KINETYKA CHEMICZNA I KATALIZA
Df.
Szybkość reakcji chemicznej określa się zmianą stężenia reagujących substancji w jednostce czasu.
Dla substratów - średnia szybkość reakcji
![]()
Dla produktów - średnia szybkość reakcji
![]()
![]()
Szybkość rzeczywista reakcji chemicznej w danej chwili gdy określa się jako
![]()
Równanie kinetyczne reakcji jest to doświadczalnie wyznaczona krzywa zależności szybkości reakcji od stężenia reagentów uczestniczących w reakcji.
Stałe szybkości reakcji są to współczynniki proporcjonalności w równaniu kinetycznym, łączące szybkość reakcji ze stężeniami reagentów.
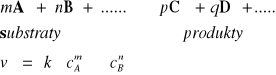
Szybkość reakcji jest proporcjonalna do iloczynu stężeń substratów.
Wykładniki potęgowe, oznaczone symbolami n i m noszą nazwę rzędów reakcji względem składnika A lub B a ich suma nazywana jest ogólnym rzędem danej reakcji.
REAKCJE CHEMICZNE RÓŻNYCH RZĘDÓW
![]()
Reakcje I rzędu
![]()
Reakcja II rzędu
CZĄSTECZKOWOŚĆ REAKCJI
Cząsteczkowość reakcji jest to liczba cząsteczek, które muszą się spotkać, aby zaszła reakcja.
Cząsteczkowość reakcji jest niezależna od rzędu reakcji.
W reakcji jednocząsteczkowej pojedyncza cząsteczka ulega rozpadowi lub przegrupowaniu atomów składowych.
W reakcji dwucząsteczkowej spotykają się dwie cząsteczki lub dwa atomy.
Wyznaczanie równania kinetycznego
Metoda izolacji
wyznacza się rząd reakcji względem wybranego substratu
przez bezpośrednią obserwację zmian jego stężenia w czasie.
Metoda szybkości początkowych
równanie kinetyczne w postaci różniczkowej
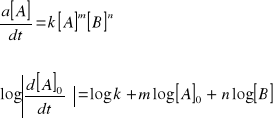
można zapisać jako
Na podstawie wykresu zależności logarytmu początkowej szybkości reakcji od [A]0 (przy ustalonym [B]0), można wyznaczyć stałą szybkości i rząd reakcji względem A. Jest to bowiem, odpowiednio, rzędna punktu przecięcia prostej z osią rzędnych i nachylenia prostej.
SZYBKOŚĆ REAKCJI ZŁOŻONYCH
REAKCJE ODWRACALNE
A Ⴎ B v1 = kA · cA
B Ⴎ A v2 = kB · cB
To w stanie równowagi dynamicznej zachodzi równość prędkości
i otrzymujemy kA / kB = cB/ cA = Kc
Dla reakcji odwracalnych typu Aြွှ B stała równowagi Kc jest równa ilorazowi stałych szybkości kA i kB
REAKCJE SZEREGOWE
Reakcje chemiczne nie przebiegają od razu lecz biegną etapami poprzez różne stany pośrednie.
A ႮB ႮC ႮD ႮE ႮF Ⴎ G
Reakcje szeregowe (następcze) są bardzo rozpowszechnione
Hydroliza estrów dwukarboksylowych, dwuestrów glikoli, niektóre procesy rozpadu promieniotwórczego.
REAKCJE RÓWNOLEGŁE
Typowym przykładem jest
Reakcja dysproporcjonoweania
3 KClO4 + KCl
4 KClO3
reakcja rozpadu soli
4 KCl + 6 O2
REAKCJE ŁAŃCUCHOWE
To obszerna grupa reakcji: spalania, rozkładu, polimeryzacji węglowodorów, niektóre reakcje fotochemiczne (foto synteza chlorowodoru z H2 i Cl2)
Początek łańcucha
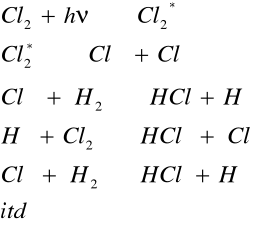
Rozwijanie
- Cl2* w stanie
wzbudzonym cząsteczka
- rodniki
Reakcje łańcuchowe dzieli się na reakcje
Proste i rozgałęzione
np. spalanie wodoru w tlenie
ZALEŻNOŚCI ENERGETYCZNE I MECHANIZMY
Zależność stałej szybkości większości reakcji chemicznych od temperatury opisuje równanie ARRRHENIUSA
![]()
w którym Ea (energia aktywacji) i A -parametry reakcji
TEORIA ZDERZEŃ
Zakłada się, że reakcja zachodzi wówczas gdy zderzają się dwie cząsteczki reagentów o energii (wzdłuż linii łączącej ich środki) większej od energii aktywacji.
Cząstki traktuje się jak sztywne kule, które oddziałują ze sobą tylko wtedy, gdy odległość między ich środkami jest mniejsza od promienia zderzenia czyli sumy promieni reagujących cząstek.
Cząsteczki podczas zderzeń musza być odpowiednio zorientowane względem siebie.
CO + NO2 Ⴎ CO2 + NO
TEORIA KOMPLEKSU AKTYWNEGO
Drugą teorią dotyczącą kinetyki reakcji jest teoria kompleksu aktywnego lub inaczej teoria stanów przejściowych.
W teorii tej zakłada się, że reagujące ze sobą cząsteczki lub atomy w czasie reakcji chemicznej tworzą aktywny kompleks, który może się rozpaść, wytwarzając produkty lub odtwarzając substraty.
Podczas zbliżania się do siebie cząstek substratów AB i CD ulegają stopniowemu osłabieniu ich wiązania chemiczne i jednocześnie zaczynają tworzyć się nowe wiązania.
Powstaje przejściowy układ cząsteczek - kompleks aktywny o duż Kompleks aktywny
jest w stanie równowagi z substratami
A B A B A B
+ +
C D C D C D
Kompleks aktywny
Produkty
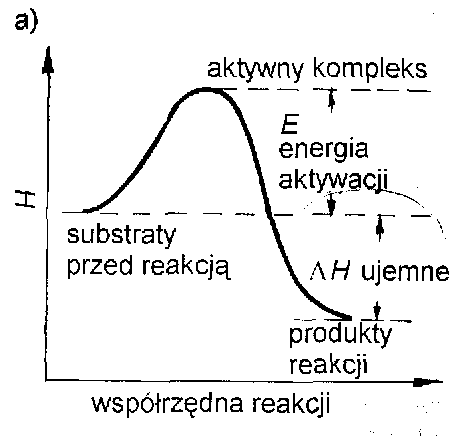
Przebieg reakcji z wytworzeniem aktywnego kompleksu
- reakcja egzotermiczna
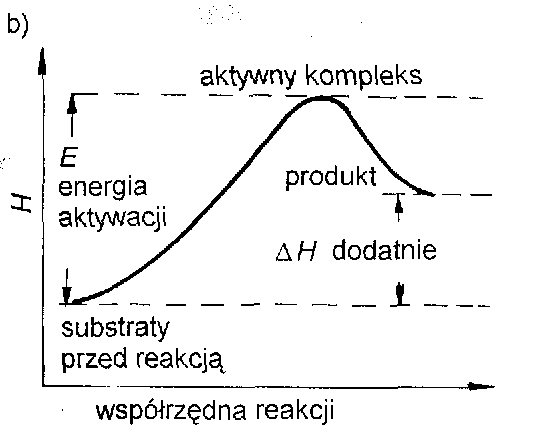
Przebieg reakcji z wytworzeniem aktywnego kompleksu
- reakcja endotermiczn
REAKCJE KATALITYCZNE
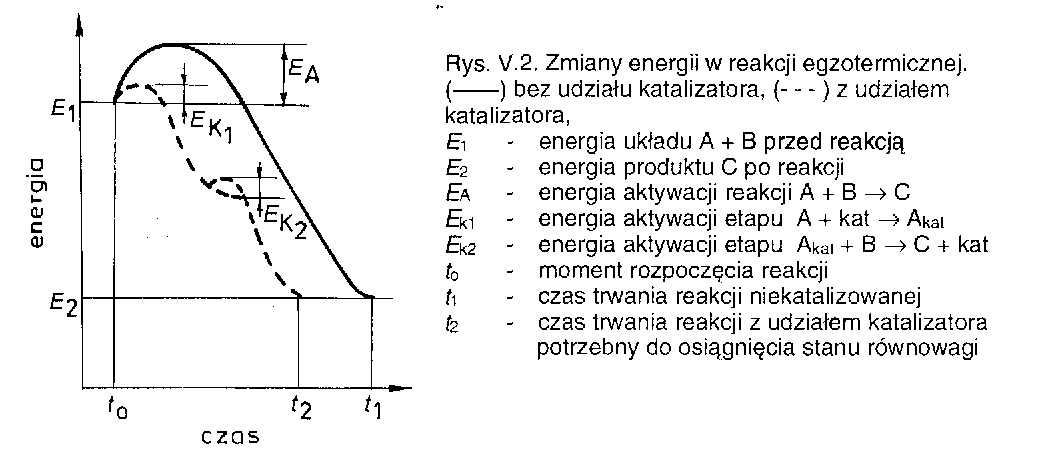
Stała szybkości reakcji zależy od temperatury, wysokości bariery aktywacji, energii aktywacji Ea i czynnika przedwykładniczego A.
Wzrost temperatury można wykorzystać do zwiększenia stałej szybkości, natomiast pozostałe parametry nie mogą zostać zmie-nione, ponieważ są charakterystyczne dla określonej drogi reakcji oraz zdeterminowane przez strukturę elektronową i konfigurację wiązań substratów w kompleksie aktywnym.
Szybkość reakcji można zwiększyć przez zastosowanie katali-zatora, który umożliwia przebieg reakcji alternatywna drogą o niższej energii aktywacji.
Katalizator powoduje jedynie zmianę mechanizmu reakcji, a więc nie jest w reakcji zużywany.
Co więcej, katalizator nie wpływa na równowagowe względne stężenie produktów i substratów, które to są określane wyłącznie przez termodynamikę, natomiast zwiększa szybkość osiągnięcia stanu równowagi.
W przypadku, gdy substrat wiąże się z katalizatorem bardzo mocno, to reakcja z katalizatorem może mieć wyższą barierę energetyczną - mamy wtedy do czynienia z katalizą ujemną a katalizator opóźniający reakcję nazywamy inhibitorem. Odpowiednie inhibitory opóźniają reakcję utleniania lub korozję metali i stopów.
Rozróżniamy katalizę zachodzącą w układach jednorodnych - homogenicznych
w układach niejednorodnych - heterogeniczną
W przypadku katalizy heterogenicznej istotną rolę odgrywają procesy adsorpcji zachodzące na powierzchni stałego katali-zatora tworzącego w układzie reagującym odrębną fazę.
Katalizator taki nazywa się często kontaktem.
Zastosowanie tego typu katalizy umożliwia otrzymywanie H2SO4 metodą kontaktową, syntezę amoniaku, otrzymywanie HNO3 przez utlenienie amoniaku
W przypadku katalizy heterogenicznej istotną rolę odgrywają procesy adsorpcji zachodzące na powierzchni stałego katali-zatora tworzącego w układzie reagującym odrębną fazę.
Katalizator taki nazywa się często kontaktem.
Zastosowanie tego typu katalizy umożliwia otrzymywanie H2SO4 metodą kontaktową, syntezę amoniaku, otrzymy-wanie HNO3 przez utlenienie amoniaku.
KINETYKA UKŁADÓW RZECZYWISTYCH
Mechanizm reakcji łańcuchowych można przedstawić następująco:
Zapoczątkowanie (inicjacja) reakcji - rozpad cząsteczki i utwo-rzenie rodników,
Propagacja (rozwijanie) - reakcje rozgałęziania, w których powstaje więcej nośników łańcucha niż jest zużywanych.
Hamowanie (spowalnianie) - np. utworzony nowy rodnik spowalnia reakcje dalsze.
Terminacja (zakończenie)- Są to reakcje elementarne, w których łączą się rodniki, tworzą się cząsteczki, które przejmują energię wyzwoloną w zderzeniach.
Wybuchy
to reakcje łańcuchowe, zawierające rozgałęzienia łańcucha, w których zwiększa się całkowita liczba nośników łańcucha.
Kinetyka reakcji enzymatycznych
Wiele reakcji w organizmach żywych katalizowanych jest
przez substancje białkowe nazywane ENZYMAMI.
Hipoteza zamka i klucza zakłada, że enzym E zawiera bardzo specyficzne miejsce wiążące, do którego pasuje jedynie określony substrat S, tworzący z nim kompleks enzym - substrat ES. Kompleks ten może ulegać rozkładowi w reakcji jednocząsteczkowej albo do S lub E lub do utworzeni P- produktu.
UKŁADY FAZOWE
df Ⴎ Fazą nazywamy jednolitą część danego układu, oddzieloną od pozostałych jego części określoną granicą.
Jeżeli w zamkniętym naczyniu znajduje się woda i nad nią para wodna, to mówimy, że układ jest dwu fazowy - ciecz i para.
Jeżeli w herbacie rozpuścimy cukier to uzyskamy roztwór dwuskładnikowy 1 czy 2 fazowy.
W przypadku gazów ze względu na ich nieograniczoną wzajemną mieszalność, istnieje zawsze tylko jedna faza
UKŁADY JEDNOSKŁADNIKOWE
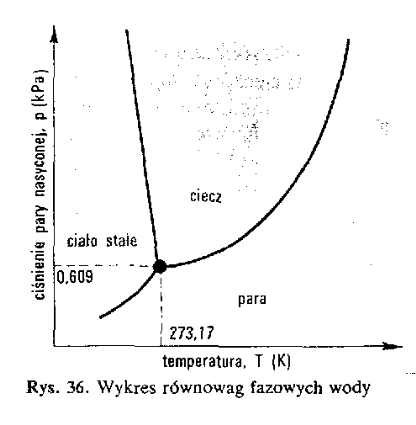
RÓWNOWAGA CIECZ - PARA
Wyobraźmy sobie, że mamy parującą ciecz znajdującą się w zamkniętym naczyniu.
Procesowi parowania w danych warunkach (T,p) towarzy-szy proces przeciwny czyli skraplanie.
Po pewnym czasie w danej temperaturze ustali się stan równowagi, w którym szybkość parowania i skraplania stają się równe.
W tym stanie ciśnienie pary nad cieczą osiągnie wartość stałą.
Krzywa zależności ciśnienia pary nasyconej od temperatury kończy się w punkcie krytycznym K w którym zanika różnica pomiędzy fazą ciekłą i gazową i tworzy się układ jednofazowy.
Aby przeprowadzić 1 kg wody w 1 kg pary należy dostarczyć 22,6 · 105 J energii na sposób ciepła a energię tę nazywamy ciepłem parowania.
Dla danej cieczy ciepło parowania jest zawsze równe co do bezwzględnej wartości ciepłu skraplania (kondensacji).
RÓWNOWAGA CIECZ - CIAŁO STAŁE
LÓD - WODA
Temperaturę, w której ciecz zamienia się w ciało stałe nazywa się temperaturą krzepnięcia.
Temperaturę, w której ciało stałe zamienia się w ciecz nazywa się temperaturą topnienia.
Temperatury krzepnięcia i topnienia zależą od ciśnienia.
Dla niektórych substancji wzrastają wraz ze wzrostem ciśnienia np. dla benzenu a dla wody maleją.
Zdarza się, że pomimo osiągnięcia temperatury niższej od temperatury krzepnięcia ciecz nadal pozostaje w stanie ciekłym.
Zjawisko nazywa się przechłodzeniem cieczy, a ciecz tworzy wtedy ciało bezpostaciowe(np. szkliwo).
RÓWNOWAGA PARA - CIAŁO STAŁE
Proces, w którym ciało stałe zamienia się w parę bez przejścia przez pośredni stan ciekły nazywamy sublimacją, a przechodzenie gazu w stan stały resublimacją np. siarka.
REGUŁA FAZ GIBBSA, PUNKT POTRÓJNY
Niektóre wielkości intensywne charakteryzujące układ fazowy jak: temperatura, gęstość, ciśnienie, współczynnik załamania światła są niezależne od objętości.
Wielkości, które zależą od objętości układu to wielkości ekstensywne np. masa.
W celu scharakteryzowania danego układu nie potrzeba podawać wszystkich wielkości.
Najmniejszą liczbę wielkości intensywnych konieczną do opisu stanu układu nazywa się liczba stopni swobody
Liczba stopni swobody z określa nam liczbę parametrów intensywnych, które można zmieniać w pewnych granicach, niezależnie od siebie, tak aby nie uległa zmianie liczba faz będących w równowadze w dany układzie.
REGUŁA FAZ GIBBSA
z = s - f + 2
s - liczba składników znajdujących się w układzie,
f - liczba faz
UKŁAD JEDNOFAZOWY z = s + 1
UKŁAD DWUFAZOWY z = s o jednym składniku np. ciecz - para -woda, lub układ dwu składnikowy - roztwór etanolu w wodzie
UKŁAD TRÓJFAZOWY jednoskładnikowy - woda
![]()
![]()
![]()
![]()
![]()
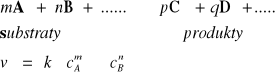
![]()
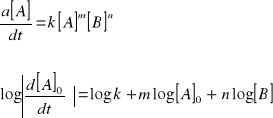
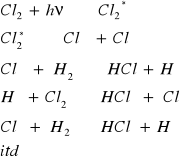
![]()
Wyszukiwarka
Podobne podstrony:
pytanianaegzamin, Biotech PWr I stopien, chemia fiz wykład Komorowski
chemia fiz spr połączone, SGGW - Technologia żywnosci, II semestr, SEMESTR 2, wyklady II rok, od ol
chemia fizyczna wykłady, sprawozdania, opracowane zagadnienia do egzaminu Sprawozdanie 9 chemia f
chemia organiczna wykład 6
Chemia medyczna wykład 1
Chemia fizyczna wykład 11
Cząsteczka (VB), CHEMIA, semestr 1, chemia ogólna, wykłady
chemia fizyczna wykłady, sprawozdania, opracowane zagadnienia do egzaminu Sprawozdanie ćw 7 zależ
Chemia ogolna wyklady 5 6 2012 Nieznany
chemia analityczna wyklad 11 i 12
fiz wyklad 05
chemia fizyczna I wykład(1)
Chemia organiczna wykłady całość(1)
Geo fiz wykład 5 03 2013
Chemia żywności wykład 7
chf wykład 6, Studia, Chemia, fizyczna, wykłady
Chemia fizyczna wykład 10
Chemia fizyczna wykład 4
więcej podobnych podstron