W życiu płodowym człowieka pierwszym narządem krwiotwórczym jest pęcherzyk
żółtkowy, następnie wątroba i przejściowo śledziona i dopiero w ostatnim trymestrze - szpik
kostny.
Masa szpiku kostnego u dorosłych wynosi 1600-3400g, średnio 2600g, co stanowi ok.
3-6% masy ciała. Jedną trzecią do połowy tej masy stanowi utkanie komórkowe a resztę
bezpostaciowa macierz.
W szpiku kostnym znajduje się ok. 1 kg komórek jądrzastych krwi, czyli 10
12
- 12
12
-
tj. 1,0 -1,2 biliona komórek (1 bilion = 1 milion milionów). Szpik kostny jest narządem o
ogromnej dynamice i największej zdolności do proliferacji, czyli mnożenia się komórek. W
ciągu każdej sekundy u dorosłego człowieka do krwiobiegu wychodzi 2,5 miliona krwinek
czerwonych i 2 miliony krwinek białych.
Wszystkie komórki układu krwiotwórczego wywodzą się z wielopotencjalnej
komórki macierzystej (komórka pnia, stem cell, colony forming unit). W mikroskopie
świetlnym wygląda ona jak zwykły limfocyt szpikowy małej i średniej wielkości. Nie ma
możliwości oceną morfologiczną odróżnić komórkę macierzystą wielopotencjalną od innych
limfocytów utkania szpiku. Badanie jej potencjalnych funkcji umożliwia wyróżnienie tej
komórki spośród innych. Służą do tego: badania in vivo - kolonizacja śledziony myszy
(polega na wstrzyknięciu szpiku zdrowej myszy - innej myszy, której śledziona została
napromieniowana w dawce niszczącej całkowicie jej układ krwiotwórczy) i metoda in vitro -
tj. hodowli komórkowych. Komórka macierzysta ma zdolność tworzenia kolonii na
odpowiednich podłożach umożliwiających jej kolejne podziały i tworzenie różnych kolonii
zależnie od dodawanych do podłoża czynników regulacyjnych; stąd pochodzi skrót
stosowany do określenia komórek macierzystych szpiku - CFU - colony forming unit
(jednostka tworząca kolonie).
Hemopoezę charakteryzują procesy: odnowy, różnicowania i dojrzewania.
Proces odnowy to zdolność do rozmnażania tj. podziału na dwie jednakowe komórki
potomne o identycznych cechach genetycznych, morfologicznych, biochemicznych i
funkcjonalnych w stosunku do posiadanych przez komórkę matczyną,
Proces różnicowania to zdolność do różnicowania; układ genetyczny komórki steruje
przemianami biochemicznymi prowadzącymi do powstawania określonych białek
warunkujących wykonywanie odpowiednich funkcji biologicznych. Różnicowanie komórek
działa w dwóch kierunkach, po pierwsze - ogranicza zakres spełnianych przez komórkę

funkcji biologicznej, po drugie przystosowuje się ją maksymalnie do pełnienia
przewidywanych czynności.
Proces dojrzewania polega na kumulowaniu związków powstałych w procesie
różnicowania i wytwarzaniu struktur subkomórkowych koniecznych do wysokiej specjalizacji
komórek. W hemopoezie można wyróżnić komórki dzielące się lub zdolne do podziału,
komórki ulegające różnicowaniu, komórki dojrzewające i komórki końcowe - dojrzałe,
przechodzące do krążenia ogólnego.
Do prawidłowego przebiegu hemopoezy niezbędne są:
-
hemopoetyczne komórki macierzyste,
-
czynniki wzrostu,
-
mikrośrodowisko hemopoetyczne.
1. Hemopoetyczne komórki macierzyste.
Krwiotwórcze komórki macierzyste odznaczają się zdolnością do samoodnawiania przez całe
niemal życie organizmu oraz do podziału i wytwarzania komórek rodzicielskich dla głównych
linii: szpikowej (CFU GEMM - colony forming unit – granulocyte, erythrocyte,
megakariocyte, monocyte) wytwarzającej kolonie granulocytowe, erytrocytowe,
megakariocytowe i monocytowe oraz limfocytowej. Hemopoetyczne komórki macierzyste
występują głównie w szpiku, ale pewne ich ilości pojawiają się także we krwi obwodowej.
Wielopotencjalna komórka macierzysta - CFU-s (tworząca kolonie w śledzionie – colony
forming unit- spleen) charakteryzują szczególne własności, a mianowicie:
a/ zdolność do samoodnawiania ( patrz wyżej),
b/ zdolność do różnicowania w odpowiedzi na różnorakie bodźce - warunkuje to powstanie
komórek rodzicielskich określonego szeregu komórek np. dla linii erytrocytarnej,
c/ pozostawanie większości komórek (ok.95%) w fazie spoczynkowej cyklu komórkowego;
jest to mechanizm ochronny dla populacji komórek macierzystych - opornych wówczas na
działanie czynników toksycznych.
2. Czynniki wzrostu
Czynniki regulujące hematopoezę nazywane są czynnikami wzrostowymi lub cytokinami. Są
to hormony tkankowe o wielokierunkowym działaniu pozytywnym (pobudzającym) lub
negatywnym (hamującym). Mogą pobudzać lub hamować komórki docelowe bezpośrednio
lub też stymulować lub hamować komórki docelowe w zakresie wydzielania innych cytokin.
Cytokiny wydzielane są przez komórki układu odpornościowego - aktywowane limfocyty i
makrofagi, ale także inne komórki ustroju. Cytokiny wydzielane przez limfocyty noszą nazwę
limfokin, a uwalniane przez makrofagi - monokin. Ich najważniejszą funkcją fizjologiczną
jest regulacja wzrostu i różnicowania różnych komórek w ustroju. Są to hormonopodobne
peptydy i niskocząsteczkowe białka.
Wśród czynników wzrostu wyróżnić można 3 grupy zależnie od rodzaju komórek
docelowych, na które wywierają swoje działanie:
1/ czynniki pobudzające (CSF colony stimulating factor) lub hamujące (CIF colony inhibiting
factor) wzrost kolonii w hodowli szpiku; CSF pobudzają powstawanie kolonii - kolonalny
wzrost komórek prekursorowych szpiku; do tej grupy należą między innymi:
- G-CSF - czynnik stymulujący kolonie granulocytarne (granulocyte colony stimulating
factor),
- M-CSF - czynnik stymulujący kolonie monocytowe (monocyte colony stimulating factor)
- GM-CSF - czynnik powodujący wzrost kolonii granulocytowo-monocytowych
(granulocyte-monocyte colony stimulating factor),

- EPO – erytropoetyna (stymuluje wzrost kolonii linii erytroidalnej) ,
- Meg-CSF - czynnik powodujący wzrost kolonii megakariocytowych (megakariocyte-
colony stimulating factor)
2/ interferony (INF) - pobudzające komórki mikrośrodowiska hemopoetycznego szpiku do
wydzielania czynników wzrostu pod wpływem działania wirusów; należą tu: INF
α
, INF -
β
, INF -
γ
. Interferony stanowią grupę białek wytwarzanych przez komórki w odpowiedzi
na zakażenia wirusowe lub stymulacje antygenową. Posiadają zdolność blokowania
replikacji wirusów oraz hamowania proliferacji komórek nowotworowych i prawidłowych.
3/ interleukiny (IL), działają bezpośrednio poprzez pobudzanie komórek do wydzielania
innych cytokin.
Interleukiny najważniejsze w regulacji hemopoezy to:
- Il-3 jest określana jako wielopotencjalny czynnik stymulujący kolonie; powoduje
proliferację wielopotencjalnych komórek macierzystych oraz wczesnych prekursorowych
komórek erytroidalnych, megakariocytarnych i granulocytarno- monocytarnych,
- Il-2 stanowi najważniejszy czynnik wzrostowy limfocytów T, odgrywa istotną rolę w
różnych typach reakcji immunologicznych,
- Il-1 jest określana jako czynnik aktywujący limfocyty,
- Il-5 jest cytokiną o dość selektywnym zakresie działania, które przejawia w stosunku do
komórek prekursorowych szeregu eozynofilowego i dojrzałych eozynofilów,
- Il-6 wywiera różnorodne efekty biologiczne, stymuluje różnicowanie limfocytów B do
plazmocytów, nasila rozwój megakariocytów.
Obok regulacji wzrostu i różnicowania hemopoezy i innych komórek ustroju, cytokiny
odgrywają rolę w patogenezie pewnych chorób, a ostatnio niektóre z nich (uzyskiwane
metodą rekombinacji niektórych genów) znalazły zastosowanie w terapii.
Ukierunkowanie wielopotencjalnych komórek macierzystych do poszczególnych linii
hemopoetycznych zależy od wpływu kilku czynników jednocześnie. Różnicowanie i
dojrzewanie komórek w określonym szeregu hemopoetycznym również jest pod kontrolą i
wpływem wielu cytokin.
3. Mikrośrodowisko hemopoetyczne.
Mikrośrodowisko hemopoetyczne (hematopoetic microenvironment, HIM) tj. struktura
anatomiczna i czynnościowa szpiku, w której przebiega hemopoeza. Podścielisko szpiku
tworzą zatoki naczyniowe z łączącą je siatką włókien retikulinowych oraz struktury
cytoplazmatyczne utworzone z komórek siateczkowych. Światło zatok wyściełają komórki
śródbłonka kontaktujące się przez cienką warstwę przydanki z komórkami siateczkowymi
znajdującymi się na zewnętrznej powierzchni zatok w ścisłym powiązaniu z ich ścianą. Do
mikrośrodowiska hemopoetycznego należą także fibroblasty i adipocyty (prawdopodobnie
powstałe w wyniku transformacji komórek siateczkowych) oraz limfocyty. Wytwarzają one
czynniki wzrostu i dostarczają składniki odżywcze komórkom hemopoetycznym.
Różne linie komórkowe układu krwiotwórczego znajdują się w określonym położeniu
w stosunku do naczyń krwionośnych. Wyspy układu czerwonokrwinkowego występują w
sąsiedztwie zatok żylnych. Erytroblasty najbardziej dojrzałe znajdują się najbliżej ścian zatok;
blisko zatok znajdują się także megakariocyty. Natomiast ogniska komórek układu
granulocytowego znajdują się z dala od zatok. Limfocyty tworzą otoczkę wokół małych
tętniczek promieniowych. Makrofagi występują w środku wysp erytroblastycznych, w
ośrodkach rozmnażania układu granulocytowego, blisko ścian zatok i w ich świetle.
Fagocytują one resztki jąder erytroblastów.
Hematopoeza jest możliwa jedynie w przypadku sprawnej czynności mikrośrodowiska
hemopoetycznego, skąd wydzielone substancje (np. restryktyny dla poszczególnych linii
komórkowych) mogą hamować rozwój jednych linii komórkowych, a promować inne. Dłużej

trwająca hodowla komórek krwiotwórczych jest możliwa tylko w obecności komórek
podścieliska. Uszkodzenie tego układu jest przyczyną jednej z postaci aplazji szpiku
kostnego, niemożliwej do wyleczenia metodą przeszczepiania szpiku, ponieważ komórki
macierzyste osiedlają się i tworzą kolonie jedynie w szpiku ze sprawnym mikrośrodowiskiem
hemopoetycznym.
Przechodzenie dojrzałych komórek ze szpiku do krwi jest regulowane za pomocą nie
do końca poznanych mechanizmów, nazywanych barierą (zaporą) szpikową. Przypuszcza się,
że bariera szpikowa jest wytworzona przez komórki mikrośrodowiska szpiku, a szczególnie
komórki zatok. Mechanizm odróżnienia przez podścielisko dojrzałych komórek od
niedojrzałych jest zależny między innymi od właściwości samych krwinek, głównie od
obecności różnych receptorów powierzchniowych. Przechodzenie np. krwinek czerwonych do
krążenia jest związane z utratą przez nie receptorów dla fibronektyny - glikoproteiny
związanej z komórkami mikrośrodowiska hemopoetycznego. Dojrzewając erytrocyty tracą te
receptory, co powoduje zmniejszenie się ilości ich powiązań z fibronektyną i decyduje o
przejściu do krwi. Młode retikulocyty posiadają ponadto większą elastyczność i mniejszą
lepkość umożliwiające im przeciskanie się przez pory w ściankach zatok.
W większości układów krwiotwórczych szpiku wydzielić można pule komórek o
wspólnych cechach:
a/ pulę proliferacji czyli namnażanie, w której komórki mnożą się przez podział mitotyczny,
b/ pulę dojrzewania, w której komórki nie dzielą się, lecz przekształcają w kierunku
dojrzałych i
sprawnych czynnościowo,
c/ pulę rezerwową utworzoną z dojrzałych morfologicznie i czynnościowo komórek
oczekujących na przejście ze szpiku do krwi,
d/ pulę czynnościową (funkcjonalną) złożoną z komórek znajdujących się we krwi i/lub w
tkankach.
4. Mielogram
Badanie bioptyczne szpiku (obok biopsji węzłów chłonnych i śledziony) należy do
podstawowych badań w diagnostyce zespołów hematologicznych. U dorosłych zdrowych
ludzi, jak już wcześniej powiedziano, czynny czerwony szpik wytwarzający komórki krwi
znajduje się jedynie w kościach płaskich: mostku, żebrach, obojczyku, kościach biodrowych,
kręgach i czaszce. Szpik pobiera się drogą nakłucia (mostka i kości biodrowej) lub
trepanobiopsji (kości biodrowej). U dzieci szpik kostny znajduje się w nasadach kości długich
i ich nakłucie pozwala na uzyskanie miazgi szpikowej.
Wskazania do uzyskania bioptatu szpiku kostnego ustala lekarz; wykonuje się go w
celu postawienia diagnozy, a także monitorowania skuteczności leczenia i ustalenia dalszego
postępowania terapeutycznego. Przeciwwskazaniem jest hemofilia i inne skazy krwotoczne
osoczowe, ostre skazy małopłytkowe i znaczna aktywacja fibrynolizy.
Nakłucie mostka lub kości biodrowej wykonuje się specjalną igłą punkcyjną z
mandrynem. Po odkażeniu skóry i wprowadzeniu igły, usuwa się mandryn i aspiruje
strzykawką miazgę szpiku w ilości 0,2-0,4 ml. Wykonuje się 8 - 10 cienkich preparatów i
barwi się metodą May-Grinvalda-Giemsy. Na miejsce nakłucia nakłada się jałowy opatrunek.
Trepanobiopsja polega na uzyskaniu przy pomocy spacjalnej igły tzw. trepanu
wycinka kości okolicy grzebienia biodrowego wraz z utkaniem szpiku kostnego. Jest
szczególnie cenna dla rozpoznawania i ustalania stopnia zaawansowania zespołów
aplastycznych, osteomielosklerozy, ostrych białaczek z hipoplastycznym szpikiem.
Rozmazy preparatów szpiku są poddawane odpowiednim barwieniom (hematoksyliną-
eozyną) pozwalającym następnie na identyfikowanie komórek szpikowych, które są zliczane i
kwalifikowane przez doświadczoną osobę do odpowiedniej linii komórkowej. Wynik badania
podawany jest w odsetkach poszczególnych komórek różnych linii, przyjmując za 100%

wszystkie znalezione komórki - liczone do 500. Poniżej podano zakres wartości
prawidłowych komórek szpiku u człowieka zdrowego.
Obok barwienia preparatów hematoksyliną-eozyną dokonuje się badań
cytochemicznych szpiku umożliwiających identyfikację poszczególnych komórek poprzez
wykazanie w nich ziarnistości cytoplazmatycznych charakterystycznych dla poszczególnych
linii komórkowych lub komórek w określonym stadium rozwojowym. Wśród nich wykonuje
się:
- odczyn peroksydozowy, - barwienie preparatów na obecność syderoblastów,- badanie na
zawartość glikogenu (reakcja PAS), -barwienie sudanem czarnym, - oznaczanie aktywności
fosfatazy alkalicznej granulocytów (FAG), - oznaczanie aktywności fosfatazy kwaśnej, -
oznaczanie aktywności esteraz.
Od niedawna stosuje się również metody immunologiczne do precyzyjnego
rozpoznawania komórek poszczególnych linii rozwojowych z zastosowaniem przeciwciał
monoklonalnych przeciwko receptorom powierzchniowym i antygenom cytoplazmatycznym.
W niektórych laboratoriach wykorzystywane są badania cytogenetyczne komórek
szpiku (obok limfocytów krwi obwodowej, komórek śledziony czy węzłów chłonnych). Są to
pracochłonne badania wymagające specjalnych pomieszczeń, gdzie w jałowych warunkach
utrzymuje się krótkie hodowle wymienionych komórek. W hematologii największą rolę
odgrywa wykazanie jedynego swoistego znacznika nowotworowego tj. chromosomu
Philadelphia.
Do precyzyjnej diagnostyki ostrych białaczek, do monitorowania ich leczenia, do
określania typów chłoniaków stosuje się w diagnostyce hematologicznej także badania
molekularne.
Odsetkowy skład komórek utkania szpikowego (Mielogram )
A. Układ czerwonokrwinkowy:
proerytroblasty
0 - 1,5
erytroblasty zasadochłonne 0,5 - 5,0
erytroblasty polichromatofilne 5,0 - 15,0
erytroblasty ortochromatyczne 5,0 - 15,0
B. Układ białokrwinkowy
mieloblasty
0,5 - 3,0
promielocyty
0,5 - 5,0
mielocyty obojętnochłonne
5,0 - 18,0
mielocyty kwasochłonne
0,5 - 2,5
mielocyty zasadochłonne
0,0 - 0,3
metamielocyty obojętnochłonne 8,0 - 25,0
metamielocyty kwasochłonne 0,0 - 2,0
metamielocyty zasadochłonne 0,0 - 0,1
granulocyty pałeczkowate
obojętnochłonne
10,0 - 30,0
kwasochłonne
0,4 - 3,0
zasadochłonne
0,0 - 0,5
granulocyty podzielone
obojętnochłonne
11,0 - 30,0
kwasochłonne
0,4 - 3,0
zasadochłonne
0,0 - 0,5

C.Utkanie chłonne
limfocyty
3,0 - 12,0
komórki limfoidalne
0,0 - 0,5
plazmocyty
0,0 - 2,5
D. Utkanie siateczki
komórki siateczki właściwe
0 - 1,5
komórki Ferraty
0 - 0,5
monocyty
0 - 2,5
E. Układ płytkotwórczy
megakarioblasty
-
megakariocyty
-
komórki nie dające się zróżnicować
Stosunek układu białokrwinkowego do czerwonokrwinkowego wynosi prawidłowo 3:1 - 4:1
Temat XIII. Patologia układu czerwonokrwinkowego
Ewa Żekanowska
Wstęp
Nieprawidłowości dotyczące układu czerwonokrwinkowego mają charakter ilościowy i/lub
jakościowy, a przyczyna tego zjawiska może być wrodzona lub nabyta. Zmiany w zakresie
liczby krwinek czerwonych, w których dochodzi do obniżenia ich liczby prowadzą do
wystąpienia niedokrwistości, natomiast nadmierny ich wzrost do stanów określanych jako
nadkrwistości.
Zmiany o charakterze jakościowym polegają na występowaniu krwinek czerwonych o
zmienionej wielkości (mikrocyty, makrocyty), zmienionym kształcie ( sferocyty, anulocyty,
krwinki sierpowate) oraz na obecności wewnątrzkomórkowych wtrętów (patologicznych
struktur w cytoplazmie komórek).
A. Niedokrwistości
Definicja
Niedokrwistość jest stanem chorobowym, w którym liczba krwinek czerwonych, stężenie
hemoglobiny i wartość hematokrytu zmniejszają się poniżej wartości zapewniających
optymalne utlenowanie tkanek i narządów. Niedokrwistości mogą mieć charakter pierwotny,
ale bardzo często rozwijają się w przebiegu innych chorób ( nowotwory, choroby o podłożu
autoimmunologicznym).
Kryteria rozpoznawcze niedokrwistości zgodnie z definicją WHO:
Parametr
Mężczyźni
Kobiety
Liczba krwinek czerwonych
Poniżej 4.5 x10
12
/l
Poniżej 4.0 x10
12
/l
Stężenie hemoglobiny
Poniżej 9.91mmol/l
Poniżej 8.7 mmol/l
Wartość hematokrytu
Poniżej 47
±
7
Poniżej 42
±
5

Podział niedokrwistości w oparciu o kryteria morfologiczne uwzględniające wielkość
krwinki czerwonej:
-
niedokrwistości mikrocytowe ( np. niedokrwistość z niedoboru żelaza, hemolityczna)
-
niedokrwistości makrocytowe ( np. niedokrwistości megaloblastyczne)
-
niedokrwistości normocytowe ( np. niedokrwistość aplastyczna)
Podział niedokrwistości w oparciu o czynniki etiopatogenetyczne:
a.nadmierna utrata krwi :
- ostry krwotok , powtarzające się, niekiedy utajone krwawienia przewlekłe,
-
wrodzone przetoki tętniczo- żylne,
b. zmniejszone i zaburzone wytwarzanie krwinek czerwonych i hemoglobiny:
-
uszkodzenie komórki macierzystej szpiku lub komórek rodzicielskich układu
czerwonokrwinkowego (np. niedokrwistości hipo- i aplastyczne),
-
niedobór substancji koniecznych do prawidłowej hematopoezy ( tzw. niedokrwistości
niedoborowe wywołane np. niedoborem żelaza, witaminy B12, kwasu foliowego,
witaminy E),
-
niedobór erytropoetyny, głównego czynnika stymulującego proces erytropoezy,
-
zaburzony metabolizm żelaza,
c. nadmierny lub przyśpieszony rozpad krwinek czerwonych:
-
wrodzone i nabyte niedokrwistości hemolityczne wywołane czynnikami
wewnątrzkrwinkowymi oraz zewnątrzkrwinkowymi,
-
hipersplenizm,
d. postaci złożone.
1. Niedokrwistość z niedoboru żelaza
Niedokrwistość z niedoboru żelaza (sideropeniczna) rozwija się na skutek niedostatecznej
syntezy hemoglobiny, wywołanej niedoborem żelaza w organizmie. Konsekwencją niedoboru
żelaza jest nieefektywna erytropoeza i zaburzenie procesu hemoglobinizacji, co prowadzi do
powstawania małych i niedobarwliwych erytrocytów. Niedokrwistość sideropeniczna jest
jedną z najczęściej występujących niedokrwistości w ogólnoświatowej populacji ludzi.
Podstawowe wiadomości na temat żelaza w organizmie człowieka
Całkowita pula żelaza w organizmie człowieka wynosi 3.5- 4.2 g. Źródłem żelaza są pokarmy
pochodzenia zwierzęcego, zawierające tzw. żelazo hemowe, znacznie lepiej przyswajalne
przez organizm człowieka w porównaniu z żelazem tzw. niehemowym występującym w
pokarmach roślinnych. Przeciętna dobowa dieta zawiera 10-20 mg żelaza, z czego zdrowy
organizm przyswaja tylko 10%. Żelazo jest wchłaniane w dwunastnicy i górnej części jelita
czczego. Występuje przede wszystkim w hemoglobinie i mioglobinie (tzw. białka
czynnościowe stanowią ok. 65%), ferrytynie i hemosyderynie (tzw. białka magazynujące,
stanowią ok. 23%) i transferynie (białko transportujące, ok. 0.15%)
Charakterystyka głównych białek zaangażowanych w metabolizm żelaza
Ferrytyna - białko magazynujące i odpowiedzialne za detoksykację żelaza; należy do tzw. puli
aktywnej, występuje w wątrobie, śledzionie, sercu, nerkach, komórkach hemopoetycznych.
Hemosyderyna- powstaje w wyniku rozpadu ferrytyny, należy do puli nieaktywnej.
Transferyna - glikoproteina odpowiedzialna za transport żelaza w organizmie, jest
syntetyzowana w wątrobie, jedna cząsteczka transferyny wiąże 2 atomy żelaza (Fe
+3
), w
warunkach fizjologicznych tylko 1/3 krążącej transferyny jest wysycona żelazem.
Laktoferyna: białko o działaniu bakteriobójczym, pochodzi głównie z ziarnistości
granulocytów obojętnochłonnych, przenosi żelazo do komórek układu siateczkowo-
śródbłonkowego.

Główne przyczyny występowania niedokrwistości na tle niedoboru żelaza:
a.
niedobory pokarmowe- nieprawidłowe odżywianie, dieta pozbawiona czerwonego
mięsa,
b.
zwiększone zapotrzebowanie – okres dojrzewania, ciąża, laktacja,
c.
upośledzone wchłanianie- niedokwaśność soku żołądkowego, zanikowy nieżyt
żołądka, stany po resekcji żołądka, zmiany zapalne jelit, celiakia,
d.
nadmierna utrata żelaza- krwawienia ostre i przewlekłe.
Okresy rozwoju niedokrwistości z niedoboru żelaza:
- niedobór żelaza w magazynach ustrojowych,
-
niedobór żelaza dostępnego dla erytropoezy,
-
jawna niedokrwistość z niedoboru żelaza.
Objawy kliniczne
Wynikają z niedotlenienia tkanek, przede wszystkim serca i OUN:
-porcelanowa bladość skóry i błon śluzowych, - bóle głowy, apatia, osłabienie, zaburzona
koncentracja, - bóle wieńcowe, arytmia, duszność, narastająca niewydolność układu krążenia,
- nasilone wypadanie włosów, włosy matowe, łamliwe, - rozdwajanie i prążkowanie
paznokci, zapalenia kącików jamy ustnej, - zaburzone łaknienie (pica syndrom)
Badania dodatkowe
1.
W badaniach biochemicznych stwierdza się:
- obniżone: stężenie żelaza w surowicy, ferrytyny, wysycenie transferyny żelazem,
- podwyższone: stężenie protoporfiryny IX oraz wolnych receptorów transferyny.
2. Zmiany hematologiczne – związane są z nieprawidłową hemoglobinizacją cytoplazmy
układu erytropoetycznego.
Krew obwodowa
Mikrocytoza ( obecność małych erytrocytów ), hipochromia ( obecność niedobarwliwych
erytrocytów), obniżona liczba retikulocytów
Szpik kostny:
Rozrost układu czerwonokrwinkowego ( hiperplazja), zahamowanie dojrzewania
erytroblastów z dominacją form młodszych, erytroblastów zasadochłonnych i
wielobarwliwych, obniżona liczba syderoblastów ( erytroblasów zawierających żelazo).
Leczenie
Leczenie niedokrwistości z niedoboru żelaza, po dokładnym ustaleniu przyczyny, polega na
podawaniu doustnych preparatów żelaza, w pewnych przypadkach ( np. po gastrektomii, w
zespołach złego wchłaniania) podaje się żelazo dożylnie.
2. Niedokrwistości megaloblastyczne
Definicja
Niedokrwistości megaloblastyczne charakteryzują się zaburzeniem procesu dojrzewania jąder
komórkowych i powstawaniem patologicznie dużych komórek (megaloblastów,
megalocytów).
Patogeneza
Główną przyczyną powyższych zaburzeń jest niedobór witaminy B12 i/lub kwasu foliowego,
substancji niezbędnych do prawidłowej syntezy kwasów nukleinowych.

Podstawowe dane o witaminie B12 i kwasie foliowym
Witamina B12
Jest syntetyzowana przez bakterie saprofitujące w przewodzie pokarmowym, jej źródłem są
również pokarmy pochodzenia zwierzęcego (wątroba, mięso, ryby). Wchłanianie wit. B12
odbywa się przy udziale czynnika wewnętrznego (Castle’a) syntetyzowanego przez komórki
okładzinowe błony śluzowej żołądka. Wit. B12 jest kofaktorem procesu aktywacji kwasu
foliowego. Magazyny ustrojowe wit. B12 wystarczają na ok. 2 lata.
Kwas foliowy
Jest syntetyzowany przez rośliny, bakterie i drożdże, zawierają go przede wszystkim świeże
zielone części roślin (sałata, natka pietruszki, cykoria). Kwas foliowy wchłaniany jest w
jelicie cienkim, jest niezbędnym czynnikiem do prawidłowej syntezy kwasów nukleinowych.
Zapasy ustrojowe zmagazynowane w wątrobie wystarczają na 3-4 miesiące.
Przyczyny
a.
niedobory pokarmowe:
-
witamina B 12: wegetarianizm, brak pokarmów pochodzenia zwierzęcego,
-
kwas foliowy: brak świeżych pokarmów roślinnych,
b.
zaburzenia wchłaniania:
-
wit. B12: choroba Addisona-Biermera, resekcja żołądka, choroby jelita cienkiego,
-
kwas foliowy: choroby jelita cienkiego, celiakia, alkoholizm,
c.
niedostateczne wykorzystanie:
-
wit. 12: zaburzenia polekowe (neomycyna, kolchicyna), wady metaboliczne (niedobory
enzymów szlaku metabolicznego wit. 12),
-
kwas foliowy: zaburzenia polekowe (metotreksat, fenobarbital), wady metaboliczne
(niedobory enzymów szlaku metabolicznego kwasu foliowego),
d.
zwiększone zapotrzebowanie:
-
wit.B12: zespoły mieloproliferacyjne, nadmierne zużycie przez bakterie lub pasożyty,
-
kwas foliowy: ciąża, zespoły mielo- i limfoproliferacyjne, niedokrwistości hemolityczne.
Objawy kliniczne
Są wyrazem niedotlenienia tkanek oraz zmian w komórkach o intensywnej proliferacji (skóra,
nabłonek przewodu pokarmowego, komórki szpiku kostnego) .
Są to:- spowolnienie psychiczne, apatia, - bóle głowy, osłabienie, duszność, - zmiany zapalne
i pieczenie języka, zaparcia, biegunki, - lekki obrzęk twarzy i kończyn, - skóra blada z
odcieniem „kawy z mlekiem”
W przypadku niedoboru witaminy B12 dominują objawy neurologiczne: parestezje, ataksje,
zaburzenia czucia głębokiego, uszkodzenie nerwu wzrokowego.
Diagnostyka laboratoryjna:
1. W badaniach biochemicznych stwierdza się obniżony poziom wit.B12 lub kwasu
foliowego, podwyższoną aktywność LDH, podwyższone stężenie żelaza.
2. Zmiany hematologiczne wynikają z asynchronii dojrzewania jąder komórkowych w
stosunku do cytoplazmy i zaburzonych podziałów komórkowych:
krew obwodowa: obecność dużych erytrocytów (makro- i megalocyty), płytek olbrzymich,
umiarkowana granulopenia, granulocyty z hipersegmentacją jąder komórkowych;
szpik kostny : rozrost układu czerwonokrwinkowego, zaburzony proces dojrzewania jąder
komórkowych prowadzący do megaloblastycznego toru odnowy układu
czerwonokrwinkowego, granulocytowego i płytkotwórczego.

2a. Niedokrwistość Addisona – Biermera
Występowanie
Należy do najczęściej występujących niedokrwstości typu megaloblastycznego, dotyczy
głównie osób starszych (50-70 lat).
Patogeneza
Jest chorobą autoimmunologiczną, w której dochodzi do powstawania przeciwciał
uniemożliwiających wchłanianie wit. B12. Rodzaje przeciwciał:
-
przeciwciała przeciwko czynnikowi wewnętrznemu Castle’a (IF- intrisinc factor)
-
przeciwciała przeciwko kompleksowi wit.B12/czynnik wewnętrzny
-
przeciwciała przeciwko komórkom okładzinowym błony śluzowej żołądka,
syntetyzującym czynnik wewnętrzny.
Objawy kliniczne
W chorobie Addisona-Biermera obserwuje się charakterystyczną triadę objawów klinicznych
ze strony układów o znacznej proliferacji komórek: układu pokarmowego (lśniący, piekący
język, zmiany zanikowo-nieżytowe błony śluzowej żołądka), układu nerwowego
(zwyrodnienie sznurów bocznych i tylnych rdzenia kręgowego, parestezje, ataksje, zmiany
psychiczne), układu krwiotwórczego (makrocytoza, leukopenia, małopłytkowość, odnowa
megaloblastyczna w szpiku).
Leczenie
Leczenie niedokrwistości megaloblastycznych ma charakter substytucyjny, polega na
podawaniu wit. B12 lub kwasu foliowego. W przypadku chorych z niedokrwistością
Addisona – Biermera podawanie wit.B12 trwa do końca życia chorego.
3. Niedokrwistości hemolityczne
Definicja
Niedokrwistości hemolityczne są to stany chorobowe wywołane skróconym okresem
przeżycia krwinek czerwonych (poniżej 30 dni). Najczęściej wzmożona hemoliza
(niszczenie krwinek czerwonych) zachodzi w układzie siateczkowo-śródbłonkowym wątroby
i śledziony, jest to hemoliza zewnątrznaczyniowa. Rzadziej rozpad erytrocytów następuje w
naczyniach krążenia ogólnego, mówimy wtedy o hemolizie wewnątrznaczyniowej.
Przyczyny
a.
wewnątrzkrwinkowe, wynikające z nieprawidłowej budowy erytrocyta, najczęściej
typu wrodzonego:
-
nieprawidłowa budowa błony komórkowej erytrocyta, np. sferocytoza, owalocytoza,
-
nieprawidłowa budowa hemoglobiny (hemoglobinopatie) np. anemia sierpowata,
-
defekty enzymatyczne (enzymopatie) np. niedobór enzymów glikolitycznych;
b.
zewnątrzkrwinkowe, wywołane czynnikami:
-
immunologicznymi: niedokrwistość autoimmunohemolityczna, błędy w przetaczaniu
krwi, żółtaczka hemolityczna noworodków, niedokrwistość immunohemolityczna
polekowa,
-
nieimmunologicznymi: środki chemiczne, bakterie, czynniki fizyczne, hipersplenizm.

Objawy kliniczne
Są wyrazem niedotlenienia tkanek i zwiększonego tworzenia bilirubiny na skutek rozpadu
erytrocytów. Objawy żółtaczki: skóra i błony śluzowe żółte, mocz i kał ciemnobrunatne. W
przypadkach bardzo nasilonej hemolizy pojawiają się dreszcze, gorączka, bóle głowy, mięśni,
stawów, powiększenie śledziony.
Badania laboratoryjne
1. W badaniach biochemicznych stwierdza się istotnie zwiększone stężenie produktów
degradacji hemoglobiny (uwolnionej z niszczonych erytrocytów): wolnej bilirubiny,
urobililinogenu, sterkobilinogenu oraz bardzo niskie stężenia haptoglobiny.
2. Zmiany hematologiczne
We krwi obwodowej w niedokrwistościach hemolitycznych typu wrodzonego występują
krwinki czerwone o charakterystycznym, zmienionym kształcie ( np. sferocyty, owalocyty,
krwinki sierpowate). Istotnie wzrasta liczba retikulocytów. Szpik jest bogatokomórkowy z
wyraźnym pobudzeniem układu czerwonokrwinkowego. Dla dokładnego określenia
przyczyny i rodzaju niedokrwistości hemolitycznej konieczne jest przeprowadzenie
specjalistycznych badań laboratoryjnych ( np. immunologicznych).
B. Nadkrwistości
Definicja
Nadkrwistości są to stany chorobowe, w których dochodzi do istotnego wzrostu liczby
krwinek czerwonych , stężenia hemoglobiny i hematokrytu.
Podział
1.
bezwzględne, inaczej prawdziwe:
-
pierwotne
-
wtórne (objawowe)
2.
względne, inaczej rzekome.
1. Czerwienica prawdziwa (Nadkrwistość pierwotna prawdziwa )
Etiopatogeneza
Zaliczana jest do przewlekłych chorób rozrostowych układu krwiotwórczego. W chorobie tej
dochodzi do klonalnego, nowotworowego rozrostu układu czerwonokrwinkowego,
granulocytowego i płytkotwórczego, z wyraźną dominacją linii erytrocytarnej.
Etiopatogeneza nie jest wyjaśniona w pełni, upatruje się znaczenia czynników genetycznych.
Objawy kliniczne
Częściej chorują mężczyźni, w wieku 50-75 lat. Charakterystycznymi objawami są:
ciemnoczerwone zabarwienia twarzy z purpurową sinicą warg, uszu i palców, duszność, bóle
wieńcowe, szum w uszach, bóle i zawroty głowy, nudności, wzdęcia, zaparcia, powiększenie
śledziony, świąd skóry, krwawienia żołądkowo-jelitowe. Wzrost we krwi obwodowej liczby
erytrocytów, granulocytów i płytek krwi, prowadzi do jej zagęszczenia i nadmiernej lepkości
co jest przyczyną częstych powikłań zakrzepowo-zatorowych.
Leczenie
Polega na stosowaniu krwioupustów oraz podawaniu leków mielosupresyjnych, hamujących
proces rozrostowy w szpiku kostnym.

2. Nadkrwistości wtórne (objawowe)
Definicja
Występuje w nich wzrost wyłącznie liczby erytrocytów, bez zmian w liczbie innych komórek
krwi. Związane są z nadmiernym wytwarzaniem erytropoetyny stymulującej proliferację
komórek układu erytroidalnego.
Przyczyny
Niedotlenienie w przewlekłych chorobach płuc, wrodzonych wadach serca, w związku z
przebywaniem na dużych wysokościach odpowiada za wzmożoną syntezę erytropoetyny.
Zwiększona synteza erytropoetyny przez guzy lub torbiele nerek, mięśniaki macicy.
3. Nadkrwistości względne (rzekome)
Powstają w wyniku zagęszczenia krwi na skutek odwodnienia ( nadmierna utrata płynów:
wymioty, biegunki), zaburzeń w regulacji gospodarki wodno-elektrolitowej lub upośledzonej
wydolności układu krążenia
Temat XIV. Patologia układu białokrwinkowego – zmiany ilościowe i
jakościowe
Ewa Żekanowska
Nieprawidłowości komórek układu białokrwinkowego dotyczą granulocytów
(obojętnochłonnych, kwaso- i zasadochłonnych), limfocytów i monocytów. Zaburzenia mogą
mieć charakter ilościowy lub jakościowy, a ich przyczyna może być wrodzona lub nabyta. W
przebiegu licznych chorób obserwuje się zmiany ilościowe, w których dochodzi do
zwiększenia liczby krwinek białych, zjawisko to określamy mianem leukocytozy lub
zmniejszenia liczby krwinek białych, co nazywamy leukopenią.
Tabela 1. Zmiany ilościowe komórek układu białokrwinkowego.
Rodzaj komórki
Zwiększenie liczby komórek Zmniejszenie liczby komórek
Neutrfile
(granulocyty obojętnochłonne)
Neutrofilia
Neutropenia
Eozynofile
(granulocyty kwasochłonne)
Eozynofilia
Eozynopenia
Bazofile
( granulocyty zasadochłonne)
Bazofilia
Bazopenia
Limfocyty
Limfocytoza
Limfopenia
Monocyty
Monocytoza
Monocytopenia
1. Zmiany ilościowe neutrofilów
1a. Granulocytoza obojętnochłonna ( neutrofilia)
Definicja
Jest to stan, w którym liczba granulocytów obojętnochłonnych (neutrofilów) wzrasta
powyżej 7,5 x 10
9
/l. W stanie zdrowia może wystąpić tzw. granulocytoza fizjologiczna (

wzrost granulocytów nawet do 20 x 10
9
/l) występuje u kobiet ciężarnych, w czasie owulacji,
po intensywnym wysiłku , pod wpływem stresu.
Przyczyny
Stany chorobowe, w których obserwujemy granulocytozę objawową to:
-
zakażenia: ostre i przewlekłe (bakteryjne, wirusowe, pasożytnicze, grzybicze),
-
urazy, oparzenia, krwotoki, stany pooperacyjne,
-
ostre choroby układu krążenia ( zawał serca, zator tętnicy płucnej),
-
choroby metaboliczne ( śpiączka cukrzycowa, zespół Cushinga),
-
leki ( kortykosteroidy, lit, acetylocholina),
-
zatrucia (tlenek węgla, ołów),
-
guzy lite: rak żołądka, płuca, trzustki,
Granulocytoza występuje również w przebiegu chorób rozrostowych układu
krwiotwórczego takich jak przewlekła białaczka szpikowa, przewlekła białaczka limfatyczna ,
mielofibroza, ziarnica złosliwa, czerwienica prawdziwa.
1b. Granulocytopenia obojętnochłonna (neutropenia)
Definicja
Jest to stan, w którym liczba granulocytów obojętnochłonnych zmniejsza się poniżej 1,7 x
10
9
/l.
Przyczyny :
a.
upośledzone wytwarzanie tzw. neutropenia ośrodkowa: uszkodzenie szpiku (leki,
zakażenia, promieniowanie), zmniejszenie czynnej masy szpiku (nacieki
nowotworowe, białaczkowe), zahamowanie procesu granulopoezy wynikające z
niedoboru witamin i hormonów (niedobór witaminy B12, alkoholizm, niedoczynność
przysadki, nadnerczy),
b.
zwiększone zużycie lub zaburzona dystrybucja neutrofilów tzw. neutropenie
obwodowe: zakażenia, kolagenozy, hipersplenizm.
1c. Agranulocytoza
Definicja
Jest to szczególnie ciężka postać neutropenii, w której liczba neutrofilów jest mniejsza niż 0.2
x 10
9
/l.
Przyczyny
Głównymi przyczynami granulocytozy są: polekowe uszkodzenie szpiku kostnego, zakażenia,
białaczki, mielofibroza.
Objawy kliniczne
Charakterystycznymi objawami neutropenii są: wzmożona podatność na zakażenia,
miejscowe i ogólnoustrojowe. Chory jest osłabiony, ma wysoką gorączkę, skarży się na bóle
mięśni i stawów. Na błonach śluzowych występują zmiany zapalno-martwicze, owrzodzenia
przełyku, odbytu, pochwy.

Leczenie
Polega na zastosowaniu antybiotyków o szerokim spektrum działania. U chorych z ciężką
postacią neutropenii i agranulocytozą podawane są krwiotwórcze czynniki wzrostu G-CSF
lub GM-CSF.
2. Zmiany jakościowe neutrofilów
Nieprawidłowości morfologiczne neutrofilów mogą mieć charakter wrodzony, występują one
jednak bardzo rzadko. Są to np. anomalia Pelgera- Hueta, polegająca na występowaniu
neutrofilów z dwoma płatami w jądrze, anomalia Maya-Hegglina, w której stwierdza się
obecność w cytoplazmie neutrofilów olbrzymich ziarnistości.
Do najczęściej występujących, nabytych, zmian jakościowych granulocytów zalicza
się stany, w których dochodzi do zwiększenia w obrazie krwi obwodowej odsetka młodszych
postaci granulocytów pałeczkowatych i dwupłatowych (zakażenia, zatrucia, po krwotokach)
lub występowania starszych granulocytów ze zwiększoną liczbą płatów w jądrze
komórkowym (niedokrwistości megaloblastyczne, choroby wątroby). Innym rodzajem
nieprawidłowości jest obecność toksycznych ziarnistości w cytoplazmie granulocytów.
Ziarnistości takie pojawiają się np. w ciężkich oparzeniach, zatruciach i zakażeniach.
Neutrofile o nieprawidłowej budowie wykazują zaburzenia funkcji fagocytarnych i
migracyjnych, co wiąże się z utratą nieswoistej odporności komórkowej organizmu i
wzmożoną podatnością na zakażenia.
3. Zmiany ilościowe eozynofilów i bazofilów
3a. Eozynofilia
Definicja
Eozynofilia jest stanem, w którym liczba granulocytów kwasochłonnych (eozynofilów) ulega
zwiększeniu powyżej 0.4 x 10
9
/l.
Przyczyny :
Najczęstszymi przyczynami eozynofilii są:
a.
choroby alergiczne (alergie pokarmowe, astma oskrzelowa, pokrzywka),
b.
choroby pasożytnicze (włośnica, toksoplazmoza wągrzyca, glistnica),
c.
choroby zakaźne (płonica, odra, cholera),
d.
choroby krwi ( białaczka eozynofilowa, przewlekła białaczka szpikowa, ziarnica
złośliwa),
e.
środki chemiczne i leki.
3b. Eozynopenia
Definicja
Eozynopenia charakteryzuje się zmniejszeniem liczby granulocytów kwasochłonnych poniżej
0.1 x 10
9
/l.
Przyczyny
Przyczyny eozynopenii sa następujące:
a. zakażenia ( czerwonka, posocznica),
b. intensywny wysiłek fizyczny, stres,
c.
adrenalina , wysokie dawki glikokortykoidów,
d.
śpiączka cukrzycowa, mocznicowa.

3c. Bazofilia
Definicja
Bazofilia występuje wtedy, gdy liczba granulocytów zasadochłonnych we krwi obwodowej
jest wyższa od 0.1 x 10
9
/l.
Przyczyny
Bazofilia może występować w przebiegu :
a.
chorób hematologicznych: nowotworowych (przewlekła białaczka szpikowa,
czerwienica prawdziwa, ziarnica złośliwa), nie nowotworowych ( niedokrwistości z
niedoboru żelaza, hemolityczne),
b.
stanów zapalnych i alergicznych,
c.
w związku ze zmianami hormonalnymi organizmu (ciąża, owulacja, niedoczynność
tarczycy).
3d. Bazopenia
Definicja
Bazopenia polega na zmniejszeniu liczby granulocytów zasadochłonnych we krwi
obwodowej poniżej 0.015 x 10
9
/l. W stanie zdrowia liczba granulocytów zasadochłonnych
jest bardzo niewielka, co może przyczyniać się do trudności w zdiagnozowaniu bazopenii.
Przyczyny
a.
choroby endokrynologiczne ( zespół Cushinga, nadczynność tarczycy)
b.
ostre zapalenie płuc, gorączka reumatyczna
c.
leki (adrenalina, glikokortykosteroidy, hormony tarczycy)
4. Zmiany ilościowe limfocytów:
4a. Limfocytoza
Definicja
Limfocytoza jest stanem, w którym obserwujemy wzrost liczby limfocytów we krwi
obwodowej powyżej 4 x 10
9
/l. Wyróżniamy tzw. limfocytozy pierwotne, które rozwijają się w
wyniku nowotworowego rozrostu układu chłonnego oraz limfocytozy wtórne, które
towarzyszą innym chorobom.
Przyczyny
Przyczyny limfocytozy pierwotnej:
a. chłoniaki,
b. ostra i przewlekła białaczka limfatyczna,
c. gammapatie monoklonalne,
Przyczyny limfocytozy wtórnej:
a. zakażenia wirusowe (różyczka, ospa, odra, świnka, grypa, HIV),
b. zakażenia bakteryjne ( krztusiec, kiła, toksoplazmoza),
c. inne : wstrząs kardiogenny, zabiegi operacyjne, rozległe urazy.

4b. Limfopenia
Definicja
Limfopenia charakteryzuje się obniżeniem liczby limfocytów we krwi obwodowej poniżej 1 x
10
9
/l.
Przyczyny :
a.
wrodzone ( niedorozwój układu chłonnego, wrodzone niedobory odpornościowe),
b.
nowotworowe choroby hematologiczne (chłoniaki, ziarnica złośliwa, białaczki
szpikowe),
c.
zakażenia (malaria).
5. Zmiany ilościowe monocytów
5a. Monocytoza
Definicja
Monocytoza, czyli wzrost liczby monocytów we krwi obwodowej, występuje wtedy gdy ich
liczba jest większa niż 0.8 x 10
9
/l.
Przyczyny:
a.
zakażenia bakteryjne ( gruźlica, kiła, bruceloza),
b.
zakażenia wirusowe (mononukleoza) i pierwotniakowe (zimnica),
c.
choroby nowotworowe układu krwiotwórczego ( ziarnica złośliwa, białaczka
monocytowa),
d.
choroby autoimmunologiczne tkanki łącznej ( toczeń układowy rumieniowaty,
reumatoidalne zapalenie stawów).
5b. Monocytopenia
Definicja
Monocytopenia jest stanem, w którym liczba monocytów we krwi obwodowej ulega
obniżeniu poniżej 0.2 x10
9
/l.
Przyczyny:
a.
niedokrwistość aplastyczna,
b.
choroby autoimmunologiczne tkanki łącznej ( toczeń układowy rumieniowaty,
reumatoidalne zapalenie stawów),
c.
zakażenie wirusem HIV,
d.
przewlekła białaczka limfatyczna.
Temat XV. Białaczki
Ewa Żekanowska
Definicja
Białaczki są to choroby nowotworowe układu krwiotwórczego, których cechą
charakterystyczną jest klonalny, niepohamowany rozrost określonego rodzaju krwinek

szeregu mieloidalnego. Komórki białaczkowe wykazują zaburzenia procesów podziału,
różnicowania i dojrzewania. Proliferacja i kumulacja komórek białaczkowych zachodzi w
obrębie układu krwiotwórczego, w narządach krwiotwórczych okresu życia płodowego
(wątroba i śledziona). Dochodzi także do naciekania innych narządów i upośledzania ich
funkcji (np. nerek, OUN, serca, skóry, węzłów chłonnych, układu rozrodczego).
Ogólny podział białaczek
1.
Ze względu na przynależność układową i stopień zróżnicowania komórek białaczkowych
wyróżniamy:
-
białaczki pochodzenia szpikowego ( mielopatie),
-
białaczki wywodzące się z układu limfatycznego (limforetikulopatie).
2.
Ze względu na intensywność zmian w układzie krwiotwórczym, stopień nasilenia i
dynamikę objawów klinicznych wyróżniamy:
-
białaczki ostre,
-
białaczki przewlekłe.
Epidemiologia
W Polsce roczna zachorowalność na białaczki wynosi: u mężczyzn 6,8 a u kobiet 4,2 na
100.000 mieszkańców. Najczęstszą postacią białaczki występującą u dzieci jest ostra
białaczka limfoblastyczna (ponad 85%), u osób dorosłych najczęściej występuje przewlekła
białaczka limfatyczna i przewlekła białaczka szpikowa. Pomiędzy 35 a 70 rokiem życia
obserwuje się wzrost zachorowalności na ostrą białaczkę szpikową.
Etiopatogeneza białaczek
Patomechanizm powstawania białaczek jest bardzo złożony i mimo ogromnego postępu
wiedzy na ten temat nadal pozostaje nie do końca wyjaśniony. W świetle aktualnej wiedzy za
najważniejsze czynniki inicjujące i/lub predysponujące uznaje się:
a.
zaburzenia cytogenetyczne,
b.
wirusy onkogenne indukujące nowotwory układu krwiotwórczego,
c.
działanie tzw. kofaktorów środowiskowych (czynniki fizyczne, chemiczne i
biologiczne),
d.
zachwianie równowagi homeostatycznej organizmu wynikające z obniżonej
sprawności układu immunologicznego.
W większości białaczek wykazano obecność nieprzypadkowych, swoistych zmian w
chromosomach. Stwierdzono ponadto w wielu przypadkach zależność pomiędzy rodzajem
aberacji chromosomalnych występujących w białaczce, a czasem przeżycia chorych. Zmiany
w budowie chromosomów powstające np. w wyniku translokacji lub delecji fragmentu
chromosomu mogą prowadzić do aktywacji tzw. protoonkogenów. W warunkach
prawidłowych protoonkogeny odpowiedzialne są za wytwarzanie białek kierujących
procesami wzrostu i różnicowania komórek. Podstawą transformacji białaczkowej jest
patologiczna aktywacja protonkogenu, która prowadzi do wytwarzania zbyt dużej ilości
białka. Ponadto białko to może być zmienione jakościowo lub powstawać w innej niż
normalnie fazie cyklu komórkowego. Ponad 30 opisanych protoonkogenów zawartych w
ludzkim genomie wykazuje homologię z onkogenami zawartymi w genomach wirusów
wywołujących nowotwory u różnych zwierząt. Za podłożem genetycznym białaczek
przemawiają również dane epidemiologiczne wskazujące na prawie 20-krotnie większą
zachorowalność na białaczki u osób z takimi, uwarunkowanymi genetycznie, chorobami jak:
zespół Downa, zespół Klinefeltera, niedokrwistość Fanconiego.

Wirusowe pochodzenie białaczek wykazano pierwotnie u zwierząt (wirusy indukujące
przewlekłą białaczkę limfatyczną u ptaków, wirus ostrej białaczki limfatycznej u myszy,
kotów). W latach osiemdziesiątych XX wieku udowodniono udział retrowirusa HTLV w
indukcji przewlekłej białaczki limfatycznej u człowieka, wykazano również udział wirusa
Epstain-Barr w patogenezie chłoniaka Burkitta oraz innych białaczek, szczególnie typu
ostrego.
Za najważniejsze kofaktory środowiskowe zaangażowane w rozwój białaczek uznaje się
promieniowanie jonizujące oraz środki chemiczne (benzen, środki ochrony roślin, środki
alkilujące, leki immunosupresyjne)
1. Białaczki ostre
Definicja
Białaczki ostre charakteryzują się występowaniem w szpiku kostnym i krwi obwodowej
młodych, często bardzo słabo zróżnicowanych komórek – blastów białaczkowych-
pochodzących od komórki, która uległa transformacji nowotworowej na jednym z wczesnych
etapów hematopoezy.
Klasyfikcja ostrych białaczek
Klasyfikacja ostrych białaczek opiera się na kryteriach morfologicznych,
cytoenzymatycznych i immunologicznych.
Klasyfikacja morfologiczna polega na ocenie w mikroskopie świetlnym takich cech jak
wielkość komórek, wielkość i kształt jądra, obecność jąderek, struktura chromatyny jądrowej,
wygląd cytoplazmy, ziarnistości wewnątrzkomórkowych. Badania cytoenzymatyczne o
najistotniejszym znaczeniu w diagnostyce różnicowej białaczek ostrych to ocena aktywności
peroksydazowej (reakcja POX), wykrywanie ziaren glikogenu (reakcja PAS), aktywności
nieswoistej esterazy alfa-naftylu (reakcja NASDA) oraz wykrywanie lipidów Sudanem
czarnym B. Diagnostyka immunologiczna polega na wykrywaniu swoistych antygenów na
powierzchni komórek, są tzw. antygeny różnicowania komórkowego (CD). W odniesieniu
do białaczek ostrych najczęściej stosowany jest podział na podstawie kryteriów
morfologicznych, zaproponowany przez francusko-amerykańsko-brytyjską grupę badaczy
(FAB) w 1976 roku, uzupełniany i modyfikowany w latach następnych w związku z
postępem diagnostyki immunologicznej.
Zgodnie z klasyfikacją FAB wyróżnia się:
a.
ostre białaczki limfoblastyczne z trzema podtypami: L
1
, L
2,
L
3
b. ostre białaczki nielimfoblastyczne (szpikowe), wśród których wyróżniono następujące
podtypy:
-
M
0
– ostra białaczka szpikowa słabo zróżnicowana,
-
M
1
– ostra białaczka mieloblastyczna bez cech dojrzewania,
-
M
2
– ostra białaczka mieloblastyczna z cechami dojrzewnia,
-
M
3
– ostra białaczka promielocytowa,
-
M
4
– ostra białaczka mielomonocytowa,
-
M
5
– ostra białaczka monocytowa,
-
M
6
– erytroleukemia,
-
M
7
– ostra białaczka megakariocytowa.

1a. Ostra białaczka nielimfoblastyczna ( szpikowa)
Definicja
Ostra białaczka szpikowa charakteryzuje się uogólnionym, niepohamowanym rozrostem
komórek pochodzenia szpikowego, które nie są zdolne do różnicowania i dojrzewania.
Występowanie
Najczęstszą postacią, stanowiącą ok. 80% wszystkich ostrych białaczek u dorosłych jest
białaczka typu M
1
i
M
2
Objawy kliniczne
Wynikają z wyparcia i kumulacji blastów białaczkowych w szpiku i innych narządach:
-niedokrwistość, -objawy skazy krwotocznej (małopłytkowość), -gorączka, częste zakażenia
dróg oddechowych, bóle kostno-stawowe, -powiększenie węzłów chłonnych, powiększenie
śledziony, - niewydolność układu krążenia będąca konsekwencją rozwijającej się
niedokrwistości jak również obecności nacieków białaczkowych w sercu.
Badania dodatkowe
1. Obraz krwi obwodowej: podwyższona leukocytoza, w obrazie dominują blasty białaczkowe
(występuje tzw. przerwa białaczkowa związana z obecnością młodych blastów białaczkowych
i pojedynczych dojrzałych granulocytów, a brakiem dojrzewających form granulocytów)
cechy niedokrwistości: obniżona liczba erytrocytów, obniżone stężenie hemoglobiny i
hematokrytu, obniżona liczba płytek krwi.
2. Szpik kostny: najczęściej bogatokomórkowy, monomorficzny, zdominowany przez blasty
białaczkowe (powyżej 30% ogólnej liczby komórek jądrzastych), znaczne obniżenie odsetka
układu czerwonokrwinkowego i płytkotwórczego. Różnicowanie blastów białaczkowych
możliwe jest w oparciu o badania cytochemiczne i fenotypowanie antygenowe.
3. W badaniach biochemicznych obserwuje się podwyższone stężenie kwasu moczowego i
LDH.
1b. Ostra białaczka limfoblastyczna
Definicja
Jest spowodowana nowotworowym rozrostem młodych, niedojrzałych komórek układu
limfatycznego. Prawie 80% ostrych białaczek limfoblastycznych wywodzi się z komórek
prekursorowych linii B, a ok. 20% z linii T-komórkowej.
Występowanie
Ostre białaczki limfoblastyczne stanowią około 85-95% wszystkich białaczek wykrywanych u
dzieci, najczęściej pomiędzy 3-7 rokiem życia. Częściej chorują chłopcy niż dziewczynki
(3:2).
Objawy kliniczne
Objawy kliniczne i laboratoryjne związane są, podobnie jak w przypadkach ostrych białaczek
szpikowych, z szybko postępującą pancytopenią (obniżenie we krwi obwodowej liczby
erytrocytów, leukocytów i płytek krwi), naciekaniem i kumulacją blastów w różnych
narządach. Poza osłabieniem, utratą masy ciała, gorączką, nocnymi potami, krwawieniami,
powiększeniem węzłów chłonnych, śledziony i wątroby, cechą charakterystyczną jest zajęcie
ośrodkowego układu nerwowego.

2. Białaczki przewlekłe
Częstość występowania
Przewlekła białaczka szpikowa stanowi 15-20% , a przewlekła białaczka limfatyczna 20 –
30% białaczek występujących u osób dorosłych. Zapadalność na przewlekłą białaczkę
szpikową wynosi ok. 1-2 osób na 100.000 mieszkańców. Najczęściej chorują osoby pomiędzy
40-55 rokiem życia.
2a.Przewlekła białaczka szpikowa
Definicja i patogeneza
Przewlekła białaczka szpikowa związana jest z nadmiernym wytwarzaniem wszystkich
komórek szeregu granulocytowego, na skutek mutacji w wielopotencjalnej komórce
macierzystej szpiku. Cechą charakterystyczną przewlekłej białaczki szpikowej jest pojawienie
się w wyniku tej mutacji chromosomu Philadelphia (Ph), powstającego w wyniku wzajemnej
translokacji pomiędzy ramionami długimi chromosomów 9 i 22. Czynnikami mutagennymi
może być promieniowanie jonizujące lub związki chemiczne, nie wykazano natomiast udziału
wirusów w patogenezie przewlekłej białaczki szpikowej.
Objawy kliniczne
Są to bóle stawowe, umiarkowana niedokrwistość, powiększenie śledziony, utrata masy ciała
(objawy narastają wraz z rozwojem choroby).
Chorobę cechuje kilkuetapowy przebieg:
-
faza przewlekła, o łagodnym przebiegu (2-3.5 roku),
-
faza przyspieszenia,
-
faza kryzy blastycznej i końcowe stadium choroby ( trwa kilka miesięcy)- na tym etapie
dochodzi do charakterystycznej transformacji mieloblastycznej , klinicznie
przypominającej ostrą białaczkę szpikową; następuje wzrost liczby mieloblastów w szpiku
i krwi, nasilenie objawów niedokrwistości i skazy krwotocznej, postępujące włóknienie
szpiku, powiększenie węzłów chłonnych, śledziony, pojawiają się nacieki białaczkowe w
różnych narządach.
Badania dodatkowe
1. Krew obwodowa: hiperleukocytoza ( 50-200 x
10
9
/l), dominują neutrocyty na wszystkich
szczeblach rozwoju począwszy od mieloblasta , często eozynofilia lub bazofilia, limfopenia,
nadpłytkowość.
2. Obraz szpiku: szpik bogatokomórkowy z hiperplazją komórek układu granulocytowego na
wszystkich etapach rozwoju, liczne megakariocyty, wyparcie układu czerwonokrwinkowego i
limfatycznego.
3. Cechą charakterystyczną jest istotne obniżenie (nawet do wartości zerowych) aktywności
fosfatazy alkalicznej granulocytów (FAG).
4. W badaniach biochemicznych stwierdza się zwiększone stężenie kwasu moczowego i
witaminy B12, związane z nasilonym rozpadem granulocytów.
2b. Przewlekła białaczka limfatyczna
Definicja
W przewlekłej białaczce limfatycznej dochodzi do nowotworowego rozrostu limfocytów,
które kumulują się w szpiku, krwi obwodowej i narządach limfatycznych. Najczęściej klon

białaczkowych limfocytów wywodzi się z linii B-komórkowej, znacznie rzadziej (ok.10%
przypadków) z limfocytów T.
Etiopatogeneza
Nie jest wyjaśniona. Nie potwierdzono w rozwoju przewlekłej białaczki limfatycznej udziału
promieniowania jonizującego, leków alkilujących oraz zakażeń wirusowych.
Objawy kliniczne
Najczęściej chorują osoby powyżej 50.roku życia, czas przeżycia chorych wynosi od kilku do
kilkunastu lat.
Na początku choroby objawy są mało charakterystyczne, takie jak ogólne osłabienie,
męczliwość. W miarę jej rozwoju następuje: powiększenie węzłów chłonnych szyi, dołów
pachowych i pachwin, powiększenie śledziony, rzadziej wątroby, niedokrwistość
autoimmunohemolityczna, spadek poziomu gammaglobulin, podatność na zakażenia
grzybicze i wirusowe, częste choroby skóry. Objawy te wynikają ze spadku odporności
immunologicznej typu komórkowego i humoralnego.
Badania laboratoryjne
1. Krew obwodowa: hiperleukocytoza z wyraźną dominacją limfocytów (80-90% wszystkich
krwinek białych), niedokrwistość i małopytkowość nasilające się wraz z rozwojem choroby.
2. Szpik kostny zajęty przez nacieki limfocytarne, hipoplazja układu granulocytowego i
erytropoetycznego.
3. Badania biochemiczne: dysproteinemia z hipogammaglobulinemią
Leczenie białaczek
Leczenie białaczek polega na leczeniu choroby podstawowej oraz jej powikłań takich jak
niedokrwistość, skaza krwotoczna, zakażenia. Leczenie procesu nowotworowego opiera się
głównie na stosowaniu chemioterapii (bardziej lub mniej agresywnej w zależności od rodzaju
białaczki). Na chemioterapię składają się złożone protokoły leków cytostostycznych, które
stosowane są w celu:
a.
indukcji remisji czyli powrotu chorego do zdrowia
b.
konsolidacji i leczenia podtrzymującego remisję, którego celem jest wzmocnienie
efektów leczenia i zapobieganie nawrotowi choroby.
Jedną z najskuteczniejszych metod leczenia białaczek jest przeszczep szpiku kostnego lub
komórek macierzystych krwi uzyskanych z krwi obwodowej. Przeszczep może być:
a.
autologiczny: szpik zostaje pobrany od samego chorego znajdującego się w fazie
remisji choroby, zostaje oczyszczony z komórek białaczkowych i ponownie
przeszczepiony choremu, przeszczep autologiczny wiąże się jednak z dużym ryzykiem
nawrotu choroby.
b.
allogeniczny: szpik zostaje pobrany od zgodnego antygenowo (w zakresie układu
HLA) dawcy.
Istotną rolę w leczeniu białaczek, w przypadkach polekowej aplazji szpiku i po
przeszczepieniu szpiku, odgrywają krwiotwórcze czynniki wzrostu stymulujące granulopoezę
(G-CSF, GM-CSM) oraz erytropoezę (EPO).
Temat XVI. Proces hemostazy.

Maria Kotschy
Definicja
Komórki śródbłonka naczyń wspólnie z krwinkami płytkowymi, układem krzepnięcia krwi i
fibrynolizy utrzymują płynność krwi w naczyniach, zapobiegają zamykaniu ich światła przez
skrzeplinę a po uszkodzeniu ściany naczyń hamują krwawienie i uczestniczą w jej naprawie.
W procesach tych ściana naczyniowa współdziała także z otaczającymi ją tkankami i
składnikami morfotycznymi krwi. Zespół tych czynności umownie nazwano procesem
hemostazy.
Podział
Ze względu na kolejność działania składowych tego układu i pewne uproszczenia
dydaktyczne można proces hemostazy podzielić na fazę:
1.
naczyniową,
2.
płytkową,
3.
osoczową (proces krzepnięcia i fibrynolizy).
1. Hemostaza naczyniowa.
Do czasu wprowadzenia hodowli komórkowej in vitro (1970r.) sądzono, że naczynia
krwionośne wraz z wyścielającym je śródbłonkiem służą jedynie do rozprowadzania krwi w
organizmie i regulacji ich przepuszczalności. Od kilkunastu lat znamy coraz więcej substancji
syntetyzowanych przez komórki poszczególnych warstw ściany naczyniowej.
Czynniki syntetyzowane przez komórki śródbłonka.
Hamujace krzepnięcie:
Heparynopodobne glikozoaminoglikany (siarczany heparanu i dermatanu)
Trombomodulina (TM)
Antytrombina (AT)
Inhibitor zewnątrzpochodnego toru aktywacji krzepnięcia (TFPI)
Białko S
Inicjujące krzepnięcie:
Czynnik von Willebranda (vWF)
Czynnik tkankowy (TF)
Czynnik aktywujący płytki (PAF)
Aktywujace fibrynolizę:
Tkankowy aktywator plazminogenu (t-PA)
Urokinazowy aktywator plazminogenu (u-PA)
Hamujące fibrynolizę:
Inhibitor aktywatorów plazminogenu typu 1 (PAI-1)
Inhibitor aktywatorów plazminogenu typu 2 (PAI-2)
Rozszerzające naczynia:
Tlenek azotu (NO)
Prostacyklina (PGI
2
), niektóre prostaglandyny np. PGE
2
Zwężające naczynia:

Endotelina (ET-1), tromboksan (TXA
2
), czynnik hiperpolaryzujący błony komórkowej
(EDHF), niektóre prostaglandyny np. PGF
2a
Adhezyny: (adhezja różnych komórek do śródbłonka błon podstawnych i składników
macierzy)
Selektyny E i P
Integryny i adhezyny należące do nadrodziny immunoglobulin (Ig)
Enzymy
Enzym konwertujący (kininaza II)
Lipaza lipoproteinowa
Ektonukleotydazy (przekształcają ATP i ADP do adenozyny i inozyny)
Cytokiny
Czynniki wzrostowe dla komórek śródbłonka, fibroblastów i komórek krwi
Interleukiny 1, 2, 4, 6, 8 i czynnik martwicy nowotworów (TNF)
Interferony
Liczne receptory błonowe dla wielu w/w i innych substancji znajdujących się we krwi.
W hemostazie naczyniowej biorą udział wszystkie trzy warstwy ściany naczynia:
wewnętrzna, środkowa i zewnętrzna. Błona wewnętrzna ze śródbłonkiem na swojej
powierzchni odpowiedzialna za utrzymanie płynności krwi krążącej hamuje aktywację płytek
krwi, leukocytów, monocytów i proenzymów układu krzepnięcia.
Duży udział w tym zjawisku mają glikozoaminoglikany (głównie siarczan heparanu i
siarczan dermatanu), które dzięki ujemnemu ładunkowi czynią powierzchnię śródbłonka
niezwilżalną i chronią ją przed działaniem tlenowych rodników i innych czynników
uszkadzających. Komórki śródbłonka syntetyzują również inhibitory enzymów krzepnięcia
takie jak antytrombinę (AT), II kofaktor heparyny (IICH), inhibitor zewnątrzpochodnego toru
krzepnięcia (TFPI) oraz inhibitor aktywatora plazminogenu typu 1 (PAI-1), które w razie
pojawienia się we krwi takich enzymów proteolitycznych jak trombina, tkankowy (t-PA) czy
urokinazowy (u-PA) aktywator plazminogenu lub ekspresji błonowego czynnika tkankowego
(TF) mogą zostać zahamowane.
Komórki śródbłonka naczyń wspomagają także organizm człowieka w stanach
niedokrzepliwości grożących krwawieniami lub w powikłaniach zakrzepowo-zatorowych, w
których dochodzi do wzmożonej syntezy i uwalniania do krwi czynnika von Willebranda,
zwiększającego adhezję krwinek płytkowych do śródbłonka lub podstawnej błony
kolagenowej oraz nasilonej ekspresji czynnika tkankowego (TF) inicjującego krzepnięcie
krwi w torze zewnątrzpochodnym. Komórki śródbłonka naczyń uczestniczą w mechanizmach
fibrynolizy; syntetyzują aktywatory plazminogenu typu tkankowego i urokinazowego, które
w razie zakrzepicy mogą aktywować znajdujący się w zakrzepie plazminogen i upłynniać
obecną tam fibrynę.
Ważną rolę spełniają obecne na powierzchni komórek śródbłonka białka adhezyjne (selektyny
E i P), integryny oraz adhezyny należące do nadrodziny immunoglobulin ułatwiające
migrację komórek poza naczynia.
Duże znaczenie w czynności naczyń mają syntetyzowane i uwalniane ze śródbłonków
substancje naczynioaktywne: rozszerzające naczynia: prostacyklina (PGI
2
), tlenek azotu
(NO), czynnik hiperpolaryzujący błony komórkowe (EDHF), bradykinina, histamina;
zwężające naczynia: endoteliny (ET), tromboksan (TXA
2
), czynnik aktywujący płytki (PAF).

Błona komórkowa komórki śródbłonka posiada liczne receptory wiążące trombinę jak
trombomodulina (TM) czy serpentynowy jej receptor, receptory aktywatorów plazminogenu
tkankowego i urokinazowego (u-PAR), receptory dla lipoprotein (LRP) wiążące także
aktywatory plazminogenu oraz receptory wiążące kininogeny i pośredniczące w wiązaniu do
błony komórkowej prekalikreiny (PK) lub XI czynnika krzepnięcia krwi - proenzymów
biorących udział w wewnątrzpochodnym torze krzepnięcia krwi.
2. Hemostaza płytkowa.
2a. Dane ogólne o płytkach krwi.
Płytki krwi (trombocyty) są bezjądrzastymi elementami o średnicy 2 – 4 m. 5 – 10%
stanowią płytki duże tzw. megatrombocyty, które są zazwyczaj płytkami młodymi, gdyż w
miarę starzenia ich objętość ulega zmniejszeniu. Płytki krwi wytwarzane są w procesie tzw.
trombopoezy w szpiku kostnym i częściowo w płucach. We krwi obwodowej znajdują się w
ilości 120 – 350 x 10
9
/l (120.000 – 350.000/l). Czas ich życia wynosi około 7 do 10 dni, a
ich usuwanie z krwi krążącej przez układ jednojądrzastych komórek fagocytujących związane
jest ze zmianami w błonie płytkowej zachodzącymi w procesie starzenia się. We krwi
krążącej znajduje się 70% całkowitej puli płytek a w śledzionie około 30%. Tworzenie płytek
odbywa się przez fragmentację cytoplazmy megakariocytów.
Na zewnątrz płytek znajduje się bogata w glikoproteiny otoczka zawierająca m. in.
receptory dla czynników aktywujących płytki i substancje niezbędne do reakcji płytkowych.
W dwuwarstwowej fosfolipidowej błonie płytkowej, zachodzą reakcje z czynnikami
krzepnięcia.
W cytoplazmie płytkowej występuje układ włókienkowych białek o różnym stopniu
polimeryzacji, utrzymujących dyskowaty kształt płytki oraz umożliwiających podczas adhezji
zmianę jej kształtu, tworzenie wypustek i wydzielanie substancji wewnątrzpłytkowych.
Białka te stanowią 30 - 50% całkowitej masy płytkowej. W skład organelli płytkowych
wchodzą ziarnistości alfa i ziarnistości gęste delta, lizosomy, mitochondria, aparat Golgiego i
ziarna glikogenu. W organellach tych zachodzą różne procesy metaboliczne oraz
magazynowane są różne białka, nukleotydy adeninowe, serotonina i jony wapnia. Do układu
błon płytkowych należą kanaliki o dużej gęstości elektronowej. Znajdują się w nich enzymy
przemiany kwasu arachidonowego oraz jony wapnia biorące udział w procesach kurczliwych.
Substancje obecne we wnętrzu płytki przedostają się do otoczenia poprzez kanaliki łączące
się z powierzchnią komórek.
2b. Udział płytek krwi w hemostazie.
Zasadnicze znaczenie płytek krwi polega na ich udziale w hemostazie pierwotnej. Już w
pierwszych sekundach po uszkodzeniu śródbłonka naczyniowego płytki ulegają adhezji do
warstwy podśródbłonkowej. W procesie tym biorą udział dwa białka: czynnik von
Willebranda, którego receptorami są płytkowe glikoproteiny I b i IX oraz fibronektyna.
Kontakt płytek z odsłoniętymi włóknami kolagenu powoduje ich aktywację. Płytki zmieniają
swój kształt z dyskowatego na kulisty i tworzą wypustki cytoplazmatyczne. Dochodzi do
uwolnienia na zewnątrz substancji zawartych w ziarnistościach płytek.
Z ziarnistości alfa uwalniane są m. in. fibrynogen, trombospondyna i płytkopochodny
czynnik wzrostu (PDGF), z ziarnistości gęstych m. in. ADP, ATP, serotonina, a z lizosomów
m. in. glukoronidaza. W wyniku zmian konformacyjnych płytki i ujawnienia na jej
powierzchni receptorów IIb/ IIIa dla fibrynogenu dochodzi do agregacji płytek. W miejscu
uszkodzenia ściany naczyniowej powstaje czop płytkowy.
Czynnikami aktywującymi płytki w stanach fizjologicznych oprócz kolagenu są
przede wszystkim trombina, tromboksan A
2
i ADP, które wiążą się ze swoistymi receptorami.

Mniejsze znaczenie dla procesu hemostazy w stanach fizjologicznych ma aktywacja płytek
przez adrenalinę, serotoninę, wazopresynę i tzw. czynnik aktywujący płytki (PAF). Podczas
uczynniania płytek m. in. przez trombinę i kolagen dochodzi do aktywacji wewnątrzpłytkowej
przemiany kwasu arachidonowego do tromboksanu A
2
oraz uwolnienia endogennego ADP,
które ten proces aktywacji potęgują. W procesie agregacji płytek wywołanej różnymi
agonistami wspólnym mechanizmem jest przejście jonów Ca
2+
z układu kanalików o dużej
gęstości do cytoplazmy płytkowej oraz fosforylacja niektórych białek płytkowych.
Po związaniu z powierzchnią płytek czynnika V i Xa powstaje kompleks, zwany
protrombinazą, który przekształca protrombinę w obecności jonów Ca
2+
do trombiny.
Utworzona przez trombinę fibryna, wzmacnia czop płytkowy i zapobiega wynaczynieniu krwi
w miejscu uszkodzenia ściany naczyniowej.
3.
Hemostaza osoczowa
Na hemostazę osoczową składają się białka obecne w osoczu a biorące udział w procesie
krzepnięcia krwi, fibrynolizy i kininogenezy.
3a. Proces krzepnięcia krwi
Istotą procesu krzepnięcia krwi jest zamiana rozpuszczalnego białka osocza fibrynogenu
(I) w przestrzenną sieć fibryny (włóknika) Ia. Za wykrzepianie fibrynogenu
odpowiedzialna jest trombina (IIa).
Tabela 2 przedstawia niektóre właściwości czynników krzepnięcia (według M. Czokało-
Plichty, Patofizjologia, red. S. Maślińskiego i J. Ryżewskiego, PZWL, W-wa 2002).
Czynnik
Masa
cząsteczko
wa
Stężenie
W osoczu
T
1/2
(dni)
Niezbędne
stężenie
%
Rola
I
II
III
IV
V
VII
VIII
a
IX
X
XI
XII
XIII
Prekalikreina
HMWK
kininogen o
dużej masie
cząsteczkowej
340 000
72 000
43 000
286 000
48 000
260 000
57 000
59 000
160 000
80 000
320 000
85 000
110 000
10 mol
1,2 mol
-
20 nmol
10 nmol
1 nmol
70 nmol
140 nmol
60 nmol
500 nmol
78 nmol
450 nmol
640 nmol
1,5 – 6,3
3
-
0,5
0,25
0,5
1
2
2 – 3
1,5
7 – 14
2
5
10 – 25
40
-
5 – 20
5 – 45
25
25
15 – 25
10 – 25
1 – 3
-
-
substrat
proenzym
kofaktor
jony Ca
2+
kofaktor
proenzym
kofaktor
proenzym
proenzym
proenzym
proenzym
proenzym
proenzym
kofaktor
a
Czynnik VIII występuje w osoczu w kompleksie z czynnikiem von Willebranda
Aktywacja protrombiny w trombinę może odbywać się 2 torami: zewnątrz i
wewnątrzpochodnym.
Ryc.3. Schemat krzepnięcia krwi aktywowanego na drodze egzo- i endogennej.

Według obecnych poglądów czynnik tkankowy (tissue factor) – TF jest
zasadniczym aktywatorem krzepnięcia krwi w torze zewnatrzpochodnym. Czynnik
tkankowy jest przezbłonową glikoproteiną, obecną na powierzchni wielu komórek, w tym
także nowotworowych. W niepobudzonych komórkach śródbłonka in vivo TF nie występuje,
ale cytokiny i endotoksyny bakteryjne indukują jego syntezę i ekspresję na powierzchni błon
komórkowych. Pobudzenie lub uszkodzenie śródbłonka naczyń odsłania TF, który łączy się z
osoczowym czynnikiem VII w stosunku 1:1, tworząc stechiometryczny kompleks TF/VIIa.
Powstaje w ten sposób aktywna postać czynnika VIIa o proteolitycznych własnościach w
stosunku do czynników IX i X, które aktywuje do IXa i Xa. Czynnik VIIa nie ma
bezpośredniego swoistego inhibitora a rolę tę w warunkach fizjologicznych spełnia inhibitor
drogi zewnątrzpochodnej (Tissue Factor Pathway Inhibitor) – TFPI. Funkcja TFPI polega na
hamowaniu czynnika Xa i wtórnej inaktywacji kompleksu TF/VIIa. Aktywny czynnik Xa
aktywuje protrombinę do trombiny przy współudziale czynnika Va.
Tor wewnątrzpochodny krzepnięcia krwi jest drogą alternatywną dla toru
zewnątrzpochodnego. Po uszkodzeniu komórek śródbłonka naczyń i odsłonięciu kolagenowej
błony podstawnej dochodzi do aktywacji czynników kontaktu: prekalikreiny, czynnika XII i
XI w obecności wysokocząsteczkowego kininogenu (HMWK).
Poprzez aktywację czynnika X w obecności aktywnego kofaktora, jakim jest czynnik
VIII dochodzi do wspólnej drogi krzepnięcia krwi obejmującej trombinogenezę (aktywację
protrombiny w trombinę i fibrynogenezę (przekształcenie fibrynogenu w fibrynę).
Generowana trombina aktywuje płytki krwi, czynnik V, VIII, XIII i wykrzepia fibrynogen.
Aktywny czynnik XIIIa stabilizuje i umacnia przez dodatkowe krzyżowe wiązania siarkowe,
strukturę włóknika.
Układ krzepnięcia znajduje się pod kontrolą białek tworzących układy inhibitorowe.
Jednym z licznych receptorów komórek śródbłonka jest trombomodulina (TM), błonowy
receptor a także inhibitor dla generowanej we krwi in vivo trombiny. W kompleksie z
trombomoduliną (TM/T) trombina traci swoje własności prokoagulacyjne (aktywacja płytek
cz. V, VIII, XIII i wykrzepiania fibrynogenu) i nabiera właściwości przeciwzakrzepowych
poprzez aktywację osoczowego białka C. Kompleks TM/T aktywuje białko „C” (PC) do jej
formy aktywnej aPC, która w obecności białka S inaktywuje na drodze proteolitycznej
czynniki Va i VIIIa. Hamuje ponadto inhibitor aktywatorów plazminogenu typu 1 (PAI-1),
zwiększając w ten sposób aktywność tkankowego aktywatora plazminogenu a ogólnie rzecz
biorąc aktywność fibrynolityczną krwi.
W inaktywacji przede wszystkim trombiny i czynnika Xa a także innych czynników
krzepnięcia XIIa, XIa, IXa bierze udział antytrombina (AT), której aktywność wzmaga
wielokrotnie obecność we krwi heparyny.
Wrodzone lub nabyte obniżenie poziomu AT poniżej 50% sprzyja powstawaniu
powikłań zakrzepowo-zatorowym.
3b. Układ fibrynolizy.
Istotą procesu fibrynolizy jest upłynnianie fibryny (włóknika) głównie przez plazminę,
ale także przez inne enzymy proteolityczne generowane we krwi, do produktów rozpadu
(degradacji) fibryny – FDP (fibrin degradation products). Istnieje też pojęcie trombolizy,
zjawiska występującego in vivo, które polega na trawieniu fibryny, obecnej w strukturze
zakrzepu naczyniowego. W organizmie człowieka istnieje powolna samoistna tromboliza,
częstsza w zakrzepach żylnych, albo tromboliza indukowana odpowiednim leczeniem
głównie aktywatorami plazminogenu (streptokinazą, tkankowym lub urokinazowym
aktywatorem plazminogenu), a także innymi lekami stosowanymi w zakrzepicy żylnej i
tętniczej. Zastosowanie terapii fibrynolitycznej w zawałach serca znacznie zmniejszyło

śmiertelność wśród leczonych tym sposobem pacjentów i od wielu lat jest leczeniem
obowiązującym.
Układ fibrynolizy składa się z aktywatorów plazminogenu tkankowego (t-PA) i
urokinazowego (u-PA), plazminogenu aktywowanego przez nie do plazminy, która
rozkłada fibrynę, a generowana w dużym nadmiarze trawi również fibrynogen.
Mechanizmem regulującym nadmierną aktywację fibrynolizy są specyficzne
substancje hamujące inhibitory aktywatorów plazminogenu przede wszystkim typu 1
znajdującego się w nadmiarze we krwi. Mniejsze znaczenie w warunkach prawidłowych mają
inhibitory PAI-2 (syntetyzowany przez łożysko) i PAI-3 oraz alfa-2 antyplazmina (alfa-2AP)
inhibitor plazminy. W stanach nadmiernej plazminogenezy i wyczerpaniu alfa-2AP,
inhibitorem plazminy drugiego rzutu jest alfa-2-makroglobulina (alfa-2M).
Proces fibrynolizy jest zjawiskiem wtórnym wobec procesu krzepnięcia krwi.
Generowana we krwi trombina poza wykrzepianiem fibrynogenu do fibryny, pobudza
komórki śródbłonka naczyń do syntezy a przede wszystkim do sekrecji t-PA, który adsorbując
się na zakrzepie aktywuje obecny tam plazminogen do plazminy, rozkładającej włóknik.
Składniki układu fibrynolizy i niektóre ich właściwości przedstawiono na tabeli 3.
Tabela 3. Główne składniki układu fibrynolizy (według M. Czokało-Plichty, Patofizjologia,
red. S. Maślińskiego i J. Ryżewskiego, PZWL, W-wa 2002).
Substancje układu fibrynolizy Skrót
Masa
cząsteczkowa
D
Stężenie w
osoczu
Okres
połowicznego
zaniku
1.
Tkankowy aktywator
plazminogenu
2.
Urokinazowy aktywator
plazminogenu
3.
Inhibitory aktywatorów
plazminogenu 1
2
3
4.
Czynnik XII
5.
Prekalikreina
6.
Plazminogen
7.
2
-antyplazmina
t-PA
u-PA
PAI-1
PAI-2
PAI-3
XII
PK
2
AP
68 000
54 000
52 000
46 000 – 70 000
57 000
80 000
85 000
92 000
70 000
70 pmol
150 pmol
1 nmol
100 pmol
35 nmol
375 nmol
450 mol
2 mol
1 pmol
4 min
8 min
20 min
2 dni
2 dni
2,2 dnia
2,2 dnia
Ryc4
Przekształcanie plazminogenu w plazminę przebiega dwoma drogami: torem zewnątrz i
wewnątrzpochodnym pokazanym na rycinie 2.
Temat XVII. Skazy krwotoczne
Maria Kotschy
Definicja
Mianem skazy krwotocznej określa się skłonność do krwawień w obręb tkanek (np. skóry i
błon śluzowych), narządów (np. nosa, stawów) oraz układów (np. pokarmowego, moczowo-
płciowego, ośrodkowego układu nerwowego).

Podział
Skazy krwotoczne, zgodnie z udziałem w procesie hemostazy różnych mechanizmów,
dzielimy na:
a.
skazy naczyniowe,
b.
skazy płytkowe,
c.
skazy osoczowe (obejmujące zaburzenia w układzie krzepnięcia krwi i fibrynolizy),
d.
skazy mieszane (złożone)
Każdy rodzaj skazy może być uwarunkowany genetycznie lub może mieć charakter nabyty
(wtórny). W wielu nabytych skazach krwotocznych dochodzi zarówno do zmian w obrębie
drobnych naczyń krwionośnych, płytek krwi i procesu krzepnięcia. W przebiegu wielu chorób
np. w ostrej białaczce, aplazji szpiku czy marskości wątroby może występować skaza
krwotoczna maskująca właściwy obraz choroby.
Objawy kliniczne
Skazy krwotoczne pochodzenia naczyniowego i płytkowego różnią się w obrazie klinicznym
zasadniczo od skaz osoczowych spowodowanych zaburzeniami krzepnięcia krwi. W
przebiegu skaz naczyniowych i płytkowych najczęściej występują wybroczyny, sinice i
wylewy podskórne a także krwawienia z nosa i z dróg rodnych, pojawiające się na ogół
bezpośrednio po urazie i dające się dość łatwo opanować miejscowym uciskiem.
W skazach spowodowanych zaburzeniami hemostazy osoczowej dominują wylewy
domięśniowe, dostawowe, duże krwawienia pourazowe występujące później, po kilku lub
kilkudziesięciu godzinach od urazu. Są trudne do miejscowego opanowania.
Skaza krwotoczna może być jawna lub utajona tj. taka, która ujawnia się dopiero po
zadziałaniu czynnika sprawczego (urazu, usunięcia zęba, zabiegu chirurgicznego, zakażenia
lub leków).
Badanie laboratoryjne
Ustalenie dokładnej przyczyny skazy krwotocznej umożliwia przeprowadzenie badań układu
hemostazy. Najpierw wykonuje się testy podstawowe (tzw. skriningowe) określając liczbę
płytek krwi, czas krwawienia, czasy krzepnięcia osocza: protrombinowy, kaolinowo-
kefalinowy (aPTT) i poziom fibrynogenu (często jakościowo określanego pomiarem czasu
trombinowego osocza). Następnie dokonuje się oceny aktywności czynników krzepnięcia.
Naczyniowe skazy krwotoczne
Są związane z zaburzeniami budowy i czynności drobnych naczyń krwionośnych – tętniczek,
naczyń włosowatych i małych naczyń żylnych. Dzielimy je na: wrodzone i nabyte.
A. Wrodzone naczyniowe skazy krwotoczne.
1. Choroba Rendu-Ostera (wrodzona naczyniakowość krwotoczna)
Charakteryzuje się wrodzonym zaburzeniem budowy ściany naczyniowej. W obrębie błony
śluzowej nosa i jamy ustnej i innych odcinkach przewodu pokarmowego występują czerwone,
okrągłe zmiany o średnicy 1 – 3 mm blednące pod wpływem ucisku. U 80% chorych
występują krwawienia z nosa pojawiające się w wieku pokwitania i nasilające się w starszym
wieku.
2. Zespół Marfana

Zespół ten charakteryzuje się zaburzeniem struktury kolagenu, powodującym jego
zwiększoną rozpuszczalność. Wzrasta wydalanie hydroksyproliny z moczem. Chorobę
dziedziczy się autosomalnie dominująco. U chorych występują długie pająkowate kończyny
często z wadami budowy klatki piersiowej i układu sercowo-naczyniowego.
3. Zespół Ehlersa-Danlosa
Jest to rzadko występujące (1:200.000) wrodzone zaburzenie tkanki łącznej, polegające na
niedoborze okołonaczyniowego kolagenu i zwiększeniu liczby włókien elastycznych. Skóra
jest rozciągliwa. Zakres ruchów w stawach jest nadmiernie zwiększony. Chorzy z tym
zespołem występowali dawniej w cyrkach jako tzw. ludzie z gumy.
B. Nabyte naczyniowe skazy krwotoczne
Patogeneza
Nabyte plamice naczyniowe mają związek:
a.
ze stanem zapalnym naczyń, zwiększającym ich przepuszczalność (np. zakażenia),
b.
procesami immunologicznymi (przeciwciała lub kompleksy odkładające się w obrębie
naczyń),
c.
zaburzeniami syntezy lub niszczeniem włókien kolagenowych wchodzących w skład
ściany naczyniowej.
Badania laboratoryjne
W naczyniowych skazach krwotocznych badania laboratoryjne układu hemostazy nie
wykazują zazwyczaj odchyleń od normy w zakresie testów dotyczących płytek krwi,
osoczowych czynników krzepnięcia krwi i fibrynolizy. Może niekiedy występować dodatni
objaw opaskowy (Rumpel-Leede’go) lub dodatni test aspirynowy (pomiar czasu krwawienia
po zażyciu aspiryny).
Do nabytych skaz należy:
1/ zespół Schönleina-Henocha,
2/ plamice w przebiegu zakażeń, polekowe, starcze,
3/ plamice w nadczynności kory nadnercza,
4/ plamica w gnilcu (awitaminoza C) oraz
5/ plamica ortostatyczna.
Ad1. Zespół Schönleina-Henocha
Patogeneza
Jest plamicą alergiczną, u podłoża której leży zjawisko autoimmunizacji. Po przebytym
zakażeniu występuje stan zapalny naczyń ze zwiększoną ich przepuszczalnością.
Objawy kliniczne
Początek choroby jest nagły. Występują objawy skórne, stawowe, brzuszne i nerkowe. Na
wyprostnych powierzchniach kończyn, szczególnie dolnych ale także i górnych, występują
symetryczne wybroczyny.
Badania dodatkowe
Nie stwierdza się odchyleń od normy w badaniach hemostazy. Można natomiast stwierdzić
dodatni odczyn opaskowy. U chorych stwierdza się często w surowicy krwi czynnik
reumatoidalny oraz kompleksy immunologiczne, w skład których wchodzi przede wszystkim
IgA. Często objawy ustępują samoistnie.

Leczenie
Przy współistnieniu zmian nerkowych podejmuje się leczenie immunosupresyjne.
Płytkowe skazy krwotoczne.
Patogeneza
Płytkowe skazy krwotoczne mogą mieć związek:
a.
z nieprawidłową liczbą płytek krwi – małopłytowości lub
b.
z zaburzeniami czynności płytek krwi – trombocytopatie
1. Małopłytkowości. (trombocytopenie)
Są najczęstszą przyczyną skaz krwotocznych. Mogą być samodzielną chorobą lub objawem
innych schorzeń lub zespołów.
Podział małopłytkowości ze względu na mechanizm powstawania.
Małopłytkowości mogą być uwarunkowane:
a/ zmniejszonym wytwarzaniem płytek krwi,
b/ nieprawidłowym ich rozmieszczeniem (sekwestracją),
c/ nadmiernym ich niszczeniem lub
d/ nadmierną utratą płytek krwi np. przy krwotokach.
Objawy kliniczne.
Kliniczne objawy krwotocznej skazy małopłytkowej są podobne, niezależnie od ich
przyczyny. Małopłytkowość rzędu 30 – 100 x 10
9
/l może przebiegać bezobjawowo z
tendencją do siniaczenia po niewielkich urazach i krwawieniu z dziąseł np. po myciu zębów.
Liczba płytek 30 x 10
9
/l stanowi tzw. „minimum” hemostatyczne, przy którym może już dojść
do samoistnych krwawień. Skaza krwotoczna ma charakter skórno-śluzówkowy. Na skórze
głównie kończyn dolnych i tułowia oraz na śluzówkach jamy ustnej występują wybroczyny i
podbiegnięcia krwawe, krwawienia z nosa, dziąseł, dróg rodnych, przewodu pokarmowego i
najbardziej niebezpieczne do ośrodkowego układu nerwowego.
a.
Małopłytkowości uwarunkowane zmniejszonym wytwarzaniem płytek krwi.
Przyczyną może być uszkodzenie szpiku, w tym układu płytkotwórczego przez
różnorodne czynniki: leki (immunosupresyjne, cytostatyki), związki chemiczne (benzen),
promieniowanie jonizujące, zakażenia wirusowe np. HIV, czynniki immunologiczne i inne.
Małopłytkowość może występować także jako wynik wyparcia układu płytkotwórczego przez
procesy rozrostowe (białaczki, szpiczak mnogi, uogólnione chłoniaki oraz przerzuty
nowotworów do szpiku). Przyczyną może być także brak witaminy B
12
, kwasu foliowego czy
żelaza.
b. Małopłytkowości wywołane zwiększonym obwodowym niszczeniem płytek.
Immunologiczne: samoistna plamica małopłytkowa, małopłytkowość poprzetoczeniowa,
toczeń rumieniowaty układowy, chłoniaki złośliwe
Nieimmunologiczne: zakażenia, rozsiane krzepnięcie śródnaczyniowe, zespół hemolityczno-
mocznicowy, zakrzepowa plamica małopłytkowa
Najczęstszą przyczyną małopłytkowości wywołanej zwiększonym obwodowym niszczeniem
płytek jest tło immunologiczne. Przeciwciała przeciwpłytkowe (głównie klasy IgG, rzadziej
IgM) prowadzą do przyspieszonego niszczenia opłaszczonych płytek i skracają ich czas
przeżycia nawet do kilku godzin.
Rodzaje małopłytkowości

1a. Małopłytkowość samoistna
Ostra postać małopłytkowości.
Występuje głównie u dzieci zimą lub na wiosnę po przebyciu wirusowej infekcji. Choroba
rozpoczyna się nagle uogólnioną skazą krwotoczną (wybroczyny skórne, krwawienie z
dziąseł, nosa i przewodu pokarmowego). We krwi występują przeciwciała wolne lub
związane z płytkami. Szpik zawiera zwiększoną liczbę megakariocytów z zaburzeniami
czynności płytkotwórczej. U 80% chorych dochodzi do samoistnego wyleczenia.
Postać przewlekła małopłytkowości.
Obserwuje się ją głównie u dorosłych, 4 razy częściej u kobiet. Nasilenie skazy bywa różne.
Najczęściej występują wybroczyny na skórze i błonach śluzowych. Liczba płytek w granicach
30 – 100 x 10
9
/l. Na płytkach i lub w surowicy występują przeciwciała przeciwpłytkowe.
Megakariocyty szpiku zachowują się tak jak w postaci ostrej. Niebezpieczne dla życia w obu
postaciach są wylewy do ośrodkowego układu nerwowego. W leczeniu obu postaci stosuje się
glikokortikoidy.
1b. Małopłytkowości polekowe.
Wiele leków (najczęściej stosowanie chinidyny, chininy, sulfonamidów, soli złota) może
wywołać małopłytkowość na tle immunologicznym. Lek lub jego metabolity działają jako
hapten i po związaniu z glikoproteinami błony płytkowej lub białkami osocza powstaje
neoantygen stymulujący syntezę przeciwciał. Rozpoznanie polekowej małopłytkowości jest
możliwe po stwierdzeniu niewystępowania skazy przed zastosowaniem leku, ujawnieniu się
jej po 1-2 tygodniach jego stosowania oraz jej ustąpieniu po odstawieniu leku.
1c. Małopłytkowość poprzetoczeniowa.
Występuje zwykle u kobiet, które otrzymywały transfuzję w czasie ciąży. Po przetoczeniu
masy płytkowej lub erytrocytarnej 5 – 8 dnia występują objawy skazy ze szczególnie
niebezpiecznymi krwawieniami śródczaszkowymi (l. płytek < 10 x 10
9
/l). Małopłytkowość ta
jest spowodowana przez swoiste alloprzeciwciała przeciwpłytkowe obecne w surowicy krwi,
natomiast na płytkach krwi chorego nie można stwierdzić odpowiadającego im antygenu.
Leczeniem z wyboru jest podawanie dożylnie immunoglobulin (1g/kg m.c./dobę) lub mniej
aktywna plazmofereza.
1d. Zakrzepowa plamica małopłytkowa.
Choroba występuje rzadko. Związana jest z obecnością w mikrokrążeniu rozsianych
zakrzepików, głównie utworzonych z płytek, będących przyczyną małopłytkowości ze
zużycia. Towarzyszy jej niedokrwistość mikroangiopatyczna oraz niedokrwienie i
niedotlenienie tkanek i narządów. Najczęstszymi przyczynami tego zespołu są infekcje
bakteryjne i wirusowe, leki i ciąża. Do częstych objawów klinicznych poza małpłytkowością i
niedokrwistością należą zaburzenia neurologiczne, uszkodzenie nerek oraz gorączka.
Śmiertelność sięga 20 – 40%. Przyczyną powstawania zakrzepów w mikrokrążeniu jest
uszkodzenie komórek śródbłonka i obecność wielkich multimerów czynnika von
Willebranda, które łącząc się z glikoproteinami płytek (Ib/IX, V, IIb/IIIa) powodują ich
agregację. We krwi stwierdza się autoprzeciwciało przeciw wytwarzanej w wątrobie
metaloproteinazie ADAMS 13, która trawi multimer von Willebranda. W okresie zdrowienia
przeciwciało to zanika i struktura czynnika von Willebranda wraca do normy. Przy
przewlekłej postaci stwierdza się predyspozycję rodzinną.
1e. Zespół hemolityczno-mocznicowy.
Objawy: ostra niewydolność nerek, mikroangiopatyczna niedokrwistość hemolityczna,
małopłytkowość. Uważa się, że zespół ten występujący u dzieci poniżej 4 lat jest jedną z
postaci zakrzepowej plamicy małopłytkowej, której objawy ograniczają się przede wszystkim
do naczyń nerek. Uszkodzenie komórek śródbłonka naczyń przez różne czynniki
chorobotwórcze zmienia ich właściwości antygenowe co powoduje powstawanie przeciwko
nim autoprzeciwciał. Przez uszkodzony śródbłonek przedostają się do krążenia nerkowego
wielkie multimery czynnika von Willebranda, które wiążąc się z glikoproteinami płytek krwi
powodują miejscowe tworzenie się agregatów płytkowych zamykających światło naczyń.
Stężenie osoczowe metaloproteinazy ADAMS 13 trawiącej czynnik von Willebranda jest
prawidłowe.
2. Trombocytopatie.
Trombocytopatie polegają na zaburzeniach czynności płytek charakteryzujących się
występowaniem skazy krwotocznej i przedłużonego czasu krwawienia. Są spowodowane
wrodzonym lub nabytym defektem funkcji hemostatycznych płytek krwi.
A/ Wrodzone:
zaburzenie adhezji płytek (choroba Bernarda-Souliera), zaburzenie agregacji płytek
(trombastenia
Glanzmanna),
zaburzenia
w
zakresie
uwalniania
substancji
wewnątrzpłytkowych, choroba puli magazynowej (niedobór gęstych ziarnistości), zespół
szarych płytek (niedobór alfa ziarnistości).
B/ Nabyte: polekowe, w mocznicy, w przewlekłych zespołach mieloproliferacyjnych, w
gammapatiach monoklonalnych, w chorobach wątroby.
Osoczowe skazy krwotoczne
Skazy osoczowe są spowodowane brakiem czynnika krzepnięcia, zmiejszeniem jego ilości
we krwi albo obecnością nieaktywnego białka. Skazy można podzielić na postaci : A.
wrodzone i B. nabyte
A. Wrodzone skazy osoczowe
Podłożem patogenetycznym wrodzonych skaz krwotocznych jest w większości przypadków
zaburzona synteza jednego czynnika krzepnięcia manifestująca się przede wszystkim jego
niedoborem lub nieprawidłową budową cząsteczki.
Występowanie wrodzonych osoczowych skaz krwotocznych.
Pod względem częstości występowania, a może ze względu na ciężkość choroby, pierwsze
miejsce wśród wrodzonych skaz krwotocznych zajmuje choroba von Willebranda, następnie
hemofilia A, hemofilia B i hipokonwertynemia (niedobór czynnika VII). Inne wrodzone
defekty krzepnięcia należą do rzadkości.
1. Hemoflie
Definicja
Hemofilia A polega na wrodzonym braku lub niedoborze VIII czynnika krzepnięcia krwi,
zwanym globuliną (czynnikiem) przeciwhemofilową A, a hemofilia B na niedoborze IX
czynnika krzepnięcia tj. globuliny przeciwhemofilowej B.
Częstość występowania
Częstość występowania hemofilii w Polsce ocenia się na 1:16.000 mieszkańców. Według
Łopaciuka (1981) obydwa rodzaje hemofilii stanowią 87% wszystkich przypadków
wrodzonych zaburzeń krzepnięcia krwi, a stosunek częstości występowania hemofilii A do B

wynosi jak 6:1. W Wielkiej Brytanii hemofilię B nazywa się chorobą „Christmas” od
nazwiska chorego, u którego wykryto ten niedobór.
Dziedziczenie
Oba rodzaje hemofilii A i B dziedziczą się jako cechy recesywne, sprzężone z chromosomem
X. Hemofilia występuje u mężczyzn a kobiety są jej przenosicielkami. Kobieta przenosicielka
jest heterozygotą, gdyż tylko w jednym z jej dwóch chromosomów X znajduje się gen
hemofilii. Wśród potomstwa hemofilika i zdrowej kobiety, wszystkie córki będą
przenosicielkami, zaś wszyscy synowie będą zdrowi i nie przekażą choroby żadnemu z
potomków. W około 30% przypadków hemofilia A występuje sporadycznie tj. po pierwszy
raz w danej rodzinie.
Obraz kliniczny
Oba rodzaje hemofilii maja taki sam obraz kliniczny a jego ciężkość zależy od stopnia
niedoboru odpowiedniego czynnika. W zależności od poziomu białka niedoborowego
wyróżnia się w hemofiliach:
a/ ciężką postać hemofilii (cz. VIII lub IX <1%),
b/ umiarkowaną (1-5%),
c/ łagodną (>5%).
Hemoflilia (zarówno A jak i B) charakteryzuje się nasilonymi krwawieniami występującymi
zazwyczaj samoistnie lub pod wpływem różnorodnych urazów lub zabiegów (np.
chirurgicznych). Krwawienia pod postacią wylewów podskórnych, domięśniowych i
dostawowych, które występują najczęściej do stawów kolanowych i łokciowych, powodują
obrzęk, ograniczenie ruchomości stawu, prowadząc w efekcie do trwałych uszkodzeń w
układzie kostnostawowym (tzw. artropatie hemofilowe).
U chorych z umiarkowaną postacią hemofilii krwawienia występują rzadziej i są mniej
nasilone. Chorzy z łagodną postacią hemofilii często nie wiedzą o istnieniu skazy
krwotocznej, która ujawnia się przypadkowo np. po usunięciu zęba lub po zabiegu
operacyjnym. Zabiegi chirurgiczne powinny być przeprowadzane po uzupełnieniu
niedoborowego czynnika VIII lub IX. Należy zwrócić uwagę, że wśród przyczyn zgonów
chorych na hemofilię pierwsze miejsce zajmują krwawienia śródczaszkowe, występujące u 2
– 8% hemofilików, głównie dzieci.
Badania laboratoryjne.
Podstawowym badaniem, występującym zarówno hemofilii A jak i B jest przedłużony czas
kaolinowo-kefalinowy (aPTT). Przedłużony czas krzepnięcia krwi pełnej występuje przede
wszystkim w ciężkiej postaci hemofilii. Po stwierdzeniu przedłużonego aPTT u chorego
podejrzanego o hemofilię należy oznaczyć poziom czynników VIII i IX w osoczach
niedoborowych w te białka (tj. w osoczach ciężkiej hemofilii A i B). U chorych z ciężką
postacią hemofilii szczególnie typu A należy wykluczyć obecność krążących
antykoagulantów. We krwi chorych na hemofilię stwierdza się prawidłową liczbę płytek krwi,
prawidłowy czas krwawienia i prawidłowy czas protrombinowy.
Leczenie.
W okresie krwawienia leczenie hemofilii polega na dożylnym podawaniu preparatów
brakującego czynnika krzepnięcia krwi. Jako źródło czynnika VIII najczęściej stosuje się
świeżo mrożone osocze, kriopecypitat, koncentraty uzyskane z osocza lub na drodze
inżynierii genetycznej. Wszystkie preparaty są obecnie odpowiednio preparowane, aby
uniemożliwić wtórne zakażenia wirusowe (wzw typu B i C oraz HIV). Rodzaj krwawienia i

jego nasilenie decydują o wielkości stosowanej dawki czynnika VIII. Ważnym elementem
leczenia hemofilików jest rehabilitacja ortopedyczna.
2. Choroba von Willebranda.
Definicja
Istotą choroby von Willebranda jest niedobór, brak lub nieprawidłowa budowa czynnika von
Willebranda (vWf).
Występowanie i dziedziczenie
Uważa się, że choroba ta zajmuje w Polsce pierwsze miejsce po hemofilii A. Wydaje się
jednak, że występuje częściej, lecz chorzy z tym niedoborem nie są rejestrowani. vWf
występuje pod postacią multimerów o różnej masie cząsteczkowej. W procesie hemostazy
najbardziej aktywne są wielkocząsteczkowe multimery. Choroba von Willebranda dziedziczy
się autosomalnie, dominująco lub recesywnie, występuje zarówno u kobiet jak i u mężczyzn
Podział
W 1994 roku zaproponowano obecnie obowiązującą klasyfikację choroby von Willebranda.
Typ 1 jest najczęstszą i najbardziej łagodną postacią choroby von Willebranda.
Występuje u 80% chorych. W tym typie obserwuje się niedobór VWf i czynnika VIII.
Typ 2 (podtypy A i B) rozpoznawany jest u 20% chorych. W podtypie 2A brak jest
wielkocząsteczkowych
multimerów
a
w
podtypie
2B
występuje
równocześnie
małopłytkowość.
Typ 3 jest najcięższą postacią tej choroby występującą bardzo rzadko. W tym typie
stwierdza się brak lub śladowe ilości osoczowego i płytkowego vWF oraz wtórny niedobór
czynnika VIII na poziomie 2 – 10% normy.
Obraz kliniczny choroby.
U chorych występują krwawienia charakterystyczne dla zaburzeń hemostazy pierwotnej
(defekt płytkowo-włośniczkowy) jak i zaburzenia krzepnięcia krwi. Osoby z tą skazą należące
do typu 1, krwawią z nosa i występuje u nich skłonność do powstawania pourazowych
sińców, szczególnie na kończynach. Kobiety mogą mieć przedłużające się miesiączki.
Występowanie krwawień po ekstrakcji zębów lub zabiegach operacyjnych może być dopiero
podstawą do rozpoznania skazy. W typie 2 a szczególnie ciężkim 2B krwawienia ujawniają
się już w dzieciństwie i są bardziej obfite. Klinicznie mogą przypominać hemofilię.
Badania laboratoryjne
Laboratoryjnie chorobę tę rozpoznaje się na podstawie:
a. przedłużonego czasu krwawienia,
b.
obniżonego stężenia czynników vWf i VIII,
c.
zmniejszonej agregacji płytek krwi pod wpływem ristocetyny,
d.
czas kaolinowo-kefalinowy (aPTT) może być przedłużony lub prawidłowy.
Leczenie
Jest podobne jak w hemofilii A. Stosuje się świeżo mrożone osocze, krioprecypitat i
koncentraty czynnika VIII zawierające czynnik vWf.
3. Niedobór czynnika VII. Hipokonwertynemia.
Występowanie i dziedziczenie

Częstość jej występowania ocenia się na 1:500.000. Choroba ta dziedziczy się w sposób
autosomalny recesywny.
Obraz kliniczny
Charakteryzuje się krwawieniami z nosa, łatwym powstawaniem sińców, u kobiet obfitymi
miesiączkami oraz krwawieniami po usunięciu zębów.
Badania laboratoryjne
W hipokonwertynemii występuje przedłużony czas protrombinowy i niedobór czynnika VII
przy prawidłowym czasie kaolinowo-kefalinowym.
Leczenie
Ze względu na krótki okres półtrwania cz. VII (4 – 6 godzin) w krwawieniach należy
przetaczać osocze mrożone lub koncentraty czynnika VII kilkakrotnie w ciągu dnia.
B/ Nabyte zaburzenia krzepnięcia krwi.
Definicja
W przeciwieństwie do wrodzonych osoczowych skaz krwotocznych, które są z reguły
spowodowane niedoborem jednego czynnika krzepnięcia, skazy krwotoczne nabyte mają
charakter złożony i wiążą się najczęściej z niedoborem wielu czynników krzepnięcia a
czasem są skojarzone z zaburzeniami płytek krwi lub naczyń.
Przyczyny
Nabyte skazy krwotoczne mogą być spowodowane:
a. zaburzoną syntezą czynników krzepnięcia krwi (choroby wątroby, awitaminoza K),
b. wzmożonym zużyciem lub niszczeniem czynników krzepnięcia (zespoły DIC, enzymy
proteolityczne),
c.
unieczynnieniem czynników krzepnięcia przez antykoagulanty (inhibitory).
Objawy kliniczne
Na ogół skaza krwotoczna na tle nabytych zaburzeń krzepnięcia ma łagodniejszy przebieg niż
np. hemofilia. Wyjątkiem są gwałtowne krwotoki w powikłaniach położniczych i
chirurgicznych wywołanych zespołem śródnaczyniowego wykrzepiania, chorobą krwotoczną
noworodków, czy przedawkowaniem stosowanych w leczeniu zakrzepic antykoagulantów
(heparyny i doustnych antykoagulantów – pochodnych dikumarolu czy indandionu).
1. Nabyte skazy krwotoczne w chorobach wątroby i niedoborze witaminy K.
1a. Choroby wątroby
Wątroba pełni ważną rolę w regulacji procesów krzepnięcia krwi i fibrynolizy.
Syntetyzuje wiele białek: I, II, V, VII, IX, X, XI, XII, PK, WK, antytrombinę, plazminogen,
alfa-2 antyplazminę, oczyszcza krew z aktywnych czynników krzepnięcia, aktywatorów
plazminogenu oraz produktów rozpadu fibryny.
W chorobach wątroby może dochodzić:
a.
do zmniejszonej syntezy białek osoczowych krzepnięcia i fibrynolizy,
b.
przyspieszonego ich rozpadu,
c.
wytwarzania nieprawidłowych białek,
d.
upośledzonego oczyszczania aktywnych form białek osoczowych,
e.
zmniejszenia liczby płytek krwi lub ich jakościowych zmian.
1b. Niedobór witaminy K

Patogeneza
Zaburzenie syntezy czynników krzepnięcia przez wątrobę może być spowodowane
niedoborem witaminy K, która jest niezbędna w końcowym okresie syntezy protrombiny
oraz czynników VII, IX i X do potranslacyjnej biosyntezy kwasu glutaminowego obecnego
w tych czynnikach. Źródłem witaminy K w organizmie człowieka jest pokarm roślinny oraz
bakterie flory bakteryjnej przewodu pokarmowego. Witamina K rozpuszcza się w tłuszczach i
wchłania w obecności kwasów tłuszczowych.
Do najważniejszych przyczyn jej niedoboru należą:
a.
jałowy przewód pokarmowy i zaburzenia wchłaniania (noworodki),
b.
niszczenie flory bakteryjnej (sulfonamidy, antybiotyki),
c.
zaburzenia wydzielania żółci oraz
d.
leczenie doustnymi antykoagulantami (pochodne dikumarolu i indandionu).
Objawy kliniczne
W niedoborze czynników zespołu protrombiny (II, VII, IX, X) występuje skłonność do
siniaczenia, krwawienia śluzówkowe, z przewodu pokarmowego oraz krwiomocz.
Leczenie
W leczeniu stosuje się domięśniowo lub dożylnie preparaty witaminy K. 1
1c. Choroba krwotoczna noworodków związana jest z niedoborem witaminy K.
U 0,1 – 1% noworodków obniżenie czynników zespołu protrombiny jest tak duże, że
drugiego lub trzeciego dnia życia występuje skaza krwotoczna manifestująca się podskórnymi
wylewami krwi, smolistym stolcem oraz krwawieniem do ośrodkowego układu nerwowego.
2. Krążące antykoagulanty
Definicja
Krążące antykoagulanty są immunoglobulinami, skierowanymi przeciw jednemu z czynników
krzepnięcia, przy czym większość z nich stanowią inhibitory czynnika VIII.
Występowanie
Inhibitory czynnika VIII występują najczęściej:
a. w przebiegu leczenia substytucyjnego hemofilii
b. u kobiet w okresie pierwszych miesięcy po porodzie
c. oraz w chorobach o podłożu immunologicznym.
Badania laboratoryjne
Za obecnością antykoagulanta czynnika VIII przemawia znacznie przedłużony czas
kaolinowo-kefalinowy badanego osocza i mieszaniny tego osocza z równą ilością osocza
prawidłowego po 30 minutach inkubacji w temperaturze 37
°
C. Inhibitory innych czynników
krzepnięcia występują bardzo rzadko.
Leczenie
Leczenie polega na stosowaniu leków immunosupresyjnych: glikokortikosteroidów,
cytostatyków lub dożylnie immunoglobulin.
3. Nabyta skaza krwotoczna występuje także w zespołach śródnaczyniowego
wykrzepiania.

Temat XVIII. Zakrzepica. Trombofilia.
Maria Kotschy
Dane ogólne
U zdrowego człowieka, dzięki naczyniowym i osoczowym mechanizmom
antykoagulacyjnym krew krąży w nieuszkodzonych naczyniach w stanie płynnym.
Uszkodzenie śródbłonka naczyń, aktywacja płytek i/lub procesu krzepnięcia krwi przez różne
czynniki chorobotwórcze prowadzi do powstania w naczyniach krwionośnych zakrzepu.
Definicja
Zakrzep jest utworzony przez usieciowaną fibrynę, w oczkach której znajdują się elementy
morfotyczne krwi i/lub przez skupisko zagregowanych płytek krwi. W rozwoju zakrzepu
uczestniczą te same mechanizmy jak w powstawaniu czopu hemostatycznego hamującego
krwawienie. W przeciwieństwie do niego zakrzep jest strukturą dynamiczną, ulegającą
stałemu narastaniu i rozpuszczaniu. Zakrzep może być przyścienny lub może całkowicie
zamykać światło naczynia, utrudniając lub uniemożliwiając przepływ krwi.
1.Ze względu na lokalizację odróżniamy zakrzepy:
a/ żylne
b/ tętnicze.
1a. Zakrzepy żylne
Są to tzw. zakrzepy czerwone o luźniejszej budowie niż zakrzepy tętnicze, mniejszym
związku ze ścianą naczyniową, łatwiejsze do upłynnienia. Niebezpieczeństwo groźnych dla
życia powikłań, związane jest z odrywaniem się fragmentów zakrzepów tzw. zatorów i ich
wędrówką do rozgałęzień tętnicy płucnej, powodujących zawały płuc, a przy niedrożności
dużych odgałęzień tętnicy płucnej, mogą one prowadzić do śmierci.
Występowanie.
Zakrzepicę żylną i związane z nią powikłania nazywamy obecnie żylną chorobą zakrzepowo-
zatorową. W USA z powodu tej choroby hospitalizowanych jest rocznie 300.000-600.000
chorych, z których około 50.000 umiera. W Polsce brak jest takich danych. Choroba ta
częściej występuje u ludzi starszych.
Przyczyny.
Do wystąpienia żylnej choroby zakrzepowo-zatorowej predysponują:
a/ wszelkiego rodzaju urazy,
b/ zabiegi operacyjne szczególnie ortopedyczne,
c/ nowotwory złośliwe,
d/ choroby serca,
e/ długotrwałe unieruchomienie,
f/ poród, połóg, leczenie estrogenami,
g/ otyłość i przebyte zakrzepice.
1b. Zakrzepy tętnicze
Mają barwę białawą, szczególnie ich przednia część, gdyż zbudowane są w przeważającej
części z płytek krwi. Mają bardziej zbitą strukturę i są trudniejsze do upłynnienia. Zakrzepy
tętnicze powodują niedokrwienie tkanek i narządów.
Rudolf Virchow już w 1855 roku podał trzy zasadnicze przyczyny zakrzepicy naczyniowej:

a. zmiany w przepływie krwi,
b. uszkodzenie ściany naczyniowej,
c. zmiany w składzie samej krwi.
Zmiany w przepływie krwi polegają na zmianach reologicznych tj. strukturalno
mechanicznych właściwościach naczyń i krwi np. na zwolnieniu przepływu krwi w sercu i
naczyniach krwionośnych (szczególnie przy ich podziale) oraz na zagęszczeniu krwi i
zwiększonej jej lepkości.
Mechanizmem zapłonowym w powstawaniu zakrzepów naczyniowych, szczególnie
tętniczych jest przede wszystkim uszkodzenie ściany naczyniowej przez:
a. proces miażdżycowy,
b. nadciśnienie tętnicze,
c. endotoksyny bakteryjne i wirusy,
d. tlenek węgla, nadtlenki lipidowe,
e. kompleksy immunologiczne,
f. hiperhomocysteinemię.
Czynnikami inicjującymi zakrzepicę zarówno żylną jak i tętniczą mogą być także
zagęszczenie krwi jak i zaburzenia hemostazy płytkowej i osoczowej:
a. nadkrwistość (>6,0 x 10
12
/l),
b. nadpłytkowość (>600 x 10
9
/l) lub zwiększona aktywność płytek,
c. hiperfibrynogenemia (> 5g/l),
d. wzrost aktywności czynnika VII,
e. obniżenie aktywności fibrynolitycznej.
2. Trombofilia.
U wielu chorych (według niektórych statystyk nawet u ~ 50%) szczególnie młodszych przed
45 rokiem życia nie udaje się ustalić związku przyczynowego między zakrzepicą a chorobą
podstawową czy leczeniem. Często zakrzepy u młodych osób pojawiają się jakby bez
uchwytnej przyczyny. Intensywne badania doświadczalne i kliniczne wykazały, że przyczyną
zakrzepicy przede wszystkim u ludzi młodych mogą być wrodzone lub nabyte zaburzenia
hemostazy osoczowej. Dla tych zaburzeń wprowadzono nazwę trombofilie.
Trombofilia oznacza wrodzone lub nabyte zaburzenia hemostazy usposabiające do
występowania zakrzepicy.
W trombofilii zakrzepica jest najczęściej następstwem zachwiania równowagi pomiędzy
czynnikami sprzyjającymi aktywacji krzepnięcia a naturalnymi układami antykoagulacyjnymi
krwi. Mimo, że żylna choroba zakrzepowo-zatorowa jest najczęstszym przejawem tych
defektów, u wielu osób występują także zakrzepy tętnicze.
Występowanie.
Badania przeprowadzone w 37 holenderskich szpitalach wykazały, że na 100.000
mieszkańców przypada 40 osób z rodzinną trombofilią. Jej częstość występowania jest zatem
pięciokrotnie większa niż w hemofilii.
A. Wrodzona trombofilia
Wieloletnie obserwacje chorych w wielu krajach wykazały, że w niektórych rodzinach
zakrzepica żył głębokich i zatorowość płucna występują znacznie częściej, niż w ogólnej
populacji. Rodzinne występowanie zakrzepicy wskazało na możliwość udziału czynników
genetycznych w jej rozwoju.

Przyczyny.
Najczęściej występujące przyczyny wrodzonej trombofilii to:
a.
niedobór antytrombiny (dawniej zwaną antytrombiną III),
b.
niedobór białka C i S,
c.
oporność czynnika V na aktywne białko C (aPC-R),
d.
mutacje genu protrombiny.
Opisywane są także inne rzadko występujące lub prawdopodobne przyczyny trombofilii:
Prawdopodobne przyczyny trombofilii:
-
dysfibrynogenemia,
-
dysplazminogenemia,
-
zwiększenie we krwi zawartości glikoproteiny bogatej w histydynę (HRGP),
-
niedobór II kofaktora heparyny,
-
niedobór TFPI (inhibitora drogi aktywacji czynnika tkankowego),
-
nadmiar PAI-1 (inhibitora aktywatorów plazminogenu typu1),
-
niedobór t-PA ( tkankowego aktywatora plazminogenu),
-
dysfunkcja TM (trombomoduliny),
-
homocysteinemia.
Wrodzoną trombofilię polegającą na niedoborze antytrombiny opisał po raz pierwszy w
1963 roku w norweskiej rodzinie Egeberg. Drugi defekt odpowiedzialny za tendencję do
powstawania zakrzepicy a mianowicie wrodzony niedobór białka C opisano w 1981 roku, a
wkrótce potem wykryto wrodzony niedobór białka S. Dopiero w 1993 roku odkryto
wrodzoną oporność czynnika V na aktywne białko C. Dotychczas poznano także wiele
innych wrodzonych defektów hemostazy mogących sprzyjać rozwojowi zakrzepicy (patrz
wyżej). Niektóre z nich występują bardzo rzadko a w innych nie wykazano jeszcze
bezpośredniego związku z powstaniem zakrzepicy. Osoby z wrodzonym niedoborem
antytrombiny, białka C i białka S są heterozygotami nieprawidłowego genu a aktywność
niedoborowego białka w ich osoczu wynosi 40 – 60% prawidłowego poziomu. Rozróżniamy
dwa typy białek niedoborowych:
-
typ I – występuje zmniejszona synteza niedoborowego białka
-
typ II – charakteryzuje się jakościowym defektem cząsteczki białka
1. Niedobór antytrombiny (AT).
Antytrombina (dawniej zwana antytrombiną III) jest glikoproteiną syntetyzowaną w wątrobie
i w komórkach śródbłonka a wydalaną przez nerki. Jest głównym inhibitorem enzymów
układu krzepnięcia krwi, hamuje trombinę, aktywny czynnik X a w mniejszym stopniu
czynniki XIIa, XIa, IXa tworząc z nimi nieaktywne kompleksy. W obrębie cząsteczki AT
znajdują się dwa miejsca wiążące jedno dla trombiny (lub innego enzymu) a drugie dla
heparyny. Gen kodujący AT znajduje się na ramieniu długim pierwszego chromosomu.
Wśród mutacji tego genu można rozróżnić dwa typy. W pierwszym typie ilościowym (I)
jeden z pary alleli nie ulega ekspresji, co prowadzi do 50% redukcji AT we krwi krążącej.
Nie obserwowano dotąd homozygot z tą mutacją, co może sugerować, że całkowity brak AT
jest letalny. W typie II (jakościowym) zmutowany gen koduje białko o zmienionych
właściwościach, nie spełniające funkcji inhibitorowych. Choroba zakrzepowa spowodowana
niedoborem AT ma zazwyczaj ciężki przebieg i często bywa wikłana zatorowością płucną.
Występowanie
U chorych z nawracającą zakrzepicą, niedobór AT występował u 0,5 do 7% badanych,
stwierdzono u nich także heparynooporność. Wrodzony niedobór AT stwierdza się u 0,02%
ludności.

2. Niedobór białka „C” (PC).
Białko C jest witamino-K-zależną proteazą serynową syntetyzowaną w wątrobie. W osoczu
krwi białko C jest nieaktywne. Dopiero trombina w kompleksie z trombomoduliną (błonowy
receptor komórek śródbłonka dla trombiny) aktywuje je do formy aktywnej (APC). APC w
obecności kofaktora białka S, unieczynnia poprzez proteolizę aktywne czynniki krzepnięcia
Va i VIIIa, zmniejszając w ten sposób generację trombiny. Równocześnie APC inaktywuje
PAI-1, nasilając proces fibrynolizy.
Nawracająca zakrzepica żył głębokich, ujawniająca się u heterozygot w wieku dojrzałym, jest
najczęstszym objawem klinicznym niedoboru PC. U homozygot objawy pojawiają się we
wcześniejszych okresach życia. U noworodków z brakiem białka C obserwowano masywną
zakrzepicę z plamicą piorunującą i martwicą skóry.
Występowanie
Częstość występowania niedoboru PC u chorych z zakrzepicą ocenia się na 1-9%. Wrodzony
niedobór białka C stwierdzono u 0,2 do 0,4% ludności.
3. Niedobór białka „S” (PS)
PS jest glikoproteiną witamino-K-zależną syntetyzowaną w wątrobie i komórkach śródbłonka
naczyń. Spełnia rolę kofaktora aktywnego białka C przy unieczynnianiu aktywnych białek
krzepnięcia krwi tj. czynników Va i VIIIa, zmniejszając w ten sposób aktywność
koagulacyjną krwi. W osoczu występuje w dwóch postaciach. Około połowa PS jest związana
z należącym do układu dopełniacza białkiem wiążącym C4b w nieaktywny kompleks. Druga
część PS krąży w formie wolnej i tylko ona posiada aktywność antykoagulacyjną. W
niedoborze PS typu I występuje zmniejszenie stężenia obu postaci PS. Typ IIa charakteryzuje
się zmniejszonym stężeniem wolnego PS a typ IIb tylko obniżoną aktywnością tego białka.
Występowanie.
U chorych z rodzinną nawracającą zakrzepicą, niedobór białka S występował z częstością 1 –
13%. Wrodzony niedobór białka S stwierdzono u 1% ludności.
4. Oporność na aktywne białko C (APC-resistance –APC-R).
Punktowa mutacja genu czynnika V (mutacja Leiden) powoduje zamianę pojedynczego
aminokwasu argininy na glicynę w pozycji 506 łańcucha aminokwasowego czynnika V.
Warunkuje to oporność tego czynnika na proteolityczne działanie APC i w efekcie prowadzi
do zwiększonej trombinogenezy. W około 85% przypadków APC-R spowodowane jest
mutacją Leiden. W pozostałych 15% przyczyna nie jest w pełni znana. W badaniach
przeprowadzonych na dużych grupach ludzi zdrowych oporność na białko C występowała u 1
- 15% ogólnej populacji, w Polsce u 5%. Natomiast u chorych z nawracającą zakrzepicą
oporność na białko C stwierdzono aż u 52%. Defekt ten jest zatem najczęstszą przyczyną
trombofilii.
Występowanie
W badaniach przeprowadzonych w Polsce oporność na PC stwierdzono u 1% osób zdrowych
i u 19% chorych na żylną chorobę zakrzepowo-zatorową.
5. Przed kilku laty opisano mutację genu protrombiny, która ma także uspasabiać do
występowania zakrzepicy żylnej. Mutację tę stwierdzono u 2,3% osób zdrowych i u 6,2%
osób z przebytą zakrzepicą żył głębokich. Ryzyko występowania zakrzepicy u osób
heterozygotycznych wydaje się być znacznie mniejsze, niż przy innych defektach.

Objawy kliniczne wrodzonej trombofilii.
Charakterystyczne cechy wrodzonej trombofilii to: występowanie zakrzepicy u członków
rodziny, występowanie zakrzepicy przed 45 rokiem życia i nawroty zakrzepicy. Klinicznie u
tych osób najczęściej występuje zakrzepica żył głębokich kończyn dolnych, prowadząca
często do zatorowości płucnej. U osób z niedoborem AT zakrzepowe zapalenie żył
powierzchniowych występuje rzadko, natomiast znacznie częściej przy niedoborze PC i PS.
Charakterystyczna dla trombofilii jest zakrzepica o nietypowej lokalizacji np. żył krezkowych
lub mózgu, ale także występowanie zakrzepicy po błahych przyczynach (długie podróże
samolotem lub samochodem). Purpura fulminans (plamica piorunująca) występuje u
homozygotycznych noworodków z niedoborem białka C, a martwica skóry po doustnych
antykoagulantach powstaje na tle niedoboru białka C lub S. Dowiedziono, że oporność na PC
stanowi mniejsze zagrożenie zakrzepicą niż niedobór AT i białka C. Okazało się, że w
trombofilii mogą występować złożone defekty hemostazy (np. niedobory AT oraz białka C
czy S), w których zagrożenie zakrzepicą jest większe i na ogół występuje w młodszym wieku.
U większości osób z wrodzoną trombofilią pierwszy epizod zakrzepicy może być wywołany
przez tzw. klasyczne czynniki ryzyka zakrzepicy (ciąża, poród, zabiegi operacyjne, leki
antykoncepcyjne, unieruchomienie).
Badania laboratoryjne.
Rutynowe badania układu hemostazy nie pozwalają na rozpoznanie trombofilii. Konieczne
jest określenie stężenia i aktywności AT, PC, PS oraz zbadanie oporności na PC przy pomocy
specjalnych odczynników w dobrze wyposażonych pracowniach hematologicznych. Nie
należy przeprowadzać badań diagnostycznych w kierunku trombofilii w okresie aktywnej
zakrzepicy (do 6 tygodni) oraz podczas leczenia przeciwzakrzepowego heparyną lub
doustnymi antykoagulantami.
Leczenie.
Przy niedoborze AT, oprócz leczenia przeciwzakrzepowego, powinno się podawać preparaty
antytrombiny. U chorych z trombofilią o dużym ryzyku wskazana jest profilaktyka
przeciwzakrzepowa.
B. Nabyta trombofilia.
Częściej niż postacie wrodzone występuje nabyta trombofilia. Do najważniejszych jej
przyczyn należą:
a. zespół antyfosfolipidowy,
b. przewlekłe zespoły mieloproliferacyjne,
c. nocna napadowa hemoglobinuria,
d. nowotwory złośliwe,
e. zespół nerczycowy,
f. chemioterapia przeciwnowotworowa,
g. leczenie estrogenami niepłodności,
h. zmniejszenie aktywności fibrynolitycznej.
1. Zespół antyfosfolipidowy. (APS)
Definicja i patogeneza
Zespołem antyfosfolipidowym nazywamy stan charakteryzujący się wspołistnieniem
przeciwciał antyfosfolipidowych (antykoagulant tocznia i przeciwciała antykardiolipinowe) z
takimi objawami klinicznymi jak zakrzepica żylna lub tętnicza, kilkakrotna utrata ciąży i
małopłytkowość.

Przeciwciała antyfosfolipidowe będące niejednorodną grupą immunoglobulin (IgG, IgM,
IgA) reagują z białkami znajdującymi się w kompleksach z fosfolipidami lub innymi ujemnie
naładowanymi powierzchniami. Najlepiej dotychczas poznanymi antygenami
rozpoznawanymi przez przeciwciała antyfosfolipidowe są beta
2
glikoproteina i protrombina.
Antykoagulant tocznia (lupus anticoagulant – LA) został wykryty we krwi chorych na
toczeń rumieniowaty układowy. Hamuje on in vitro proces krzepnięcia krwi w obecności
fosfolipidów. LA przedłuża czas kaolinowo-kefalinowy osocza (aPTT).
Przeciwciała antykardiolipinowe (aCL) są przeciwciałami, które można wykryć w teście
ELISA z użyciem ludzkiej kardiolipiny. W 60% przypadków występują wspólnie z
antykoagulantem tocznia. Przeciwciała antykardiolipinowe A (aCL typu 1), w odróżnieniu od
częściej wykrywanych aCL typu B, wykazują aktywność antykoagulantu tocznia hamując
krzepnięcie krwi zależne od fosfolipidów. Aktywność ta jest związana z beta
2
glikoproteiną.
Kompleks aCL +beta
2
GPI hamuje adsorpcję X czynnika krzepnięcia na powierzchni
fosfolipidów. Mechanizm trombogennego działania aCL nie został dotychczas wyjaśniony.
Ostatnio opisano, że aCL hamuje inaktywację czynnika Va przez aktywne białko C.
Objawy kliniczne.
Najważniejszym objawem klinicznym jest zakrzepica:
a.
żył głębokich kończyn dolnych,
b.
żyły głównej dolnej,
c.
żyły wrotnej,
d.
żył wątrobowych,
e.
zatok żylnych mózgu.
Najczęstszym tętniczym powikłaniem w zespole antyfosfolipidowym jest udar niedokrwienny
mózgu wykrywany u 36% chorych poniżej 50 r. ż. U około 10% kobiet z przeciwciałami
antyfosfolipidowymi występują poważne powikłania położnicze: poronienia, zahamowanie
rozwoju i śmierć płodu. Powikłania te są związane z zakrzepicą naczyń łożyska. W zespole
antyfosfolipidowym w 80% przypadków występuje także sinica siateczkowata (livedo
reticularis), a u około 25% chorych z APS występuje małopłytkowość. Jest ona
prawdopodobnie spowodowana obecnością przeciwciał skierowanych przeciwko
glikoproteinom płytkowym (GP IIb/IIIa, Gb Ib/IX) stwierdzanych u 40% chorych z APS.
Liczba płytek waha się od 50-100G/l i na ogół nie występują krwawienia. APS o dużym
nasileniu występuje rzadko. Spowodowany bywa licznymi zakrzepami w małych naczyniach
tętniczych, które prowadzą do niewydolności wielonarządowej przypominającej zakrzepową
plamicę małopłytkową lub zespół hemolityczno-mocznicowy.
Badania laboratoryjne:
1. czas kaolinowo-kefalinowy (aPTT) - w zespole antyfosfolipidowym bywa przedłużony
2. antykoagulant tocznia (LA) w obecności fosfolipidów w teście aPTT - obecny
3. przeciwciała antykardiolipinowe (aCL) metodą immunoenzymatyczną (ELISA) z użyciem
ludzkiej kardiolipiny - obecne.
Leczenie
W zakrzepicy stosuje się leki przeciwzakrzepowe, szczególnie doustne antykoagulanty (INR
3-3,5), przy powikłaniach położniczych: glikokortikosteroidy, kwas acetylosalicylowy,
podskórnie drobnocząsteczkowe heparyny.

Temat XIX. Rozsiane krzepnięcie śródnaczyniowe
Maria Kotschy
Definicja.
Rozsiane krzepnięcie śródnaczyniowe ( Disseminated intravascular coagulation – DIC) nie
jest odrębną jednostką chorobową, lecz powikłaniem wielu chorób i stanów klinicznych.
Nazwa ta została wprowadzona przez Mc Kay’a w 1965 roku. Zespół ten był już znany
wcześniej i opisywany pod innymi nazwami jak reakcja Sanarelli-Schwartzmanna (1948 r.),
zespół defibrynacji (1951 r.), zespół zakrzepowo-krwotoczny (1966 r.) oraz koagulopatia ze
zużycia (1967 r.).
Występowanie.
Częstość występowania tego zespołu nie jest dokładnie znana. U 3% zmarłych osób podczas
sekcji, stwierdza się jego cechy. Inne kliniczne obserwacje wykazały częstość zespołu DIC u
1 chorego na 1000 hospitalizowanych, przy czym u 1/3 chorych przebieg bywa bezobjawowy,
możliwy do wykrycia tylko przy pomocy badań laboratoryjnych.
Patogeneza.
W następstwie aktywacji procesu krzepnięcia krwi spowodowanej różnymi
chorobotwórczymi czynnikami, na drodze różnych patomechanizmów, dochodzi do
powstania w mikrokrążeniu wielu zakrzepów zamykających światło naczyń. Zmiany te są
przyczyną niedokrwiennego uszkodzenia tkanek i narządów.
Aktywacja układu hemostazy w zespole DIC może odbywać się przez pobudzenie:
-
toru zewnątrzpochodnego układu krzepnięcia krwi przez kompleks TF/VIIa. Aktywacja
komórek ujawnia na zewnątrz i uwalnia czynnik tkankowy TF dawniej zwany
tromboplastyną tkankową. Ta droga aktywacji występuje najczęściej przy odklejeniu
łożyska, w pourazowych wstrząsach i nowotworach.
-
toru wewnątrzpochodnego układu krzepnięcia krwi przez aktywację fazy kontaktu to
jest czynnika XII, kalikreiny z wysokocząsteczkowym kininogenem. Ten sposób
aktywacji odbywa się przede wszystkim podczas stosowania krążenia pozaustrojowego i
przy uszkodzeniu subendotelium naczyń krwionośnych.
-
Istnieje także droga bezpośredniej aktywacji czynnika X i protrombiny przez różne
enzymy proteolityczne (obecne w ślinie niektórych wężów i różnych rodzajach bakterii
chorobotwórczych, także w białaczkach, w zapaleniu trzustki)
Zakrzepy powstają znacznie rzadziej w obrębie większych naczyń. Krwinki czerwone
uwięzione w sieci włóknika i przepływające przez zmieniony obszar mikrokrążenia ulegają
uszkodzeniu i hemolizie. Jednocześnie na wytworzenie zakrzepików zużyte zostają płytki
krwi, fibrynogen, protrombina oraz przede wszystkim takie czynniki krzepnięcia jak czynnik
V, VIII i XIII. Na zakrzepach może dochodzić do wtórnej aktywacji fibrynolizy ze zużyciem
plazminogenu, alfa 2-antyplazminy oraz pojawieniem się podwyższonego stężenia produktów
degradacji fibryny FDP (najczęściej oznaczanych D-dimerów). Niedobór we krwi krążącej
chorego z zespołem DIC płytek krwi, fibrynogenu, protrombiny i innych czynników
krzepnięcia może doprowadzić do skazy krwotocznej zwanej koagulopatią ze zużycia.
Dołączająca się niekiedy nadmierna aktywacja fibrynolizy może nasilać skazę krwotoczną.
(Ryc.5)
Przyczyny.

Do najczęstszych chorób i stanów klinicznych prowadzących do zespołu śródnaczyniowego
wykrzepiania należą:
a. powikłania ciąży i porodu: przedwczesne odklejenie łożyska, obumarła ciąża, zator wodami
płodowymi (50%),
b. uogólnione zakażenia: bakteryjne (posocznica), wirusowe, grzybicze, riketsjowe,
pierwotniakowe (39%),
c. rozległe uszkodzenie tkanek: urazy powodujące wstrząs, kwasicę, oparzenia, zabiegi
operacyjne także w krążeniu pozaustrojowym, na gruczole krokowym i w płucach. (16,9%),
d. nowotwory: rak gruczołu krokowego, trzustki, płuc, żołądka, okrężnicy i innych narządów
(6,8%),
e. choroby układu krwiotwórczego: ostre białaczki, hemoliza wewnątrznaczyniowa (np.
podczas przetoczenia krwi niezgodnej grupowo),
f. choroby naczyń krwionośnych i serca: olbrzymie naczyniaki, wady serca, tętniaki, zawał
serca, wstrząsy różnego pochodzenia,
g. choroby wątroby: ostra niewydolność, marskość wątroby z nadciśnieniem wrotnym,
h. choroby noworodków i niemowląt i inne.
Stany powodujące zwiększone ryzyko występowania zespołu DIC w tych chorobach:
a.
wstrząs, kwasica, niedotlenienie, zwolnienie przepływu krwi, odwodnienie,
b.
gorączka, stres, przewlekła niewydolność nerek, dekompensacja wątroby,
niedożywienie,
c.
zmniejszona ochrona przeciwzakrzepowa, aktywność przeciwpłytkowa i
fibrynolityczna,
d.
dysfunkcja układu siateczkowo-śródbłonkowego.
Objawy kliniczne
Do głównych objawów zespołu DIC należą:
a/ skaza krwotoczna,
b/ zmiany zakrzepowe i niewydolność różnych narządów,
c/ niedokrwistość mikroangiopatyczna.
Skaza krwotoczna powstaje na tle zużycia płytek i czynników krzepnięcia krwi. Występują
krwawienia z miejsc wkłuć, z ran pooperacyjnych, pourazowych i z różnych układów a także
wybroczyny i podbiegnięcia krwawe na skórze i błonach śluzowych.
Zmiany zakrzepowe umiejscawiają się najczęściej w obrębie małych naczyń (w
mikrokrążeniu) prowadząc do niedokrwiennego uszkodzenia narządów (nerek, płuc,
nadnerczy, wątroby, mózgu lub martwicy skóry) i ich niewydolności. Niedokrwistość
mikroangiopatyczna.
Krwinki czerwone przepływając przez zmieniony zakrzepami obszar mikrokrążenia ulegają
uszkodzeniu i hemolizie. Powstają schizocyty i fragmentocyty widoczne w rozmazach krwi
obwodowej.
Rozsiane krzepnięcie śródnaczyniowe może mieć przebieg kliniczny:
a/ ostry,
b/ podostry,
c/ przewlekły.
Postać ostra występuje przede wszystkim w posocznicach, wstrząsach o różnej etiologii, w
powikłaniach położniczych i pooperacyjnych,
Postać podostrą stwierdza się zazwyczaj w chorobach nowotworowych

Postać przewlekła występuje natomiast w chorobach wątroby i w olbrzymich naczyniakach.
Przebieg rozsianego śródnaczyniowego wykrzepiania zależy:
a. od intensywności czynników chorobotwórczych,
b. od czasu trwania aktywacji układu hemostazy,
c. od rodzaju leczenia i stopnia regeneracji składników układu hemostazy w ustroju chorego.
W razie silnego czynnika wywołującego i szybkiego zużycia składników hemostazy
występują silne wtórne krwawienia. Przy obecności słabszych czynników i dłuższym ich
działaniu powstają przede wszystkim zakrzepy w mikrokrążeniu prowadzące do
niewydolności narządowej, która stanowi trudny problem terapeutryczny. Najczęstszymi
objawami niewydolności narządowej są:
a.
niewydolność nerek (oliguria, anuria, azotemia),
b.
niewydolność płuc: dyspnoe (zaburzenia oddychania), ostry zespół ARDS (acute
respiratory distress syndrome), ostra hipoksemia i postępujący obrzęk miąższu płuc,
c.
niewydolność mózgu: mikrozatorowość naczyń mózgu może powodować zmiany w
stanie psychicznym, śpiączkę mózgową i krwawienia do OUN.
Zespół DIC może mieć postać:
a. miejscową np. w zespole Kassabach-Merrita (olbrzymie naczyniaki),
b. uogólnioną.
W przebiegu zespołu DIC możemy odróżnić kilka okresów:
1.
Faza aktywacji:
a. zwiększona agregacja płytek krwi,
b. nadkrzepliwość,
c. zwiększona fibrynoliza,
d. brak objawów klinicznych.
2a. Wczesna faza zużycia elementów hemostazy:
a.
aktywacja hemostazy jest kontynuowana,
b.
tworzenie zakrzepików w mikrokrążeniu nasilone,
c.
czynniki krzepnięcia bardziej zużywane,
d.
zmniejszenie krzepliwości krwi.
2b. Późna faza zużycia:
a.
dalsza nasilona aktywacja hemostazy,
b.
większy obszar naczyń z mikrozakrzepami,
c.
postępujące zaburzenia hemostazy i krwawienie,
d.
pogorszenie stanu pacjenta związane z ostrym przebiegiem choroby podstawowej.
3. Faza ustępowania (regresji) zespołu DIC ( przy pomyślnym zejściu choroby):
a.
czynniki aktywujące hemostazę są eliminowane z krążenia,
b.
funkcje hemostatyczne normalizują się,
c.
krwawienia ustępują,
d.
funkcja narządów ulega poprawie lub ustępuje.
Konsekwencje koagulopatii ze zużycia
Skaza krwotoczna:
a. zmniejszony potencjał hemostatyczny,
b. zmniejszenie liczby płytek,

c. zmniejszenie osoczowych czynników krzepnięcia krwi,
d. wewnątrznaczyniowe krzepnięcie z wytworzeniem zakrzepików w mikrokrążeniu,
e. kompensacyjny wzrost aktywności fibrynolitycznej z rozwojem kombinowanego defektu
hemostazy,
f. zespół defibrynacji (odwłóknienia).
Zaburzenia w miejscowym mikrokrążeniu z martwicą i manifestacją narządową:
a.
martwica obustronna kory nerek,
b.
martwica przysadki – zespół Sheehana,
c.
martwica kory nadnerczy – zespół Waterhouse-Friedrichsena,
d.
martwica wątroby – ostra dystrofia,
e.
mikroembolizacja krążenia płucnego – ostre serce płucne.
Uogólnione zaburzenia w mikrokrążeniu:
a. nieodwracalny wstrząs
Badania laboratoryjne:
1.liczba płytek krwi - zmniejszona,
2. stężenie fibrynogenu - zmniejszone,
3. czasy: protrombinowy, kaolinowo-kefalinowy (aPTT) i trombinowy osocza - przedłużone,
4. stężenie produktów degradacji, fibrynogen/fibryna szczególnie fibryny (PDFy, d-dimery) -
zwiększone,
5. dodatnie testy parakoagulacji (z siarczanem protaminy i etanolem) służące do wykrywania
rozpuszczalnej fibryny. Obecnie należą do rzadziej przeprowadzanych badań.
6. obecność schizocytów i fragmentocytów stwierdzanych w rozmazach krwi obwodowej
Leczenie:
Najważniejszą zasadą leczenia zespołu DIC jest:
a/ intensywne leczenie choroby podstawowej, w przebiegu której rozwinął się ten zespół,
b/ w stanach wstrząsu obowiązują przyjęte zasady jego leczenia tj. wyrównywanie wolemii,
gospodarki kwasowo-zasadowej, zwalczanie bólu i stosowanie antybiotyków o szerokim
spektrum działania (wstrząs septyczny),
c/ we wstrząsie anafilaktycznym stosuje się steroidy nadnerczowe,
d/ we wczesnych okresach zespołu DIC, kiedy laboratoryjnie stwierdza się nadkrzepliwość
krwi bez występowania objawów klinicznych można stosować heparynę.
e/ w późniejszych okresach DIC z ostrymi krwawieniami stosowanie heparyny jest
kontrowersyjne,
f/ jeżeli nie dochodzi do nasilenia krwawień a pojawia się hiperfibrynogenemia, można
powrócić do stosowania heparyny,
g/ przy dużych niedoborach czynników krzepnięcia stosuje się osocze mrożone, preparaty
antytrombiny.
Koncentraty płytek mogą nasilać ten zespół.
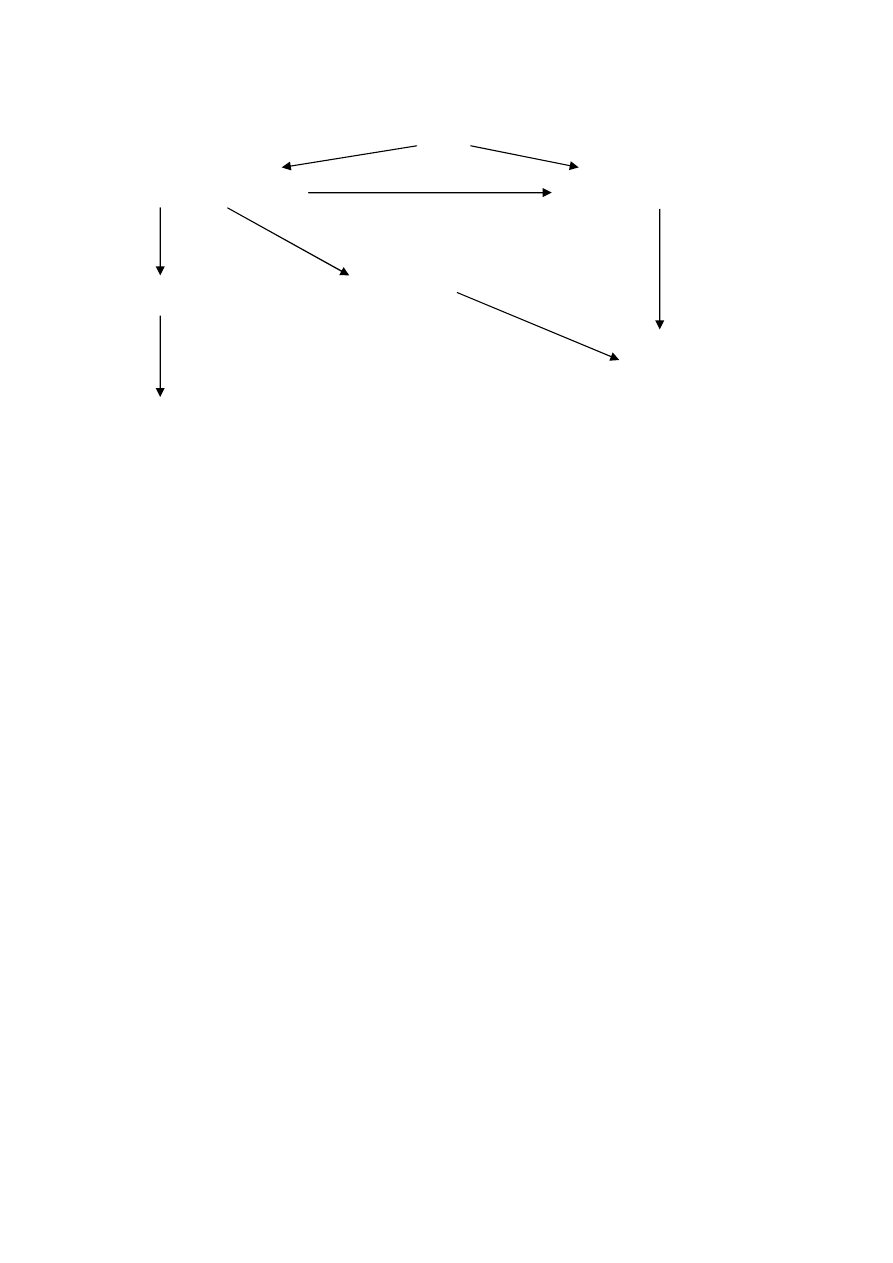
Rozsiane krzepnięcie śródnaczyniowe
DIC
Odkładanie zakrzepów
Zużycie płytek krwi
w mikrokrążeniu
i czynników krzepnięcia
Niedokrwienne
Wtórna
uszkodzenie narządów
fibrynoliza
Skłonność do
krwawień
Uszkodzenie erytrocytów
I hemoliza
Ryc.5. Najważniejsze mechanizmy w rozsianym krzepnięciu śródnaczyniowym
Wyszukiwarka
Podobne podstrony:
skrypt z patofizjologii
skrypt z patofizjologii na ostatnie kolo, III rok, Patofizjologia, 4 koło hematologia
PATOFIZJOLOGIA Skrypt
PATOFIZJOLOGIA WSTRZĄSU
3 Seminarium Patofizjologia chorób rozrostowych
patofizjologia układu odpornościowego
06 pamięć proceduralna schematy, skrypty, ramyid 6150 ppt
W08 Patofizjologia zaburzeń gospodarki węglowodanowej
W19 Patofizjologia narządów zmysłów
Wykład 5 Patofizjologia zaburzeń odporności AIDS2
Patofizjologia zaburze ä jonowych
Podstawowe pojęcia patofizjologii
Patofizjologia nerek i układu moczowego
Patofizjologia układu dokrewnego
W07 Patofizjologia komunikacji międzykomórkowej
więcej podobnych podstron