Chromatografia
Z Wikipedii
Chromatografia (gr. chromatos = barwa + grapho = pisze) to technika analityczna lub preparatywna służąca do rozdzielania lub badania składu mieszanin związków chemicznych.
W każdej technice chromatograficznej najpierw rozdziela się badaną mieszaninę, a następnie przeprowadza się detekcję poszczególnych składników. Rozdział substancji następuje w wyniku przepuszczenia roztworu badanej mieszaniny przez specjalnie spreparowaną fazę rozdzielczą (złoże), zwaną też fazą stacjonarną. Fazą rozdzielczą są substancje wykazujące zdolności sorpcyjne lub zdolne do innych oddziaływań na substancje przepływające. Podczas przepływu eluenta (fazy ruchomej) przez fazę rozdzielczą następuje proces wymywania zaadsorbowanych (lub związanych) substancji. Intensywność tego procesu jest różna dla poszczególnych składników mieszaniny. Jedne składniki są więc zatrzymywane w fazie dłużej, a inne krócej, dzięki czemu może następować ich separacja. Czas przebywania danego składnika w kolumnie określany jest mianem czasu retencji.
W zależności od rodzaju eluentu czyli substancji w której rozpuszcza się badaną mieszaninę rozróżnia się następujące techniki chromatograficzne:
W zależności od rodzaju i sposobu przygotowania fazy rozdzielczej:
TLC - thin layer chromatography, chromatografia cienkowarstwowa - w której fazę rozdzielczą stanowi cienka warstwa żelu naniesiona na sztywną płytkę. Na tak spreparowaną płytkę nanosi się próbkę roztworu, po czym na skutek działania sił kapilarnych, grawitacji lub pola elektrycznego następuje przepływ i rozdział mieszaniny;
W zależności od parametrów procesu:
FPLC - fast protein/performance liquid chromatography - szybka, białkowa/szybkosprawna chromatografia cieczowa - odmiana HPLC działająca na niższych ciśnieniach, stosująca prócz złóż sorpcyjnych, także zwykłe złoża typu sit molekularnych, służąca głównie do rozdziału białek i polipeptydów. Opatentowana i wyłączna nazwa dla firmy Pharmacia.
Związek chromatografii z badaniem struktury substancji jest dość luźny, niemniej czasami ustalenie struktury związku, szczególnie naturalnego, bez wyizolowania go metodą chromatografii z jego naturalnego otoczenia (tzw. matrycy) byłoby niemożliwe.
Pojęciem chromatografii obejmujemy techniki analityczne wykorzystujące zjawisko różnej szybkości migracji składników analizowanej mieszaniny. Różnica w szybkości migracji poszczególnych składników mieszaniny spowodowana może być przez różne czynniki - rozpuszczalność, współczynnik podziału, adsorpcje, reakcje chemiczne itp. - stąd podstawowy podział technik chromatograficznych na adsorpcyjne, podziałowe i jonowymienne. Nazwa chromatografia ("pisanie barwami") wzięła się od pierwszych doświadczeń w tej dziedzinie rosyjskiego botanika Cwieta, który na początku XX wieku przeprowadzał tą metodą rozdział barwnych związków naturalnych.
Klasyczny układ chromatograficzny to nieruchoma faza stacjonarna, w stosunku do której przesuwa się faza ruchoma. Składniki rozdzielanej mieszaniny wykazują powinowactwo zarówno do fazy stacjonarnej jak i do fazy ruchomej, jednak wielkość tego powinowactwa jest różna dla różnych składników. W efekcie tych różnic konkretny składnik mieszaniny przesuwa się wzdłuż drogi rozwijania chromatogramu tylko wtedy, kiedy znajduje się w fazie ruchomej. Składniki mieszaniny o większym powinowactwie do fazy ruchomej poruszają się zatem szybciej ("częściej" przebywają w fazie ruchomej), składniki o większym powinowactwie do fazy stacjonarnej - wolniej ("częściej" tkwią nieruchomo na fazie stacjonarnej). Ponieważ szybkość migracji każdego składnika mieszaniny jest wypadkową działania dwóch sił (powinowactw do dwóch faz chromatograficznych), dla większości mieszanin dość łatwo dobrać układ faz (układ chromatograficzny), który zapewni takie różnice w szybkości migracji, by możliwe było fizyczne odseparowanie cząsteczek poszczególnych substancji od siebie.
|
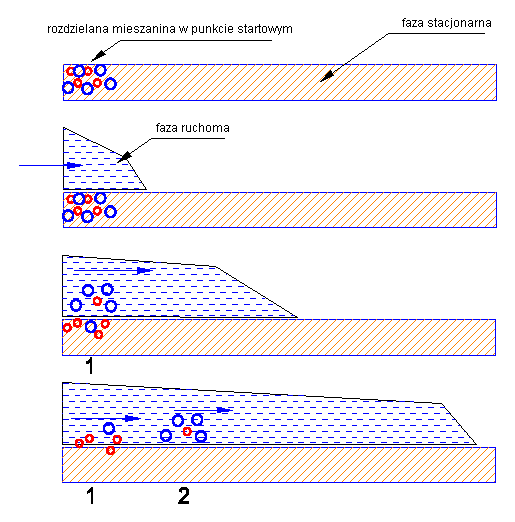
Mieszanina rozdzielanych substancji (czerwone i niebieskie cząsteczki) zostaje naniesiona na punkt startowy na fazie nieruchomej (stacjonarnej)
|
|
Rozpoczyna się ruch fazy ruchomej. Na punkt startowy napływa czysta faza ruchoma. |
|
Lepiej rozpuszczalna substancja niebieska w większości przechodzi do fazy ruchomej i wraz z nią wędruje wzdłuż drogi rozwijania chromatogramu. Substancja czerwona o większym powinowactwie do fazy stacjonarnej i słabej rozpuszczalności w fazie ruchomej w większości pozostaje w punkcie startowym na fazie stacjonarnej |
|
Ruch fazy powoduje, że nad punkt 1 napływa czysty rozpuszczalnik, co przyspiesza rozpuszczanie się w nim cząsteczek czerwonych, zaś cząsteczki które wcześniej poruszały się z faza ruchoma znalazły się nad punktem 2, co spowodowało ich częściowe przejście do fazy stacjonarnej, aż do uzyskania stanu równowagi |
|
Kolejne etapy rozwijania chromatogramu to przechodzenie cząsteczek z fazy ruchomej do stacjonarnej w miejscach, gdzie roztwór w fazie ruchomej styka się z "nie zajętą" fazą stacjonarną, oraz przechodzenie cząsteczek z fazy stacjonarnej do fazy ruchomej tam, gdzie "czysta" faza ruchoma styka się z faza stacjonarną obsadzoną cząsteczkami mieszaniny. |
|
Elementarne procesy przechodzenia z jednej fazy układu chromatograficznego do drugiej odbywają się pod wpływem dwóch czynników - powinowactwa do fazy ruchomej (zdolności rozpuszczania w fazie ruchomej) i powinowactwa do fazy stacjonarnej. |
|
Wypadkowa wartość tych dwóch przeciwstawnych czynników określa średni czas przebywania cząsteczek każdej substancji rozdzielanej mieszaniny w fazie ruchomej. Czas przebywania w fazie ruchomej określa natomiast średnią szybkość migracji wzdłuż drogi rozwijania chromatogramu, bowiem cząsteczki migrują tylko wraz z fazą ruchomą. |
|
Odpowiednio dobrany układ chromatograficzny powoduje uzyskanie takich różnic w szybkości migracji poszczególnych składników rozdzielanej mieszaniny, że odległości między skupiskami poszczególnych cząsteczek po zakończeniu procesu, są wystarczająco duże dla fizycznego odseparowania ich od siebie - dokonania rozdziału chromatograficznego |
W najprostszym, schematycznym ujęciu można to przedstawić następująco - cząsteczki czerwone mające silniejsze powinowactwo do fazy stacjonarnej poruszają się "krótszymi krokami" (linia czerwona) niż cząsteczki niebieskie, które szybciej przechodzą do fazy ruchomej i trudniej są z niej wychwytywane przez fazę stacjonarna (linia niebieska)
W chwili obecnej proces chromatograficznego rozdzielania możemy przeprowadzać na wiele sposobów, wieloma technikami. Podziału technik chromatograficznych można dokonywać na wiele sposobów, biorąc pod uwagę różne czynniki procesu. Wszystkie podziały są dość umowne i mają jedynie służyć łatwiejszemu porozumiewaniu się "chromatografistów", a nowym adeptom sztuki chromatograficznej maja ułatwić poruszanie się w gąszczu technik i ich odmian.
Ze względu na podstawowe zjawisko fizykochemiczne prowadzące do uzyskania rozdziału składników chromatografowanej mieszaniny wyróżniamy chromatografię adsorpcyjną, podziałową i jonowymienną. Ze względu na rodzaje stosowanych faz dzielimy chromatografię na gazową (faza ruchoma jest gazem), cieczową grawitacyjną (fazą ruchoma jest ciecz przepływająca przez złoże fazy stacjonarnej pod wpływem siły grawitacji) i cieczową ciśnieniową (przepływ fazy wymuszony jest wysokim ciśnieniem wytwarzanym przez specjalne pompy). Inny podział wyróżnia techniki planarne (np. cienkowarstwowa lub bibułowa) i kolumnowe (faza stacjonarna wypełnia szklana lub metalową rurkę - kolumnę). W ostatnich latach coraz częściej stykamy się z pojęciem chromatografii z użyciem odwróconych faz. Ponieważ do tej pory najczęstszym układem chromatograficznym był układ, w którym faza stacjonarna była bardziej polarna (bądź miała większe powinowactwo do substancji polarnych) niż faza ruchoma - układ, w którym faza ruchoma jest bardziej polarna niż stacjonarna uzyskał miano układu faz odwróconych (RP - reverse phase). Konkretną technikę można zazwyczaj zaliczyć do kilku grup jednocześnie, jako że kryteria poszczególnych podziałów najczęściej nie wykluczają się nawzajem.
O chromatografii adsorpcyjnej mówimy wówczas, gdy głównym zjawiskiem determinującym przebieg procesu chromatograficznego jest zjawisko adsorpcji. Zjawisko to polega z grubsza na tym, że cząsteczka substancji łączy się w sposób nietrwały (głównie siłami o charakterze wiązania wodorowego) z powierzchnia adsorbenta (np. żel krzemionkowy, tlenek glinu). W chwili gdy zaadsorbowana cząsteczka styka się z "czystą" fazą ruchomą następuje zjawisko desorpcji i cząsteczka przechodzi ponownie do fazy ruchomej.
W chromatografii podziałowej zjawiskiem decydującym o przebiegu procesu jest zjawisko podziału substancji między dwie ciekłe, nie mieszające się fazy. Ciekłą fazę stacjonarną uzyskujemy przez osadzenie na ziarnach stałego nośnika cienkiej warstewki odpowiednio dobranej cieczy (również pod względem parametrów fizycznych, głównie lepkości i związanej z tym siły przylegania - adhezji). Zgodnie z prawem podziału Nernsta, stosunek stężeń w obu fazach w stanie równowagi jest wielkością stałą i charakterystyczną w danych warunkach dla danej substancji. Tak więc, gdy stężenie składnika w fazie ruchomej jest większe, niż to wynika z prawa podziału, substancja przechodzi do fazy ciekłej stacjonarnej. Jeśli po chwili napłynie faza ruchoma o mniejszym stężeniu tej substancji nastąpi częściowe przejście z fazy stacjonarnej do fazy ruchomej (aż do ustalenia odpowiedniego stosunku stężeń w obu fazach).
Jest to typ chromatografii dość znacznie odbiegający od innych. Tu decydującą rolę pełni reakcja chemiczna, i to reakcja z zakresu oddziaływań elektrolitów. Prawie zawsze jest to technika kolumnowa, gdzie wypełnienie kolumny stanowi najczęściej granulowano drobno żywica jonowymienna. Jest to substancja z grupy polimerów, posiadająca na swej powierzchni grupy aktywne chemicznie - najczęściej o charakterze kwaśnym (kwasy sulfonowe) lub zasadowym. Te aktywne grupy chemiczne wychwytują z przepływającego przez kolumnę roztworu kationy lub aniony (w zależności od rodzaju wypełnienie), które później można wymyć z kolumny odpowiednim eluentem. Stosując odpowiednie wypełnienia i eluenty lub łącząc szeregowo kolumny o różnym wypełnieniu można doprowadzić do rozdzielenia mieszaniny kationów lub anionów.
Większe znaczenie żywice jonowymienne maja w przypadku stosowania ich do demineralizacji wody. Woda przepuszczana przed złoże kationitu a później anionitu, zostaje pozbawiona elektrolitów, co w niektórych przypadkach jest konieczne dla sprawnego działania urządzeń (kotły parowe, ogrzewanie wodne itp.). Jest to dość prosta i stosunkowo tania metoda uzdatniania wody. Zużyte kationity i anionity można następnie regenerować, przemywając je kwasem lub zasadą.
Chromatografia gazowa (Gas Chromatography - GC) ma jedno ważne ograniczenie - można ją stosować tylko dla związków lotnych w temperaturze chromatografowania (do ~350°C) i trwałych w tej temperaturze. W praktyce oznacza to najczęściej związki o masie cząsteczkowej poniżej 400 D.
Chromatografowanie przeprowadza się w kolumnie wypełnionej odpowiednim adsorbentem lub ciekłą fazą stacjonarną osadzoną na nośniku. Kolumna w czasie procesu chromatograficznego ogrzana jest do odpowiedniej temperatury. Nastrzyknięta na czoło kolumny mieszanina pod wpływem wysokiej temperatury przechodzi w postać gazową i wraz z gazem nośnym (wodór, azot, hel, itp.), stanowiącym w tej technice fazę ruchomą, przepływa przez kolumnę. Faza stacjonarna wychwytuje cząsteczki z fazy ruchomej poprzez adsorpcje lub rozpuszczanie w fazie ciekłej, a następnie oddaje do fazy ruchomej, zgodnie z zasadami zjawiska adsorpcji-desorpcji lub rozpuszczania gazów. Różna lotność składników analizowanej mieszaniny w połączeniu z różnym powinowactwem do fazy stacjonarnej daje w efekcie rozdzielenie składników.
W chromatografii gazowej dużą role odgrywają detektory, przy pomocy których wykrywamy i oznaczamy opuszczające kolumnę w gazie nośnym składniki mieszaniny. Ze względu na dużą różnorodność chemiczną i fizyczną analizowanych substancji wykorzystuje się wiele detektorów o różnych zasadach działania i bardzo różnej konstrukcji.
Wysokosprawna chromatografia cieczowa (HPLC - High Performance Liquid Chromatography; High Pressure Liquid Chromatography) jest dziś najpopularniejszą i najbardziej uniwersalną techniką chromatograficzną. Jest to technika kolumnowa, ciśnieniowa, w której faza ruchoma przepływa przez kolumnę wypełnioną drobnoziarnistą fazą stacjonarną pod dużym ciśnieniem (najczęściej 200-400 atm). Dzięki temu faza stacjonarna może być drobnoziarnista i gęsto upakowana (duża powierzchnia styku z fazą ruchomą, ale i duże opory dla przepływu fazy ruchomej), a faza ruchoma przepływać z prędkością kilku- kilkunastu mililitrów na minutę (w standardowych kolumnach) co pozwala skrócić czas analizy do sensownych rozmiarów (od kilkunastu minut do paru godzin). Stałość przepływu i wysokie ciśnienie zapewnia specjalna pompa, a jako detektor najczęściej stosuje się spektrofotometr UV, który sprzężony z rejestratorem lub komputerowym urządzeniem rejestrującym wykreśla chromatogram, na podstawie którego można jakościowo (na podstawie czasów retencji, czyli czasu między rozpoczęciem analizy a opuszczeniem przez składnik kolumny chromatograficznej) i ilościowo (na podstawie wielkości pola pod analizowanym pikiem chromatogramu) określić konkretny składnik mieszaniny. Kolumny w HPLC to grubościenne rurki stalowe o średnicy wewnętrznej 2- mm i długości 10-30 cm. Przewody doprowadzające fazę ruchomą pod ciśnieniem też są wykonane z metalu i są grubościennymi kapilarami o średnicy wewnętrznej rzędu 0,1 mm
HPLC jest stosowana jako chromatografia podziałowa, adsorpcyjna, w układzie faz odwróconych, zarówno izokratycznie (taki sam skład fazy ruchomej przez cały proces) jak i gradientowo. Ten ostatni sposób polega na tym, że w czasie procesu faza ruchoma zmienia swój skład (w sposób kontrolowany), tak aby wszystkie składniki uległy rozdzieleniu a czas analizy nie był zbyt długi.
Chromatografia cienkowarstwowa (TLC), mimo wielu mankamentów, ze względu na taniość i prostotę jest wciąż bardzo popularna techniką jakościowej analizy mieszanin. Często wykorzystuje się ją do szybkiej analizy na obecność niepożądanych zanieczyszczeń. Przy jej zastosowaniu możliwa jest również analiza ilościowa, lecz tu znacznie przewyższa ją technika HPLC.
Rozwijanie chromatogramu odbywa się na płytkach szklanych lub z folii aluminiowej pokrytych cienką warstewką adsorbentu (stąd nazwa). Na suchą płytkę nanosimy na linię startową niewielką ilość rozdzielanej mieszaniny, nakrapiając kilka miniaturowych kropel roztworu mieszaniny i odparowując rozpuszczalnik. Następnie płytkę wstawiamy do naczynia z fazą ruchomą, tak by poziom fazy był około 1 cm poniżej linii startowej. Całość wstawiamy do komory chromatograficznej, naczynia szczelnie zamkniętego i nasyconego parami fazy ruchomej, aby w czasie procesu chromatograficznego, jako skutek parowania poszczególnych składników, nie zmieniał się skład fazy ruchomej.
Faza ruchoma, dzięki siłom kapilarnym, samorzutnie pełznie po płytce do góry, co nazywamy rozwijaniem chromatogramu. Zakończenie procesu następuje zazwyczaj po osiągnięciu przez czoło rozpuszczalnika wysokości 10 - 15 cm od linii startowej (wartość parametru d). Mierząc na płytce wartości a i b obliczamy dla poszczególnych substancji wartości tzw. współczynnika retencji Rf = droga substancji / droga czoła fazy ( Rf =a/d dla substancji niebieskiej i Rf = b/d dla substancji fioletowej - patrz rysunek poniżej). Współczynnik ten jest charakterystyczny dla danej substancji w danych warunkach procesu chromatograficznego i może służyć do wstępnej identyfikacji substancji.
Jeżeli rozdzielane substancje nie są barwne, plamy możemy obserwować w świetle ultrafioletowym (wykorzystujemy tu zjawisko fluorescencji) lub spryskujemy płytkę odpowiednim odczynnikiem, z którym składniki rozdzielanej mieszaniny dają barwne produkty reakcji. Prostym i dość skutecznym sposobem uwidaczniania bezbarwnych plam jest wstawienie płytki z rozwiniętym chromatogramem na parę minut do komory zawierającej pary jodu. W celu otrzymania par jodu wystarczy nasypać na dno komory parę kryształków pierwiastkowego jodu i poczekać kilka minut (sublimacja jodu).
Chromatografia jonowymienna
Chromatografia jonowymienna to rodzaj cieczowej chromatografii kolumnowej. Jest to metoda preparatywna używana do wydzielenia z mieszaniny żądanej substancji.
W tej metodzie chromatografii faza stacjonarna, złoże, jest obdarzona ładunkiem. Stanowi je zazwyczaj żywica jonowymienna, zawierająca obdarzone ładunkiem grupy funkcyjne, oddziałujące z przeciwnie naładowanymi grupami związków, które mają zostać zatrzymane przez nośnik:
Związki związane z jonowymieniaczem mogą być wymyte z kolumny przez stopniową elucję, a także poprzez zmianę stężenia soli lub pH.
Tego rodzaju chromatografii używa się do oddzielania takich związków jak aminokwasy, peptydy i białka. Chromatografia jonowymienna jest powszechnie stosowana do oczyszczania białek w systemie FPLC.
TLC (chemia)
{kind=link}
Chromatogram z wynikiem rozdziału barwników atramentu leżący na walcowatej komorze chromatograficznej. Linia startu znajduje się po lewej stronie.
TLC (ang. thin layer chromatography) - cienkowarstwowa chromatografia cieczowa - technika analityczna, lecz także preparatywna, służąca do oczyszczania i identyfikacji mieszanin związków chemicznych [1].
Faza rozdzielcza, złoże, czyli faza stacjonarna o właściwościach sorbcyjnych (silikażel, tlenek glinu) jest umieszczona jako cienka (do 1-2 mm) warstwa na płytce szklanej, metalowej lub z tworzywa sztucznego.
Substancje rozdzielane nakrapia się punktowo przy dolnej krawędzi płytki, po czym płytkę umieszcza się w komorze chromatograficznej zanurzonej na kilka milimetrów w eluencie, tak by nakropione substancje nie zostały zanurzone. Eluent stopniowo wspina się po płytce dzięki zjawisku kapilarnemu. Powoduje to przemieszczanie się rozdzielanych substancji ku górze. Prędkość ruchu poszczególnych składników rozdzielanej mieszaniny jest zależna od oddziaływań międzycząsteczkowych między związkami chemicznymi obecnymi w analizowanej próbce, a fazą rozdzielczą i eluentem. Gdy czoło eluenta dotrze do górnej krawędzi płytki rozdział jest zakończony.
Zależnie od swojej natury fizykochemicznej - składniki rozdzielanej mieszaniny docierają w tym czasie na różną wysokość płytki (współczynnik Rf), a równomierność rozmieszczenia plam substancji można charakteryzować ilościowo tzw. funkcjami CRF (chromatographic response functions). W przypadku, gdy substancje nie są naturalnie barwne, ich obecność można stwierdzić używając, zależnie od ich właściwości, światła UV, roztworów wywołujących (w których płytka jest zanurzana lub nimi spryskiwana - reagują one specyficznie i barwnie z rozdzielanymi substancjami) lub innych metod (np. wywoływanie w parach jodu).
W preparatywnej TLC rozdzielone związki chemiczne można wydrapać z płytki razem ze złożem, po czym je odzyskać przez wymywanie z niego.
{kind=link}
Chromatogram 10 olejków eterycznych. Położenie związków na chromatogramie po jego rozwinięciu zwizualizowano w reakcji z waniliną (reakcja barwna). Poszczególne związki można zidentyfikować nie tylko na podstawie położenia (Rf), ale również koloru w reakcji wywołującej.
Prawo podziału Nernsta
Prawo podziału Nernsta albo krótko prawo podziału określa sposób, w jaki dowolna substancja chemiczna ulega podziałowi pomiędzy dwie oddzielone od siebie, ale pozostające w kontakcie fazy objętościowe (ośrodki). Układy, w których może zaistnieć równowaga podziałowa (rodzaj równowagi dynamicznej), to np. gazy lub pary rozdzielone membraną półprzepuszczalną, gaz i ciecz oraz dwie ciecze niemieszające się lub oddzielone membraną.
Prawo podziału Nernsta wyraża się wzorem:
gdzie: KX(12) - stała podziału substancji X pomiędzy fazy "1" i "2" (zwana też współczynnikiem podziału), [X]i - stężenie substancji X w fazie "i"
Należy podkreślić, że równanie określa równowagowe stężenia w obu fazach, natomiast ilości substancji w obu fazach zależą od objętości (stosunku objętości) obu faz.
W powyższym wzorze stężenie [X] (do wyrażenia stężenia często używa się symbolu c) można składnika obecnego w fazie gazowej zastąpić ciśnieniem p wg wzoru wynikającego z równania Clapeyrona (równania stanu gazu idealnego):
oraz
W praktycznych zastosowaniach przyjęło się, że dla podziału substancji w układzie gaz/ciecz oraz ciecz/ciecz w liczniku równania umieszcza się stężenie (lub ciśnienie) składnika w górnej fazie (gaz lub ciecz o niższej gęstości). Jeżeli układ składa się z cieczy organicznej i wody, wówczas najczęściej faza organiczna ("o") jest fazą górną, a faza wodna ("w") jest fazą dolną i prawo podziału możemy zapisać jako:
- w układzie ciecz organiczna/woda:
Jeżeli mamy do czynienia z równowagą podziału X pomiędzy fazę gazową "g" oraz fazę ciekłą "l" możemy w najprostszy sposób opisać ją równaniem:
- w układzie gaz/ciecz:
Należy zwrócić uwagę na fakt, że to ostatnie wyrażenie jest matematycznie identyczne z prawem Henry'ego dotyczącym absorpcji - mechanizmem absorpcji jest podział pomiędzy dwie fazy objętościowe.
Współczynniki podziału (stałe podziału) substancji pomiędzy dwie niemieszające się ciecze można często oszacować wykorzystując dane nt. rozpuszczalności tej substancji w obu cieczach.
Należy podkreślić, że prawo podziału Nernsta dotyczy identycznych form substancji X w obu fazach. Jeżeli cząsteczki podlegające podziałowi ulegają takim procesom jak asocjacja (zależy nieliniowo od stężenia) czy dysocjacja (zależy od pH, zależy nieliniowo od stężenia), prawo podziału będzie nadal słuszne dla identycznych form tej substancji w obu fazach, ale nie dla całkowitego jej stężenia.
Przykładem układu, w którym prawo Nernsta nie będzie w prosty sposób zachowane, może być np. podział kwasu benzoesowego czy salicylowego (aromatyczne kwasy karboksylowe) pomiędzy fazę wodną i fazę organiczną. W wodzie - rozpuszczalniku polarnym - kwasy ulegają dysocjacji, w roztworze obecne są obojętne cząsteczki i aniony. W słabo polarnym rozpuszczalniku organicznym (np. benzen lub toluen), w którym nie występuje dysocjacja elektrolityczna, budowa cząsteczek kwasów karboksylowych powoduje, że mają one tendencję do dimeryzacji na skutek oddziaływań typu wiązania wodorowego. Jeżeli jesteśmy w stanie obliczyć skład roztworu wodnego (musimy znać stałą dysocjacji) oraz skład roztworu organicznego (musimy określić stałą równowagi procesu asocjacji), wówczas możemy wykorzystać prawo podziału Nernsta działające dla obojętnych cząsteczek, aby opisać równowagę podziałową w takim układzie.
Należy również zwrócić uwagę na fakt, że nawet gdy substancja nie zmienia swojej formy cząsteczkowej przechodząc pomiędzy fazami, te proste równania dotyczą raczej niskich stężeń (ciśnień).
Zjawisko podziału-Substancja,która znajdzie się w układzie dwóch nie mieszających ze soba rozpuszczalników ulega podziałowi między te rozpuszczalniki.Po osiągnięciu stanu równowagi stosunek stężeń substancji w obu fazach ciekłych będzie stały w określonej temperaturze.
Adsorpcja-Jeżeli roztwór lub gaz zawierają pewną substancję graniczyć będzie z silnie rozwiniętą powierzchnia ciała stałego,na granicy faz można stwierdzic podwyższone stężenie tej substancji w porównaniu z jej stężeniem w punktach oddalnych od granic faz.Rozrożniamy adsorbcje fizyczną i chemiczna(chemisorpcje).Adsorpcja fizyczna-spowodowana jest działaniem niewysyconych sił przyciągania międzycząsteczkowego występujących na granicy faz.Chemisorpcja-uzależniona jest od sił natury chemicznej(wiązania chemiczne pomiędzy subst. a powierzchnią ciała stałego)
Desorpcja-jest to zjawisko odwrotne do adsorpcji,występuje w przypadku zmiany warunków (temperatura,ciśnienie) albo zakłócenia równowagi przez zmiane stężenia w poblizu granic faz
Wymiana jonowa-następuje w wyniku zetkniecia się roztworu zawierajacego jony zdolne do wymiany z jonami związanymi z powierzchnią ciał stałych tzw.jonitów
Dyfuzja-polega na wzajemnym przenikaniu się substancji.Zjawisko to jest związane z ruchem cząsyeczek obojętnych elektrycznie lub zjonizowanych.Dwie subst.stykające się ze sobą a zawierające cząsteczki w różnych stężeniach będą ulegały dyfuzji w kierunku gradientów stężeniowych.Prawo Ficka,które mówi,że szybkość przechodzenia substancji przez jednostke powierzchni w jednostce czasu w określonym kierunku jest proporcjonalna do gradientu stężeniowego w tym kierunku
Proces chromatograficzny:w każdym procesie chromatograficznym rozdzielaną mieszaninę wprowadza się w zasięg działania 2 faz:ruchomej i nieruchomej.Faza ruchoma stanowi siłe napędową procesu,natomiast faza nieruchoma odgrywa role siły hamującej proces migracji składników.Rozdział subst. nastepuje w przypadku różnicy współczyników podziału składników mieszaniny pomiędzy obie fazy.Szybkość migracji subst. jest tym większa im mniejszy jest współczynnik podziału.Proces chromatograficzny jest procesem dynamicznym,w którym mają miejsce jednostkowe akty absorbcji i desorpcji składników w fazie nieruchomej.
W chromatografi adsorpcyjnej fazę stacjonarną stanową różnego typu ciała stałe o rozwiniętej powierzchnij,zwane adsorbentamitakie jak węgiel aktywny,żel krzemionkowy itp..O ich działaniu decyduje przede wszystkim sposób aktywacji powierzchni i obecnosc zanieczyszczeń.
W chromatorafi podziałowe ciekła faze stacjonarną nanosi się cienkim filmem na powierzchnie specjalnych nośników.W podziałowej chromatogafi gazowej jako fazy stacjonarne stosuje się oleje silikonowe oraz różnego typu niskolotne cieczetypu polieterów itp.
W chromatografi jonowymiennej proces chromatograficzny prowadzi się w kolumnach wypełnionych jonitem albo na plytkach pokrytych warstwą jonitu.
Chromatografia bibułowa-Cały proces przeprowadza się na pasku bibuły.Substancje badane nanosi się punktowo na bibułe za pomocą kapilarki.Przepływ fazy ruchomej przez pasek bibuły osiąga się przez zanurzenie krawedzi bibuły w rozpuszczalniku w taki sposób żeby naniesiona na bibułe próbka znajdowała się tuż przed lustrem cieczy spełniajacej role fazy ruchomej.Po upływie określonego czasu bibułe wyciąga się i suszy doładnie i przystepuje do wywolywania chromatogramu.Na podstawie połozenia plamek na chromatogramie określamy jakościowy skład badanej próbki korzystając z wielkości retencyjnych.Podstawowa wielkoscia jest współczynnik Rf
Chromaografia cienkowarstwowa-Fazę nieruchoma nanosi się cienką warstwa na płtyke szklaną lub metalowa w postaci drobnoziarnistego proszku.Analizowane mieszaniny wprowadza się na płytke,do rozwijania chromatogramów stosuje się technike wstępującą.Wywoływanie przeprowadza się po wysuszeniu płytki czylo po odporwaniu zawartego w niej rozpuszczalnika.Interpretacj taka jak wyzej.
Są to metody oparte głównie na zastosowaniu jonitów. Jonity albo wymieniacze jonowe są to ciała stałe nieorganiczne lub organiczne nierozpuszczalne w wodzie, które mają zdolnośc wymiany własnych jonów z jonami otaczającego je roztworu. Reakcja przebiega na powierzchni ziaren jonitu. Jonity zdolne do wymiany kationów nazywamy kationitami, a jonity zdolne do wymiany anionów nazywamy anionitami.
Kationity wymieniają swe jony wodorowe na kationy metali znajdujące się w wodzie, według reakcji
R-A-H+ + Me+ <=> R-A-Me+ + H+
gdzie: R - szkielet polimeru, A- - grupa anionowa związana z polimerem (-SO3-, -COO-)
W procesie wymiany jonowej rozpuszczone w wodzie jony metali wypierają z kationu jony wodorowe. Jony metali są zatrzymywane na powierzchni ziaren kationitu, a jony wodorowe przechodzą do wody powodując wzrost jej kwasowości.
Woda po przejściu przez kationit zostaje wprowadzona na anionit, na którym związane zostają aniony, zgodnie z reakcją;
R-B+OH- + A- <=> R-B+A- + OH-
gdzie: R - szkielet polimeru, B+ - grupa kationowa atomowo związana z polimerem (- NH3+), =NH2+)
Znajdujące się w wodzie aniony zatrzymywane są na powierzchni anionitu, a równoważna ilość jonów wodortlenowych OH- przechodzi do wody. Jony te reagują z jonami wodorowymi H+, pochodzącymi z wymiany kationów tworząc cząsteczki wody.
Jonity regeneruje się przepuszczając przez kationity dostatecznie stężony roztwór kwasu, a przez anionity roztwór zasady. procesy regeneracji jonitów można opisać równaniami:
- kationit R-A-Me+ + H+ <=> R-A-H+ + Me+
- anionit R-B+A- + OH- <=> R-A-+OH- + A-
Wstęp teoretyczny:
Chromatografia to technika analityczna lub preparatywna służąca do rozdzielania lub badania składu mieszanin związków chemicznych. W każdej technice chromatograficznej najpierw rozdziela się badaną mieszaninę, a następnie przeprowadza się detekcję poszczególnych składników. Rozdział substancji następuje w wyniku przepuszczenia roztworu badanej mieszaniny przez specjalnie spreparowaną fazę rozdzielczą (złoże), zwaną też fazą stacjonarną. Fazą rozdzielczą są substancje wykazujące zdolności sorpcyjne lub zdolne do innych oddziaływań na substancje przepływające. Podczas przepływu eluenta (fazy ruchomej) przez fazę rozdzielczą następuje proces wymywania zaadsorbowanych (lub związanych) substancji. Intensywność tego procesu jest różna dla poszczególnych składników mieszaniny. Jedne składniki są więc zatrzymywane w fazie dłużej, a inne krócej, dzięki czemu może następować ich separacja. Czas przebywania danego składnika w kolumnie określany jest mianem czasu retencji.
Chromatografia - różnica w rozdziale substancji pomiędzy fazę stacjonarną i fazę ruchomą
Techniki chromatograficzne:
- kolumnowa (chromatografia adsorpcyjna, podziałowa, jonowymienna, żelowa, powinowactwa)
- cienkowarstwowa (TLC; chromatografia adsorpcyjna, podziałowa, powinowactwa, elektroforeza)
- bibułowa (chromatografia podziałowa)
- wysokociśnieniowa (HPLC; chromatografia adsorpcyjno-podziałowa, jonowymienna, żelowa)
- gazowa (GC, chromatografia podziałowa)
Rodzaje chromatografii
- adsorpcyjna
- podziałowa (rozpuszczalność)
- jonowymienna:
rozdział ze względu na wymianę jonów ze złożem zawierającym grupy kwaśne (sulfonowe, karboksylowe, fosforanowe) w wymieniaczach kationów (kationity), lub zasadowe (aminowe) w wymieniaczach anionów (anionity) związane ze złożem (np. polistyren, celuloza, Sephadex)
służy do rozdziału związków jonowych, kwasów i zasad (aminy), szczególnie do aminokwasów, peptydów, białek i kwasów nukleinowych
- stosuje się techniki kolumnowe
- żelowa (sączenie molekularne):
rozdział ze względu na wielkość związku, małe związki silnie penetrują pory złoża (na ogół Sephadex, wielkość jego porów (G) decyduje o możliwości rozdzielenia określonych makromolekuł), dzięki czemu są na nim silniej zatrzymywane
- służy do rozdziału związków wielkocząsteczkowych (białka, kwasy nukleinowe), jak i do ich odsalania
- stosuje się techniki kolumnowe
- powinowactwa (afinitywna):
- specyficzne oddziaływania ligand-receptor, przeciwciało-antygen, enzym-substrat
- służy głównie do wyodrębniania określonego białka z ekstraktów komórkowych
- stosuje się techniki kolumnowe i cienkowarstwowe
- elektroforeza (rozdział ze względu szybkość migracji jonów w polu elektrycznym)
Chromatografia kolumnowa
Kolumnę wypełnia się zawiesiną złoża (wysokość złoża powinna być co najmniej 10 razy
większa od średnicy kolumny; minimum 25 razy więcej złoża od próbki w chromatografii
adsorpcyjnej, 100 razy w chromatografii podziałowej), nanosi próbkę od góry i eluuje układem rozwijającym zbierając frakcje; związki we frakcjach wykrywa się za pomocą chromatografii cienkowarstwowej
Dobieranie eluentu dla chromatografii adsorpcyjnej: próby za pomocą chromatografii
cienkowarstwowej - różnica Rf rozdzielanych związków powinna być większa niż 0.3,
a jedna z substancji nie powinna mieć Rf wyższego od 0.2
Typowe błędy podczas rozdziału:
- zbyt szeroka kolumna - zbyt duża dyfuzja, nierówny przepływ
- zbyt wąska kolumna lub za małe ziarna lub zbyt mało polarny eluent - za wolny przepływ, dyfuzja
- za mało nośnika lub za grube ziarna - zły rozdział
- mokry eluent, kolumna, nośnik - utrata aktywności nośnika, zły rozdział
- zbyt mało aktywny nośnik - zły rozdział
- złe upakowanie kolumny lub nierówne nałożenie próbki - nierówny rozdział
- zbyt polarny eluent - brak rozdziału
- zbyt szybki przepływ - brak rozdziału, nie ustalają się równowagi międzyfazowe
- zbieranie zbyt dużych frakcji - złe rozdzielenie substancji
Rozdzielanie oraz analiza jakościowa węglowodorów aromatycznych metodą wysokosprawnej chromatografii cieczowej (HPLC)
Wiadomości ogólne
Chromatografia to metoda analityczna polegająca na rozdzielania substancji między dwie fazy, które się ze sobą nie mieszają. Proces rozdzielania jest ciągły, ma miejsce wielokrotni. Wspomniane wcześniej fazy różnią się m.in. wielkością. Faza stacjonarna charakteryzuje się duża powierzchnia i jest nieruchoma, zaś faza ruchoma jest mniejsza i przepływa wyłącznie w jednym i tym samym kierunku. Chromatografia jest znacznie lepsza metodą rozdzielania substancji niż np. destylacja czy ekstrakcja. Jeżeli faza ruchoma jest cieczą, to mamy do czynienia z następującymi technikami chromatograficznymi:
- chromatografia bibułowa;
- chromatografia cieczowa;
- chromatografia cienkowarstwowa;
- chromatografia jonowymienna.
Jeżeli fazą ruchoma jest gaz, to wówczas mamy do czynienia z chromatografią gazową.
Rozdział substancji wchodzących w skład badanej próbki ma miejsce dlatego, że substancje te mają różne współczynniki podziału pomiędzy faza stacjonarną i fazą ruchomą. Współczynnik podziału obliczamy w następujący sposób:
K = Cs/Cm
gdzie:
Cs to stężenie substancji w fazie stacjonarnej, zaś Cm to stężenie składnika w fazie ruchomej. Współczynnik podziału to wielkość charakterystyczna dla każdej substancji. W głównej mierze zależy od fazy stacjonarnej. Mamy do czynienia z równowagą dynamiczną. Proces migracji substancji ma miejsce tylko w fazie ruchomej. Do tego celu wykorzystywany jest eluent lub gaz nośny. Jeżeli współczynnik podziału jest bliski. 1, to oznacza że substancja przebywa dłużej w fazie stacjonarnej, później opuszcza kolumnę. Czas retencji zwiększa się. Należy wiedzieć, ze ustala się następujące fakty:
- nie ma miejsca dyfuzja;
- prędkość z jaka porusza się faza ruchoma ma wartość stałą;
- równowaga dynamiczna ustala się bardzo szybko;
Bardzo istotnym, wspomnianym wcześniej, pojęciem jest retencja. Prędkość z jaką porusza się składnik, a prędkość z jaką porusza się faza ruchoma różnią się między sobą. Dzieje się tak dlatego, że poszczególne składniki badanej próbki oddziałują z faza stacjonarną. Prędkość badanych składników jest mniejsza niż prędkość eluentu lub gazu nośnego. Gdy K zmierza do wartości 0, oznacza to, że składniki w badanej próbce nie oddziałują z faza stacjonarną. Taka substancja także potrzebuje określonego czasu do opuszczenia kolumny chromatograficznej. Czas od wstrzyknięcia do pojawienia się piku na rejestratorze nazywany jest zerowym czasem retencji. Zerowy czas retencji jest inny dla każdej substancji. Musimy go odjąć od całkowitego czasu retencji. Równanie retencji ma następującą postać:
Vr = Vm + KVs
gdzie:
Vr to objętość retencji
Vm to objętość ruchomej fazy
Vs to objętość fazy stacjonarnej
K współczynnik podziału.
Proces rozdzielenia substancji jest uzależniony od prędkości migracji. Po rozdzieleniu substancji bardzo ważnym elementem jest to, żeby substancje ponownie nie uległy zmieszaniu. Chromatograf, czyli wykres, który otrzymujemy po zarejestrowaniu rozdzielenia na rejestratorze, składa się z kilku pików.
Czas analizy przy użyciu chromatografu jest równy całkowitemu czasowi retencji ostatniej substancji na chromatografie.
To że dane substancje mogą rozdzielić się chromatograficznie bierze się stąd, że poszczególne substancje wchodzące w skład badanej próbki ulegają podziałowi w różnym stopniu. Podział ma miejsce pomiędzy dwiema nie mieszającymi się wzajemnie fazami. Obie fazy różnią się między sobą. Faza ruchoma nazywana jest eluentem, druga faza jest nieruchoma, nazywana jest fazą stacjonarną, i jej zadaniem jest wypełnianie kolumn chromatograficznych. Składniki wchodzące w skład badanej próbki poruszają się tylko będąc w fazie ruchomej, tzn. wraz z eluentem. Prędkość z jaką poruszają się składniki wchodzące w skład badanej próbki to funkcja podziału znajdująca się w stanie równowagi. Substancje charakteryzujące się większym powinowactwem do fazy nie zawierającej eluent, czyli fazy stacjonarnej, poruszają się znacznie wolniej, niż substancje, które wykazują powinowactwo do fazy zawierającej eluent, czyli fazy ruchomej. Rozdzielenie substancji zachodzące w kolumnie chromatograficznej wynika właśnie z różnic w szybkości poruszania, a co za tym idzie różnic w ilości tej samej substancji między dwiema fazami. W chemii mamy do czynienia z różnymi metodami chromatograficznymi. Najczęściej różnice pomiędzy nimi wynikają z różnych typów fazy stacjonarnej i fazy ruchomej. Chromatografia cieczowa charakteryzuje się tym, że faza ruchomą jest ciecz, zaś chromatografia gazowa charakteryzuje się tym, że faza ruchomą jest gaz. W ostatnich latach bardzo rozwija się chromatografia cieczowa. Polepszeniu wciąż są poddawane kolumny chromatograficzne, zaś szybkość migracji poszczególnych składników jest coraz większa. Obecnie jesteśmy w stanie rozdzielić bardzo wiele substancji, co kilka lat temu było niemożliwe do osiągnięcia. Wyróżniamy kilka typów chromatografii cieczowej. Najbardziej popularną jest wysokosprawna chromatografia cieczowa (HPLC).
Wysokosprawna chromatografia cieczowa (HPLC) jest lepsza w stosunku do innych typów chromatografii cieczowej dlatego że:
- stosowane kolumny w tej metodzie możemy stosować kilkukrotnie, nie musimy ich później regenerować;
- rozdzielenie w wysokosprawnej chromatografii cieczowej jest najlepsze w porównaniu z innymi metodami;
- otrzymane wyniki w metodzie wysokosprawnej chromatografii cieczowej nie sa uzależnione od sprawności obsługującej aparat. Operator nie odgrywa już takiej ważnej roli, jak w przypadku innych metod, co nie oznacza, że chromatograf HPLC może być obsługiwany przez nie przeszkolonego chemika;
- aparaty w wysokosprawnej chromatografii cieczowej są prawie w całości zautomatyzowane, co znacznie ułatwia analizę ilościową;
- czas przeprowadzanego doświadczenia w wysokosprawnej chromatografii cieczowej jest znacznie krótszy niż w przypadku innych metod.
Podział chromatografii cieczowej ze względu na typ wypełnień
Chromatografię cieczowa ze względu na typ wypełnień dzielimy na:
1. Chromatografia podziałowa LLC (chromatografia ciecz-ciecz)
Ten typ chromatografii po raz pierwszy zastosowali Martin i Synge w roku 1941. Badanymi substancjami były ecetylowane aminokwasy. W chromatografii podziałowej LLC stosujemy ciekłe fazy stacjonarne. Są one w pewien sposób osadzone na obojętnych drobnoziarnistych nośnikach, mogą także być chemicznie związane z powierzchnią. Substancje wchodzące w skład badanej próbki i rozpuszczone w fazie zawierającej eluent (faza ruchoma) rozdzielają się na fazę stacjonarną i fazę ruchomą. Współczynnik podziału określa ilościowy rozdział danej substancji pomiędzy dwiema fazami. Składniki wchodzące w skład próbki różnią się między sobą współczynnikami podziału, a co za tym idzie migrują z różna prędkością wzdłuż kolumny chromatograficznej. W chromatografii podziałowej LLC fazę stacjonarna najczęściej tworzy polarna ciecz, taka jak np. woda, glikol. Faza ruchoma nie wykazuje polarności, może to być: heksan, chloroform, benzen. W przypadku, gdy faza stacjonarna nie wykazuje polarności, np. jest to węglowodór, zaś faza ruchoma jest polarna, np. jest to woda, to tego typu metodę nazywamy chromatografię z odwróconymi fazami.
Na szczególną uwagę zasługuje chromatografia PIC, czyli podziałowa par jonowych. Jest to specyficzna odmiana chromatografii ciecz-ciecz. Składniki rozdzielane ta metodą to najczęściej związki jonowe lub związki, które z łatwością mogą ulec jonizacji (sulfoniany, aminofenole, sole amoniowe, amonofenole, aminokwasy. Podczas asocjacji tworzą się pary jonowe w środowisku wodnym. Po krótkim czasie od asocjacji pary jonowe muszą przejść do wodno-organicznej fazy, która charakteryzuje się przeciętnymi zdolnościami do solwatacji.
Chromatografia podziałowa LLC (chromatografia ciecz-ciecz) jest bardzo popularną i na szeroką skalę stosowana metodą rozdziału substancji. Dzieje się tak dlatego, że w tej technice możemy stosować rożnego rodzaju fazy stacjonarne. Możemy rozdzielać substancje polarne, jak i substancje pozbawione takich właściwości. Podstawą do rozdziału jest typ i ilość podstawników wchodzących w skład cząsteczki. Masy cząsteczkowe oczywiście także się pomiędzy sobą różnią. Niektóre grupy związków rozdzielają się bardzo dobrze przy zastosowaniu zwykłej chromatografii ciecz- ciecz. Inne preferują chromatografię ciecz- ciecz z odwróconymi fazami. Zwykła chromatografia ciecz-ciecz rozdziela takie substancje jak: barwniki, pestycydy, plastyfikatory, steroidy, glikole, związki aromatyczne, alkaloidy, kompleksy metali, fenole, Chromatografie ciecz-ciecz z odwróconą faza stosujemy do rozdzielania takich substancji jak: związki aromatyczne, alkohole, antybiotyków, alkaloidów, barbituranów, witamin, pestycydów. Chromatografia PIC, czyli par jonowych stosujemy do rozdziału sulfonamidów, amin biogennych, związków z grupami karboksylowymi.
Zjawisko podziału-Substancja,która znajdzie się w układzie dwóch nie mieszających ze soba rozpuszczalników ulega podziałowi między te rozpuszczalniki.Po osiągnięciu stanu równowagi stosunek stężeń substancji w obu fazach ciekłych będzie stały w określonej temperaturze.
Adsorpcja-Jeżeli roztwór lub gaz zawierają pewną substancję graniczyć będzie z silnie rozwiniętą powierzchnia ciała stałego,na granicy faz można stwierdzic podwyższone stężenie tej substancji w porównaniu z jej stężeniem w punktach oddalnych od granic faz.Rozrożniamy adsorbcje fizyczną i chemiczna(chemisorpcje).Adsorpcja fizyczna-spowodowana jest działaniem niewysyconych sił przyciągania międzycząsteczkowego występujących na granicy faz.Chemisorpcja-uzależniona jest od sił natury chemicznej(wiązania chemiczne pomiędzy subst. a powierzchnią ciała stałego)
Desorpcja-jest to zjawisko odwrotne do adsorpcji,występuje w przypadku zmiany warunków (temperatura,ciśnienie) albo zakłócenia równowagi przez zmiane stężenia w poblizu granic faz
Wymiana jonowa-następuje w wyniku zetkniecia się roztworu zawierajacego jony zdolne do wymiany z jonami związanymi z powierzchnią ciał stałych tzw.jonitów
Dyfuzja-polega na wzajemnym przenikaniu się substancji.Zjawisko to jest związane z ruchem cząsyeczek obojętnych elektrycznie lub zjonizowanych.Dwie subst.stykające się ze sobą a zawierające cząsteczki w różnych stężeniach będą ulegały dyfuzji w kierunku gradientów stężeniowych.Prawo Ficka,które mówi,że szybkość przechodzenia substancji przez jednostke powierzchni w jednostce czasu w określonym kierunku jest proporcjonalna do gradientu stężeniowego w tym kierunku
Proces chromatograficzny:w każdym procesie chromatograficznym rozdzielaną mieszaninę wprowadza się w zasięg działania 2 faz:ruchomej i nieruchomej.Faza ruchoma stanowi siłe napędową procesu,natomiast faza nieruchoma odgrywa role siły hamującej proces migracji składników.Rozdział subst. nastepuje w przypadku różnicy współczyników podziału składników mieszaniny pomiędzy obie fazy.Szybkość migracji subst. jest tym większa im mniejszy jest współczynnik podziału.Proces chromatograficzny jest procesem dynamicznym,w którym mają miejsce jednostkowe akty absorbcji i desorpcji składników w fazie nieruchomej.
W chromatografi adsorpcyjnej fazę stacjonarną stanową różnego typu ciała stałe o rozwiniętej powierzchnij,zwane adsorbentamitakie jak węgiel aktywny,żel krzemionkowy itp..O ich działaniu decyduje przede wszystkim sposób aktywacji powierzchni i obecnosc zanieczyszczeń.
W chromatorafi podziałowe ciekła faze stacjonarną nanosi się cienkim filmem na powierzchnie specjalnych nośników.W podziałowej chromatogafi gazowej jako fazy stacjonarne stosuje się oleje silikonowe oraz różnego typu niskolotne cieczetypu polieterów itp.
W chromatografi jonowymiennej proces chromatograficzny prowadzi się w kolumnach wypełnionych jonitem albo na plytkach pokrytych warstwą jonitu.
Chromatografia bibułowa-Cały proces przeprowadza się na pasku bibuły.Substancje badane nanosi się punktowo na bibułe za pomocą kapilarki.Przepływ fazy ruchomej przez pasek bibuły osiąga się przez zanurzenie krawedzi bibuły w rozpuszczalniku w taki sposób żeby naniesiona na bibułe próbka znajdowała się tuż przed lustrem cieczy spełniajacej role fazy ruchomej.Po upływie określonego czasu bibułe wyciąga się i suszy doładnie i przystepuje do wywolywania chromatogramu.Na podstawie połozenia plamek na chromatogramie określamy jakościowy skład badanej próbki korzystając z wielkości retencyjnych.Podstawowa wielkoscia jest współczynnik Rf
Chromaografia cienkowarstwowa-Fazę nieruchoma nanosi się cienką warstwa na płtyke szklaną lub metalowa w postaci drobnoziarnistego proszku.Analizowane mieszaniny wprowadza się na płytke,do rozwijania chromatogramów stosuje się technike wstępującą.Wywoływanie przeprowadza się po wysuszeniu płytki czylo po odporwaniu zawartego w niej rozpuszczalnika.Interpretacj taka jak wyzej.
Wyszukiwarka
Podobne podstrony:
cxhromatog+ elektrofilowa, eeeeeeeeh, Sem 2, Chemia org
amidy, eeeeeeeeh, Sem 2, Chemia org
chemia- substytucja, eeeeeeeeh, Sem 2, Chemia org
hynrydyzacja atowmow węgla, eeeeeeeeh, Sem 2, Chemia nieorg
chromatografia jonowymienna, studia, studia I rok, chemia org, 2s, chemiczna analiza instrumentalna,
org-chemia, SEM 2, chemia
chromatografia jonowymienna 2, Rok I, chemia fizyczna, chemia fizyczna-protokoły
metody otrzymywania soli, ENERGETYKA AGH, sem 2, chemia
Woda zarobowa, budownictwo pk, sem 1, chemia
Korozja betonu, budownictwo pk, sem 1, chemia
sprawozdanie z miareczkowania, UP Wrocław, IŚ I SEM, Chemia
Chemia org 2 b
laboratorium5pop, Inżynieria Środowiska [PW], sem 2, Chemia, 2, sprawka
konduktometria, farmacja, II sem, chemia ilościowa
egzamin chemia org
Opracowanie - chemia ogólna i nieorganiczna, Nanotechnologia, sem I, chemia
Zasady nazewnictwa wybranych klas zwi-zk-w organicznych, STUDIA PŁ, TECHNOLOGIA ŻYWNOŚCI I ŻYWIENIA
rozpiska analityczna wykłady, farmacja, II sem, chemia ilościowa
więcej podobnych podstron