1a). Oddziaływania międzycząsteczkowe to inne niż wiązania chemiczne siły wiążące atomy i cząsteczki. Podstawowa różnica między oddziaływaniami międzycząsteczkowymi a wiązaniami chemicznymi, polega na tym, że nie wiążą one atomów na tyle trwale, aby umożliwiało to uznanie powstałych w ten sposób struktur za związki chemiczne w pełnym znaczeniu tego terminu. Granica między oddziaływaniami międzycząsteczkowymi i wiązaniami jest jednak płynna. Np: wiązanie wodorowe - jeśli występuje w obrębie jednej cząsteczki jest często traktowane jak słabe wiązanie chemiczne, jeśli jednak wiąże ono dwie lub więcej cząsteczek w duże konglomeraty o zmiennym składzie, można je traktować jako oddziaływanie międzycząsteczkowe. Tworzeniem się tego rodzaju konglomeratów powiązanych rozmaitymi oddziaływania międzycząsteczkowymi zajmuje się chemia supramolekularna
Rodzaje oddziaływań międzycząsteczkowych: (wg wykładów)
a) jon jon;
np. kryształ NaCl; energia tego oddziaływania jest odwrotnie proporcjonalna do odległości
Energię tego typu oddziaływań można przestawić wyrażeniem wynikającym ze wzoru na siłę Coulomba.
![]()
gdzie: U - energia; ε - przenikalność elektryczna próżni, q1,q2 - wielkości ładunków jonów, r - odległość pomiędzy środkami ładunków
Jest to oddziaływanie stosunkowo dalekosiężne, o charakterze niekierunkowym, występujące pomiędzy jonami lub grupami jonowymi w cząsteczkach.
b) jon dipol indukowany;
np. pierścień benzenowy i jon NH3+
Pole elektryczne jonu znajdującego się blisko cząsteczki niepolarnej indukuje w niej moment dipolowy.
gdzie: α - polaryzowalność cząsteczki niepolarnej
c) dipol dipol;
np. grupy karbonylowe
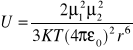
K - stała Boltzmana
d) dipol dipol indukowany;
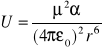
e) dipol indukowany dipol indukowany;
np. dwa pierścienie benzenowe
![]()
gdzie: α1 α2 - polaryzowalnośc cząsteczek, I1, I2 - energie jonizacji cząsteczek. Oddziaływania te nazywamy siłami Londona lub dyspersyjnymi, występują w układach z niepolarnymi cząsteczkami i atomami wykazującymi zawsze chwilowe wartości momentów dipolowych w wyniku ruchów elektronów (ładunków). Przyciągające siły pomiędzy chwilowymi momentami dipolowymi powstają w wyniku synchronizacji ruchów w obu niepolarnych cząsteczkach (atomach).
Oddziaływania międzycząsteczkowe - inne niż wiązania chemiczne siły wiążące atomy i cząsteczki.
Do oddziaływań tych zalicza się (w kolejności od najsilniejszych do najsłabszych):
oddziaływania jon-jon (kulombowskie lub elektrostatyczne) - zachodzą między dwiema różnoimiennie naładowanymi cząsteczkami; od wiązań jonowych różni je to, że ładunek w oddziałujących ze sobą cząsteczkach nie jest skoncentrowany na jednym atomie, lecz jest zdelokalizowany na kilku-kilkunastu atomach. Siła ich oddziaływania jest proporcjonalna do 1/r2 (gdzie r - odległość między cząsteczkami). W przypadku ośrodka zawierającego inne ładunki (np. roztworu elektrolitu) efekt oddziaływania jest mniejszy.
wiązanie wodorowe - tworzy się, gdy atom wodoru z cząstkowym ładunkiem dodatnim jest współdzielony przez dwie cząsteczki, które posiadają atomy z cząstkowym ładunkiem ujemnym. Wiązanie wodorowe, jeśli występuje w obrębie jednej cząsteczki, jest często traktowane jak słabe wiązanie chemiczne; jeśli jednak wiąże ono dwie lub więcej cząsteczek, można je traktować jako oddziaływanie międzycząsteczkowe
oddziaływania trwały dipol - trwały dipol - tworzą się między cząsteczkami posiadającymi trwałe momenty dipolowe. Cząsteczki takie posiadają w jednych miejscach nadmiar ładunku ujemnego, a w innych jego niedomiar. Oddziałują one ze sobą tak jak jony - tyle, że oddziaływanie to jest słabsze, gdyż w grę wchodzą cząstkowe, a nie całkowite ładunki elektryczne, a także przyciąganiu pomiędzy ładunkami różnoimiennymi towarzyszy zawsze odpychanie pomiędzy ładunkami jednoimiennymi.
oddziaływania van der Waalsa, zwane też oddziaływaniami Londona lub oddziaływaniami dyspersyjnymi - są to oddziaływania między trwałym dipolem i wzbudzonym dipolem lub między dwoma wzbudzonymi dipolami. W cząsteczkach, które nie posiadają trwałego momentu dipolowego, może on być wzbudzany przez cząsteczki z trwałym momentem; następnie taki wzbudzony dipol i trwały dipol oddziałują na siebie podobnie jak dwa trwałe dipole, tyle że znacznie słabiej. W cząsteczkach bez trwałego momentu dipolowego występują natomiast stochastyczne fluktuacje ich chmur elektronowych, powodujące powstawanie chwilowych momentów dipolowych. Cząsteczka posiadająca chwilowy moment dipolowy może go wzbudzić w cząsteczce sąsiadującej, wskutek czego obie cząsteczki mogą się nazwajem chwilowo przyciągać lub odpychać. Uśrednienie sił odpychających i przyciągających daje w wyniku oddziaływanie przyciągające proporcjonalne do 1/r6. Oddziaływania van der Waalsa wynikają m.in. z korelacji ruchów elektronów pomiędzy oddziałującymi atomami - dlatego w metodach obliczeniowych nieuwzględniających korelacji elektronowej sił tych praktycznie nie ma.
Oddziaływania międzycząsteczkowe
Oddziaływania między atomami w cząsteczkach należą do oddziaływań silnych, których energie są większe niż 100 kJ/mol. Istnieją też znacznie słabsze oddziaływania o energiach znacznie niższych od przytoczonej wartości. Przyciąganie międzycząsteczkowe jest odpowiedzialne za łączenie się atomów lub cząsteczek, ale jest ono ograniczone przez zjawisko odpychania między jądrami oraz rdzeniami elektronowymi sąsiadujących atomów. Oddziaływania międzycząsteczkowe dzieli się na:
Pierwszy typ oddziaływań - siły van der Waalsa - został zasugerowany przez holenderskiego fizyka, od nazwiska którego pochodzi ich nazwa, na podstawie badania zachowania się gazów niedoskonałych. W ujęciu mechaniki kwantowej siły te można podzielić na trzy rodzaje:
oddziaływanie dipol-dipol
przyciąganie dipol-dipol indukowany
oddziaływanie dipol chwilowy- dipol indukowany
Sens fizyczny oddziaływań dipol-dipol i dipol-dipol indukowany można wyrazić następująco. Dwie cząsteczki mające momenty dipolowe mogą przyjąć takie wzajemne położenie, że dodatni ładunek dipola jednej cząsteczki zbliży się do ujemnego ładunku dipola innej cząsteczki. W wyniku takiej orientacji nastąpi przyciąganie cząsteczek. Przyciąganie typu dipol-dipol indukowany wynika z polaryzacji niepolarnej cząsteczki w polu elektrycznym cząsteczki dipolowej co prowadzi do przyciągania obu cząsteczek.
Oddziaływanie dipol chwilowy- dipol indukowany jest wywołane przez ciągłą fluktuację ładunku w cząsteczce czy atomie. Takie ciągłe zmiany rozkładu ładunku powodują powstawanie chwilowych momentów dipolowych, które mogą powodować przyciąganie innych cząsteczek zgodnie z mechanizmem dipol-dipol indukowany. Zjawisko to występuje np. w gazach szlachetnych umożliwiając ich skraplanie i tłumacząc wzrost temperatury wrzenia tych gazów wraz ze wzrostem ich liczby atomowej (oddziaływanie to jest tym większe im większa jest liczba elektronów w rozważanej cząsteczce czy atomie i im łatwiej są one polaryzowalne). Przyciąganie wynikające z tej przyczyny określa się jako siłę dyspersyjną Londona; oddziaływanie to jest słabe zmieniające się z odwrotnością odległości pomiędzy jądrami w szóstej potędze. Oddziaływani dyspersyjne odgrywają dużą rolę w enzymach. Enzym zawiera pewne niepolarne ale polaryzowalne grupy tzw. kieszenie, które stanowią pułapkę dla niepolarnych lecz polaryzowalnych grup jak fragmenty łańcuchów węglowych ale nie przyciągają cząsteczek polarnych jak woda.
Kompleksy z przeniesieniem ładunku.
Znane są związki, w których dwie cząsteczki wykazują słabe wzajemne przyciąganie, ale jest ono silniejsze niż siły van der Waalsa i słabsze niż wiązanie wodorowe. Układy takie nazywa się kompleksami z przeniesieniem ładunku, gdyż następuje w nich przeniesienie ładunku z jednego układu do drugiego. Stan taki odpowiada utworzeniu bardzo słabo wiążącego orbitalu cząsteczkowego, w którym niewielki udział orbitalu akceptora jest domieszany do funkcji falowej orbitalu donora. Oddziaływania tego typu są zazwyczaj tak słabe, że nie daje się wyizolować czystego związku kompleksowego. Jako przykład może posłużyć tu cząsteczkowy jod rozpuszczony w benzenie.
Wiązanie wodorowe
Badając wiele związków chemicznych tak w stanie stałym jak i cieczy czy pary stwierdzono, że wodór jest przykoordynowany do dwóch atomów, przy czym chemicznie związany jest z jednym atomem ale w stosunku do drugiego jest położony znacznie bliżej niż wynikało by to z sumowania promieni van der Waalsa. Wiązanie takie można przedstawić następująco A-H.....B, przy czym odległości A-H i H-B są z reguły różne (wyjątkiem jest F-H-F). Wiązanie to powstaje gdy pierwiastkami A i B są: C, N, O, P, F, S, Cl, Se, Br, I. Rozpatrując to wiązanie trzeba uwzględnić cztery czynniki:
1. Przyciąganie elektrostatyczne - gdy atom A jest bardziej elektroujemny niż wodór, to nastąpi polaryzacja wiązania A-H i atom A będzie miał ładunek ujemny, a wodór dodatni. Teraz jeżeli elektroujemny atom B ma ładunek ujemny to nastąpi przyciąganie pomiędzy nim a wodorem.
2. Elektrostatyczne odpychanie - zachodzi głównie pomiędzy atomami A i B ponieważ wodór nie ma rdzenia atomowego. Atomy A i B mogą mieć ładunki ujemne.
3. Przeniesienie ładunku - atom B może zachować się jako donor elektronów do grupy A-H, w wyniku czego powstanie kompleks z przeniesieniem ładunku.
4. Siły dyspersyjne Londona - będą dawały niewielki dodatni wkład do mocy wiązania A-H.....B.
Najbardziej oczywistym przejawem występowania wiązania wodorowego jest podwyższenie temperatury wrzenia związków.
1b).Wiązanie chemiczne według klasycznej definicji to każde trwałe połączenie dwóch atomów. Wiązania chemiczne powstają na skutek uwspólnienia dwóch lub więcej elektronów pochodzących bądź z jednego, bądź z obu łączących się atomów lub przeskoku jednego lub więcej elektronów z jednego atomu na atom i utworzenia w wyniku tego tzw. pary jonowej.
Klasyfikacja wiązań międzycząsteczkowych ( wg wykładów):
W literaturzew opisującej międzycząsteczkowe wiązania chemiczne wyróżnia się ich następujące typy:
a) niespecyficzne
wiązania jonowe (z udziałem jonów)
wiązania van der Waalsa (z udziałem dipoli)
efekty hydrofobowe (gdy dużo cząsteczek, więcej niż2)
b) specyficzne (cząsteczki są bliżej siebie, energia jest skwantowana)
wiązania wodorowe
wiązania w kompleksach z przeniesieniem ładunku („charge transfer”)
Wiązania wodorowe (mostek wodorowy / protonowy) ( wg wykładów):
X-H•••:Y
Wiązanie wytworzone między atomem wodoru połączonym kowalencyjnie z atomem X, a atomem Y, posiadaj,ącym swobodnyą parę elektronów. Oba atomy X i Y muszą być silnie elektroujemne (np. O, N, F, Cl).
Wiązania wodorowe modyfikują własności fizykochemiczne substancji (wzrasta temperatura wrzenia i topnienia, rośnie przenikalność elektromagnetyczna).
Wiązania wodorowe występują między innymi w: H2O, NH3, białkach α i β, kwasach nukleinowych, KHF2, kwasach karboksylowych.
W wyniku występowania wiązań wodorowych pomiędzy cząsteczkami wody, substancja ta ma anomalne właściwości; lód jest strukturą bardzo regularną, w fazie ciekłej wiązania występują nieregularnie.
Wiązanie wodorowe formalnie rzecz biorąc nie jest wiązaniem chemicznym, w tym sensie, że nie powstaje ono na skutek wymiany elektronów i jest zwykle dużo mniej trwałe od "prawdziwych" wiązań, jednak ten rodzaj oddziaływania również łączy ze sobą atomy. Wiązanie wodorowe polega na "dzieleniu" między dwoma atomami (np. tlenu) jednego atomu wodoru, tak, że atom wodoru jest częściowo połączony z nimi oboma. Można to też ująć w ten sposób, że atom wodoru jest powiązany z oboma atomami wiązaniami "połówkowymi", gdyż jedno normalne pojedyncze (czyli dwuelektronowe) wiązanie wodór-inny atom jest dzielone na dwa slabsze "półwiązania" inny atom-wodór i wodór-inny atom.
Wiązania w wyniku przeniesienia ładunku (EDA - elektrono - donorowo - akceptorowe) (wg wykładów):
Podobnie jak wiązania wodorowe, są to wiązania specyficzne, o charakterze w znacznym stopniu kowalencyjnym, tworzące najczęściej kompleksy o stechiometrii 1:1, zwane też kompleksami EDA (elektrono - donoro - akceptorowe).
D + A [ D•••A D+ •••-A ]
Wiązanie tworzy się w wyniku oddziaływania chmur elektronowych cząsteczek; zachodzi częściowe przemieszczenie gęstości elektronowej od donora do akceptora. Zaangażowane są tu najczęściej wolne pary elektronowe lub elektrony π.
Energia tego wiązania zależy od warunków środowiska; dobrym przykładem występowania tego wiązania jest tzw. efekt solwatacyjny (lub hydratacyjny, jeśli rozpuszczalnikiem jest woda)
2. Lepkość, (tarcie wewnętrzne) właściwość płynów i plastycznych ciał stałych charakteryzująca ich opór wewnętrzny przeciw płynięciu. Lepkością nie jest opór przeciw płynięciu powstający na granicy płynu i ścianek naczynia. Lepkość jest jedną z najważniejszych cech olejów.
Zgodnie z laminarnym modelem przepływu lepkość wynika, że zdolności płynu do przekazywania pędu pomiędzy warstwami poruszającymi się z różnymi prędkościami.
Różnice w prędkościach warstw są charakteryzowane w modelu laminarnym przez szybkość ścinania. Przekazywanie pędu zachodzi dzięki pojawieniu się na granicy tych warstw naprężeń ścinających. Wspomniane warstwy są pojęciem hipotetycznym, w rzeczywistości zmiana prędkości zachodzi w sposób ciągły, a naprężenia można określić w każdym punkcie płynu. Model laminarny lepkości zawodzi też przy przepływie turbulentnym, powstającym np. na granicy płynu i ścianek naczynia. Dla przepływu turbulentnego jak dotąd nie istnieją dobre modele teoretyczne.
Płyn nielepki to płyn o zerowej lepkości.
Istnieją dwie miary lepkości:
Lepkość dynamiczna wyrażająca stosunek naprężeń ścinających do szybkości ścinania:
Jednostką lepkości dynamicznej w układzie SI jest: kilogram·metr-1·sekunda-1
Lepkość kinematyczna czasami nazywana też kinetyczną jest stosunkiem lepkości dynamicznej do gęstości płynu:
Jednostką lepkości kinematycznej w układzie SI jest: metr2·sekunda-1 Jej nazwa pochodzi od tego, że jest wyrażona jedynie przez wielkości właściwe kinematyce.
Dziedziną nauki zajmującą się badaniami nad lepkością jest reologia. Pomiary lepkości prowadzi się na wiskozymetrach i reowiskozymetrach.
Lepkość, tarcie wewnętrzne, wiskoza, cecha płynów, pojawienie się siły tarcia (tarcie) pomiędzy warstwami cieczy lub gazu, poruszającymi się równolegle względem siebie z różnymi co do wartości prędkościami. Warstwa poruszająca się szybciej działa przyspieszająco na warstwę poruszającą się wolniej i odwrotnie. Pojawiające się wtedy siły tarcia wewnętrznego skierowane są stycznie do powierzchni styku tych warstw.
Określana ilościowo współczynnikiem η równym wartości siły stycznej, która przyłożona do jednostki powierzchni spowoduje jednostajny, laminarny przepływ z jednostkową prędkością:
gdzie F/S - naprężenie ścinające, dv/dz - poprzeczny gradient prędkości.
Wzór powyższy podany już przez I. Newtona odnosi się do cieczy nieściśliwej. Ogólnie (z uwzględnieniem ściśliwości płynu) lepkość definiowana jest poprzez związek składowych tensora naprężeń pij i tensora opisującego pole prędkości vij:
gdzie p - ciśnienie, v - prędkość odkształcenia, η - współczynnik lepkości, η' - drugi współczynnik lepkości (tzw. lepkość druga lub objętościowa, określa ona stopień dyssypacji energii w procesach zachodzących ze zmianą gęstości cieczy), δij − Kroneckera symbol symetryczny.
W układzie CGS jednostką lepkości jest puaz, w SI jest to niutonosekunda na metr kwadratowy [N⋅s/m2].
Współczynnik lepkości η wyznacza się wiskozymetrami. Oprócz powyżej określonej lepkości stosuje się pojęcia lepkości właściwej (stosunku lepkości danej cieczy do lepkości wody w temperaturze 0°C), lepkości względnej (lepkości danej cieczy względem lepkości wody w tej samej temperaturze).
Wielkość φ=1/η nazywana jest płynnością. Dla polimerów i układów dyspersyjnych definiuje się pojęcie lepkości strukturalnej, opisującej powstawanie struktur w cieczy w zależności od prędkości przepływu.
Tarcie wewnętrzne - opór powstający między elementami jednego ciała. W ciele stałym tarcie jest uzależnione od właściwości tłumiących, natomiast w płynach od lepkości. Opory tarcia wewnętrznego wynikają z istnienia sił kohezji i zależą od swobody przemieszczania się tych cząsteczek.
3. Wiskozymetry, przyrządy służące do pomiaru lepkości. Ich konstrukcja jest różna, w zależności od metody badawczej (wiskozymetria).
Typowe rozwiązanie to wirujące naczynie z badaną cieczą, w którym, wzdłuż osi obrotu, zanurzony jest na sprężystej nici próbnik - pomiar kąta skręcenia nici z próbnikiem pozwala wyznaczyć lepkość.
Innym typowym rozwiązaniem są wiskozymetry kapilarne (np. wiskozymetr Ostwalda), gdzie podstawą pomiaru jest czas wypływu określonej objętości cieczy z kapilary.
Często wykorzystywanym jest wiskozymetr Hoeplera, w którym pomiaru lepkości dokonuje się mierząc prędkość opadania kulki zanurzonej w cieczy (prawo Stokesa).
Wiskozymetr, lepkościomierz - przyrząd pomiarowy służący do pomiaru lepkości płynów (głównie cieczy).
Najczęściej stosowane lepkościomierze służą do pomiarów względnych, tj. wyznaczania lepkości badanej substancji względem substancji wzorcowej o znanej lepkości, zwykle wody.
Rozróżnia się lepkościomierze kapilarne — lepkość wyznacza się na podstawie czasu przepływu określonej ilości płynu przez odpowiednio skalibrowane rurki kapilarne pod działaniem znanej różnicy ciśnień. Na takiej zasadzie działają m.in.: lepkościomierz Englera, stosowany głównie do wyznaczania lepkości olejów i smarów w stopniach Englera oraz lepkościomierz Ostwalda.
W lepkościomierzach rotacyjnych (szeroko stosowanych w miernictwie przemysłowym) miarą lepkości jest wartość siły działającej między dwoma współosiowymi cylindrami: cylindrem zewnętrznym i obracającym się względem niego cylindrem wewnętrznym (badana ciecz wypełnia szczelinę między cylindrami).
W lepkościomierzach z opadającą kulką miarą lepkości jest prędkość opadania kulki (o znanych wymiarach i gęstości) w badanym ośrodku pod wpływem stałej siły zewnętrznej (zwykle siły ciężkości), jak to ma miejsce np. w lepkościomierzu Hőpplera. Lepkościomierze takie wykorzystują w sposób bezpośredni prawo Stokesa.
Wyszukiwarka
Podobne podstrony:
fala świetlna-cechy, Studia, Chemia, fizyczna, ćwiczenia
napięcie powierzchniowe, Studia, Chemia, fizyczna, ćwiczenia
ćwiczeniee 43, materiały naukowe do szkół i na studia, chemia fizyczna moja, Chemia fizyczna, Opraco
ćwiczenie 42, materiały naukowe do szkół i na studia, chemia fizyczna moja, Chemia fizyczna, Opracow
ćwiczenie 42Piotr Osuch, materiały naukowe do szkół i na studia, chemia fizyczna moja, Chemia fizycz
ćwiczeniee 43Aneta Łoboda, materiały naukowe do szkół i na studia, chemia fizyczna moja, Chemia fizy
ćwiczenie 43, materiały naukowe do szkół i na studia, chemia fizyczna moja, Chemia fizyczna, Opracow
Pytania z wejściówek, analityka medyczna UMP 2014, chemia fizyczna, ćwiczenia
chf wykład 6, Studia, Chemia, fizyczna, wykłady
ćw7 - Refrakcja i wyznaczanie momentu dipolowego, studia, chemia fizyczna
chf wykład 3, Studia, Chemia, fizyczna, wykłady
chemia fizyczna-ćwiczenie 22, chemia w nauce i gospodarce Uł, semestr V, sprawozdania chemia fizyczn
chf wykład 8, Studia, Chemia, fizyczna, wykłady
Kolokwium zaliczeniowe 1, Technologia chemiczna, Chemia fizyczna, Ćwiczenia
Przykładowe zadania z równowagi chemicznej CD CH 2010 2011, Technologia chemiczna, Chemia fizyczna,
Kolokwium zaliczeniowe, Technologia chemiczna, Chemia fizyczna, Ćwiczenia, Kolokwium nr 1
Kolokwium nr 2, Technologia chemiczna, Chemia fizyczna, Ćwiczenia, Kolokwium nr 1
chf wykład 1, Studia, Chemia, fizyczna, wykłady
więcej podobnych podstron