
p r a k t y k a m e d y c z n a
Przewodnik
Lekarza
86
Mukowiscydoza (ang. cystic fibro-
sis (CF) jest najczêœciej wystêpuj¹c¹
jednogenow¹ chorob¹ rasy bia³ej, dzie-
dzicz¹c¹ siê w sposób autosomalny re-
cesywny. Czêstoœæ jej wystêpowania
w Europie wynosi œrednio 1:2 500 ¿y-
wo urodzonych noworodków. Jednak-
¿e w niektórych krajach choroba wy-
stêpuje znacznie rzadziej, nale¿¹ do
nich m.in.: Szwecja (1:8 000), W³ochy
(1:15 000) i Finlandia (1:40 000).
W Polsce czêstoœæ wystêpowania
mukowiscydozy szacuje siê na
1:2 300. Choroba znacznie rzadziej
obserwowana jest w innych popula-
cjach, np. 1:17 000 u Afroameryka-
nów i 1:90 000 na Hawajach. Mu-
kowiscydoza jest chorob¹ ogólno-
ustrojow¹, o ró¿norodnej ekspresji
klinicznej. W klasycznej postaci ob-
jawia siê zapaleniami oskrzeli
i p³uc, zewn¹trzwydzielnicz¹ nie-
wydolnoœci¹ trzustki, niep³odnoœci¹
mêsk¹ oraz podwy¿szonym stê¿e-
niem chlorków w pocie.
W po³owie lat 80. zlokalizowano
na d³ugim ramieniu chromosomu
7 gen odpowiedzialny za wystêpowa-
nie mukowiscydozy. Ostatecznie zo-
sta³ on sklonowany i scharakteryzo-
wany w roku 1989 [1]. Jest to jeden
z wiêkszych genów ludzkich, zbudo-
wany z 250 tys. par zasad i zawiera-
j¹cy 27 aksonów (fragmentów kodu-
j¹cych). Jego produktem bia³kowym
jest bia³ko CFTR (ang. Cystic Fibro-
sis Transmembrane Conductance Re-
gulator), zbudowane z 1 480 reszt
aminokwasowych i wystêpuj¹ce
w komórkach nab³onka wydzielnicze-
go p³uc, trzustki, jelita grubego, gru-
czo³ów potowych, w¹troby i œlinianek.
W strukturze bia³ka CFTR wyró¿niæ
mo¿na 2 hydrofobowe fragmenty
œródb³onowe TM1 i TM2, zakotwi-
czaj¹ce bia³ko oraz du¿y hydrofilowy
region cytoplazmatyczny, w którym
obecne s¹ domeny wi¹¿¹ce nukleoty-
dy NBF1 i NBF2 (krytyczne dla funk-
cjonowania bia³ka) oraz domena re-
guluj¹ca R. Badania elektrofizjolo-
giczne sugeruj¹, ¿e bia³ko CFTR pe³ni
funkcjê niskowolta¿owego kana³u
chlorkowego zale¿nego od cAMP. Nie
mo¿na jednak rozpatrywaæ kana³u
CFTR, jako izolowanej struktury ko-
mórki. Ca³y model funkcjonalny wy-
daje siê byæ bardziej skomplikowany,
gdy¿ CFTR wspó³dzia³a z innymi ka-
na³ami – m.in. ORCC i ENAC (ang.
outwardly rectified chloride channel,
epithelial sodium channel), a jego
funkcjonowanie wydaje siê byæ zale¿-
ne równie¿ od szeregu dodatkowych
determinant [2]. Za wyst¹pienie mu-
kowiscydozy odpowiedzialne s¹ mu-
tacje genu CFTR, prowadz¹ce do
zmiany struktury i/lub funkcji bia³ka
CFTR. Ostateczny defekt polega na
zablokowaniu wyp³ywu jonów chlo-
ru (tym samym równie¿ wody) oraz
dodatkowym wch³anianiu jonów so-
du, co prowadzi do powstania gêste-
go, lepkiego œluzu (wywo³uj¹cego ob-
jawy chorobowe mukowiscydozy).
Najistotniejsze objawy kliniczne
wystêpuj¹ce w mukowiscydozie zesta-
wiono w tab. 1. Czêstoœæ ich wystêpo-
wania jest ró¿na, a do podstawowych
nale¿¹: przewlek³a choroba p³ucna
oraz niewydolnoœæ zewn¹trzwydziel-
nicza trzustki. Wiêkszoœæ objawów
opisana zosta³a ju¿ wiele lat temu. Jed-
nak¿e pojawi³y siê równie¿ nowe ob-
jawy, które wydaj¹ siê mieæ zwi¹zek
nie tylko z mukowiscydoz¹ (jako cho-
rob¹ podstawow¹), ale tak¿e ze stoso-
wanym leczeniem. Do takich w³aœnie
nale¿y opisana niedawno kolonopatia
Jarosław Walkowiak, Wojciech Cichy
TTaabb.. 11..
PPooddssttaaw
woow
wee
zzeessppoo³³yy//oobbjjaaw
wyy
kklliinniicczznnee
w
wyyssttêêppuujj¹¹ccee
w
w m
muukkoow
wiissccyyddoozziiee
Uk³ad
Zespo³y/objawy
oddechowy
nawracaj¹ce i przewlek³e zapalenia p³uc, zapalenia oskrzelików, obturacyjne zapalenia oskrzeli,
rozstrzenia oskrzeli, niewydolnoœæ oddechowa, przewlek³y i napadowy kaszel, polipy nosa,
przewlek³e zapalenie zatok obocznych nosa
pokarmowy
obrzêk/kamica œlinianek, owrzodzenia jamy ustnej, kandydoza jamy ustnej, próchnica zêbów, choroba refluksowa
prze³yku, niedro¿noœæ smó³kowa, zespó³ czopa smó³kowego, ekwiwalenty niedro¿noœci smó³kowej, zespó³ z³ego
wch³aniania, wypadanie œluzówki odbytnicy, wg³obienie jelit, kolonopatia w³ókniej¹ca, ostre/przewlek³e zapalenie
trzustki, niewydolnoœæ zewn¹trzwydzielnicza trzustki, przed³u¿aj¹ca siê ¿ó³taczka noworodków,
kamica ¿ó³ciowa, ma³y, niefunkcjonuj¹cy pêcherzyk ¿ó³ciowy, marskoœæ ¿ó³ciowa, st³uszczenie w¹troby
rozrodczy
agenezja nasieniowodów, zwiêkszenie lepkoœci œluzu szyjkowego
wewn¹trzwydzielniczy
cukrzyca, opóŸnienie dojrzewania
kostno-stawowy
napadowe zapalenie stawów, p³ucna przerostowa osteoartropatia
inne
s³ony pot, niedobór wysokoœci i masy cia³a, odwodnienie hiponatremiczne i zasadowica hipochloremiczna,
hipoproteinemia i obrzêki w niemowlêctwie, niedokrwistoœæ hemolityczna, acrodermatitis enteropathica
Mukowiscydoza
– nadal aktualny problem
diagnostyczny i terapeutyczny
W pracy przedstawiono podstawy genetyczne oraz patofizjo-
logiczne mukowiscydozy. Podano również najczęstsze
sposoby jej ujawniania się. Wskazano aktualnie istniejące
możliwości terapii, zarówno przyczynowej, jak i objawowej.
Szczególny nacisk położono na problem często niedocenia-
nego postępowania żywieniowego. Podkreślono znaczenie
współdziałania wielospecjalistycznego zespołu w komplekso-
wym leczeniu mukowiscydozy, a także pełnego zaangażowa-
nia w ten proces pacjenta i jego rodziny.

p r a k t y k a m e d y c z n a
Przewodnik
Lekarza
87
w³ókniej¹ca, której pojawienie siê za-
le¿y najprawdopodobniej od stosowa-
nia zbyt du¿ych dawek wysokoaktyw-
nych enzymów trzustkowych oraz
obecnoœci polimerów kwasu metakry-
lowego w otoczce preparatu. Czynni-
kiem sprzyjaj¹cym wyst¹pieniu kolo-
nopatii jest zmniejszenie spo¿ycia
b³onnika. Jednak¿e pe³ne wyjaœnienie
etiopatogenezy tego powik³ania wy-
maga dalszych badañ [5].
Zgodnie ze stanowiskiem Polskiej
Grupy Roboczej Mukowiscydozy
(PGRM) [3], podejrzenie mukowiscy-
dozy opiera siê na podstawie stwier-
dzenia:
3 jednego lub wiêcej objawów kli-
nicznych wystêpuj¹cych w choro-
bie lub
3 obci¹¿aj¹cego wywiadu rodzinne-
go (rodzeñstwo chore na mukowi-
scydozê) lub
3 dodatniego wyniku badania prze-
siewowego noworodków w kie-
runku mukowiscydozy.
Wstêpne rozpoznanie mukowiscy-
dozy nale¿y potwierdziæ jednym
z badañ wykrywaj¹cych dysfunkcjê
genu CFTR:
3 testem potowym wykazuj¹cym
znamiennie wysokie wartoœci
chlorków w pocie (Cl
–
>60 mmol/l)
w co najmniej dwóch odrêbnie wy-
konanych badaniach,
3 wykryciem mutacji w genie CFTR
w obu allelach,
3 wysokimi wartoœciami przezna-
b³onkowej ró¿nicy potencja³ów
w jamie nosowej.
U chorych z rozpoznan¹ mukowi-
scydoz¹ nale¿y wykonaæ badanie
molekularne. Identyfikacja mutacji
w obydwu allelach genu CFTR jest
potwierdzeniem rozpoznania choro-
by. Ze wzglêdu na heterogennoœæ
mutacji, negatywny wynik badania
molekularnego nie wyklucza rozpo-
znania mukowiscydozy.
Wa¿nym aspektem w zakresie prac
nad patofizjologi¹ mukowiscydozy jest
badanie i próba ustalenia genetycznych
podstaw zmiennoœci fenotypowej tej
choroby. Wœród badanych tu zagadnieñ
wymienia siê pierwszo- i drugorzêdo-
we determinanty genetyczne. Do gru-
py determinant pierwszorzêdowych na-
le¿¹ wszystkie znane dotychczas mu-
tacje genu CFTR. Na drugorzêdowe
determinanty genetyczne mukowiscy-
dozy sk³adaj¹ siê czynniki genetyczne
zwane równie¿ modyfikatorami muko-
wiscydozy. S¹ to z regu³y inne geny ni¿
CFTR, zlokalizowane w ró¿nych rejo-
nach genomu. Badania na myszach
transgenicznych CF wskazuj¹, ¿e naj-
prawdopodobniej region wystêpowa-
nia jednego z takich modyfikatorów
choroby znajduje siê na chromosomie
19. O ile znajomoϾ mutacji w genie
CFTR jest ju¿ znacz¹co zaawansowa-
na, to badania drugorzêdowych deter-
minant uwa¿ane s¹ za znajduj¹ce siê
w stanie pocz¹tkowym [4].
Dotychczas opisano ok. 1 tys. ró¿-
nych mutacji w genie CFTR. Wystê-
powanie tak wielkiej liczby zró¿nico-
wanych mutacji genu CFTR odpowie-
dzialne jest przede wszystkim za
zmiennoœæ fenotypow¹, zarówno sa-
mej mukowiscydozy, jak i tzw. chorób
podobnych. Wykrycie i wyizolowanie
genu CFTR oraz analiza jego sekwen-
cji umo¿liwi³a u chorych z podejrze-
niem CF na bezpoœrednie badania mo-
lekularne DNA i zastosowanie ich do
weryfikacji rozpoznania klinicznego
choroby. Tak wiêc analiza genotypu
CFTR (od dawna wykonywana w Pol-
sce) w³¹czona zosta³a jako wa¿ny ele-
ment sk³adowy w ostatecznym ustala-
niu diagnozy choroby. Ma to szczegól-
ne znaczenie w rozpoznawaniu postaci
atypowych, w których objawy klinicz-
ne nie zawsze pozwalaj¹ na jedno-
znaczne dokonanie rozpoznania. Ba-
daniami molekularnymi genu CFTR
objête zosta³y wiêc równie¿ choroby
o czêœciowym symptomatologicznym
podobieñstwie do mukowiscydozy.
Nale¿¹ do nich: obustronna/jednostron-
na niedro¿noœæ przewodów nasien-
nych, azoospermia, oligospermia, bron-
chiektazje, alergiczna aspergiloza
oskrzelowo-p³ucna, przewlek³a hiper-
sekrecja oskrzelowa, polipy nosa, czy
wreszcie przejœciowa noworodkowa
hipertrypsynemia. U osób obci¹¿onych
tymi chorobami stwierdzono wy¿sz¹
czêstoœæ wystêpowania mutacji i wa-
riantów genu CFTR w porównaniu
z populacj¹ ogóln¹. Obserwacja ta
oznaczaæ mo¿e, ¿e istnieje znacznie
szerszy, ni¿ dotychczas s¹dzono, zakres
chorób powodowanych defektem ge-
nu CFTR. Wiele z chorób, w których
wykazano podwy¿szon¹ czêstoœæ wy-
stêpowania mutacji w genie CFTR jest
obecnie klasyfikowanych jako tzw. ró¿-
ne postaci CF (np. p³ciowa, p³ucna) lub
te¿ okreœlane jako tzw. mukopatie, zaœ
nazwê mukowiscydozy rezerwuje siê
wy³¹cznie dla jej klasycznej postaci.
Badania zwi¹zku miêdzy typem mu-
tacji oraz ró¿nymi objawami kliniczny-
mi w CF wykaza³y zró¿nicowany sto-
pieñ korelacji genotyp-fenotyp. Najbar-
dziej œcis³y zwi¹zek wykazano – jak
dotychczas – pomiêdzy genotypem
a czynnoœci¹ zewn¹trzwydzielnicz¹
trzustki. W przesz³oœci uwa¿ano, ¿e cho-
rzy z co najmniej jednym tzw. allelem
³agodnym (np. R117H, A455E itd.) ko-
reluj¹ prawie wy³¹cznie z wydolnoœci¹
zewn¹trzwydzielnicz¹ trzustki (ang.
pancreatic sufficient – PS), zaœ chorzy
z dwiema ciê¿kimi mutacjami (np.
∆
F508) prawie zawsze wykazuj¹ niewy-
dolnoœæ tego narz¹du (ang. pancreatic
insufficient – PI). W przeprowadzonych
ostatnio badaniach w³asnych wykazano,
¿e o ile obecnoœæ dwóch ³agodnych mu-
tacji rzeczywiœcie daje wydolnoœæ ze-
wn¹trzwydzielnicz¹ trzustki, to obec-
noœæ jednej ³agodnej mutacji nie wyklu-
cza ciê¿kiej niewydolnoœci narz¹du.
Próby znalezienia podobnej korelacji po-
miêdzy genotypem a objawami i prze-
biegiem choroby p³ucnej lub zmianami
w zakresie poziomów jonów chlorko-
wych w pocie nie spe³ni³y zamierzonych
oczekiwañ. Dowodem mog¹ byæ cho-
rzy z genotypem
∆F508/R117H, którzy
prawie bez wyj¹tku zwi¹zani s¹ z form¹
PS mukowiscydozy, lecz w zakresie
przebiegu zmian chorobowych w p³u-
cach reprezentuj¹ zakres od postaci ³a-
godnych do ciê¿kich. Brak œcis³ej kore-
lacji pomiêdzy genotypem CFTR,
a zmianami w uk³adzie oddechowym
t³umacz¹ niektórzy kompleksowoœci¹
zmian wystêpuj¹cych w obrêbie p³uc
oraz znaczniejszym wp³ywem modulu-
j¹cym zarówno czynników zewnêtrz-
nych (œrodowiskowych), jak i drugorzê-
dowych determinant genetycznych [4].
Z przedstawionych powy¿ej rozwa¿añ
na temat genetycznych determinant
zmiennoœci fenotypowej w mukowiscy-
dozie zapamiêtaæ nale¿y, ¿e mimo bra-
ku (poza funkcj¹ zewn¹trzwydzielnicz¹
trzustki) œciœlejszej korelacji genotyp–fe-
notyp dla objawów klinicznych muko-
wiscydozy, przewa¿nie obserwuje siê ³a-
godniejsze postacie CF u chorych z co
najmniej jedn¹ ³agodn¹ mutacj¹. Muko-
wiscydoza wystêpuje pod wieloma po-
staciami klinicznymi, zaœ postêp w zro-
zumieniu mechanizmów molekularnych
bêd¹cych podstaw¹ zmiennoœci fenoty-
powej w CF ma wa¿ne implikacje dla
diagnostyki, poradnictwa, opieki nad
chorymi oraz opracowania nowych stra-
tegii leczenia choroby. Po zlokalizowa-
niu, scharakteryzowaniu i sklonowaniu
genu CFTR wydawa³o siê, ¿e lekarze
p r a k t y k a m e d y c z n a
Przewodnik
Lekarza
88
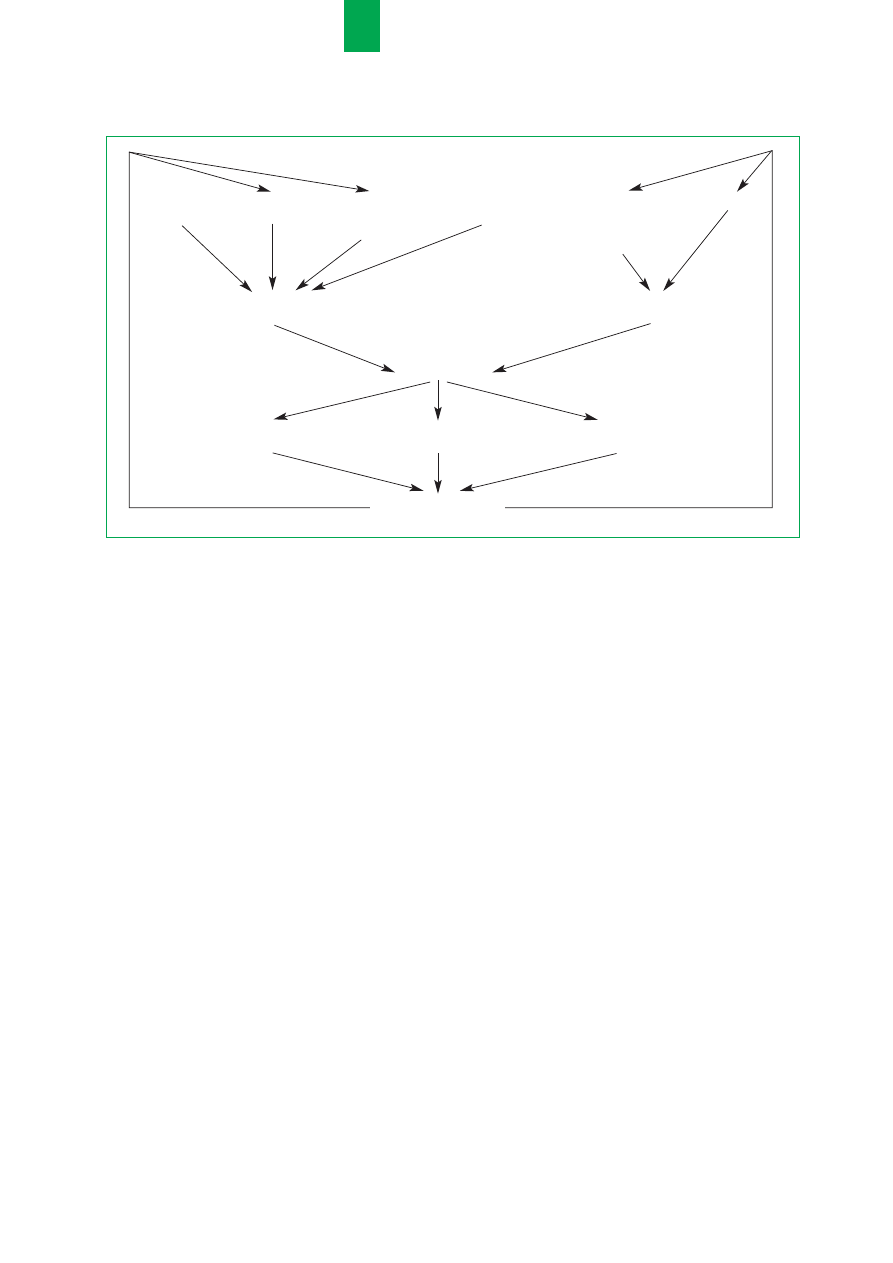
RRyycc..
M
Meecchhaanniizzm
m
bb³³êêddnneeggoo kkoo³³aa
zzaabbuurrzzeeññ
eenneerrggeettyycczznnyycchh
w
w m
muukkoow
wiissccyyddoozziiee
zajmuj¹cy siê t¹ chorob¹ otrzymaj¹ do
rêki broñ – terapiê genow¹ – daj¹c¹
mo¿liwoœæ przyczynowego leczenia
choroby. Niestety, pierwsze próby kli-
niczne terapii genowej nie przynios³y
spektakularnego sukcesu. Nadal prowa-
dzone s¹ intensywne prace nad stworze-
niem efektywnego modelu terapii geno-
wej i trzeba mieæ nadziejê, ¿e zakoñcz¹
siê one sukcesem. W chwili obecnej na-
le¿y uznaæ, ¿e postêp w leczeniu muko-
wiscydozy, daj¹cy wyd³u¿enie œrednie-
go czasu prze¿ycia oraz poprawê ogól-
nego komfortu ¿ycia, uzyskaæ mo¿na
g³ównie dziêki tzw. aktywnemu diagno-
zowaniu i leczeniu kompleksowemu.
Stworzenie w³aœciwego modelu (stan-
dardu), zarówno wczesnego wykrywa-
nia choroby, jak i efektywnego jej lecze-
nia, wymaga bezwzglêdnie wspó³pracy
specjalistów wielu dziedzin. Zrzeszeni
w PGRM pulmonolodzy, gastroentero-
lodzy (w tym autorzy niniejszego arty-
ku³u), genetycy, laryngolodzy, aneste-
zjolodzy, dietetycy, fizjoterapeuci oraz
psycholodzy opracowali polskie stan-
dardy rozpoznawania i leczenia muko-
wiscydozy [3].
Jednym z podstawowych elemen-
tów postêpowania terapeutycznego
u chorych na mukowiscydozê jest le-
czenie ¿ywieniowe. Niestety, zbyt czê-
sto zapomina siê o jego istotnym zna-
czeniu. Codzienna dieta powinna byæ
wysokokaloryczna, wysokobia³kowa
i wysokot³uszczowa. Podstawowym
celem stosowania diety wysokoener-
getycznej jest zapobieganie niedo¿y-
wieniu, w zwi¹zku z tym poda¿ kalo-
ryczna dla chorego z mukowiscydoz¹
powinna byæ zazwyczaj wiêksza o ok.
30–50 proc. w stosunku do zdrowych
rówieœników. Bia³ko w po¿ywieniu
powinno dostarczyæ ok. 15 proc. ener-
gii, t³uszcz 35–40 proc. energii, zaœ
wêglowodany 45–50 proc. energii. Za-
leca siê jak najd³u¿szy okres karmie-
nia piersi¹. Dzieci ¿ywione sztucznie
powinny otrzymywaæ dietê typow¹ dla
wieku. Poda¿ diety opartej na hydro-
lizatach bia³kowych nale¿y zarezerwo-
waæ dla dzieci ze znacznymi zaburze-
niami wch³aniania. Jednoczeœnie ko-
niecznym staje siê utrwalenie
w³aœciwej postawy chorego zwi¹zanej
z przyjmowaniem pokarmów. W mia-
rê trwania choroby opisywany w pod-
rêcznikach wilczy g³ód zamienia siê
w zdecydowan¹ niechêæ do jedzenia.
Tak wiêc wypracowanie w³aœciwych
i skutecznych metod podawania cho-
remu wiêkszych, ni¿ wynika to z jego
wieku, iloœci energii jest podstaw¹ suk-
cesu terapeutycznego. Stan od¿ywie-
nia dziecka z mukowiscydoz¹ stano-
wi bowiem istotny czynnik progno-
styczny co do przebiegu choroby,
w tym przede wszystkim zachowania
wydolnoœci uk³adu oddechowego.
Niezwykle istotne staje siê wiêc dla le-
karza praktyka zrozumienia podstaw
zaburzeñ energetycznych wystêpuj¹-
cych w CF (ryc.) oraz praktycznych
zasad prawid³owego postêpowania ¿y-
wieniowego [5].
Chocia¿ ogólne zasady ¿ywienia s¹
proste, to jednak niezwykle wa¿ne jest
dostosowanie diety dla ka¿dego pa-
cjenta w
sposób indywidualny
z uwzglêdnieniem stanu klinicznego,
apetytu, poziomu aktywnoœci ¿yciowej,
nawyków ¿ywieniowych i stylu ¿ycia.
Specjalny nacisk powinien zostaæ po-
³o¿ony na zaanga¿owanie rodziców
i pacjenta w rozmowê na temat ¿ywie-
nia, postawienie wspólnego celu i jego
realizacjê. Najlepiej wykorzystaæ co-
dzienne praktyki ¿ywieniowe, adaptu-
j¹c je do wymaganych zasad. Pacjenci
powinni spo¿ywaæ 4–5 posi³ków
dziennie oraz przek¹ski pomiêdzy po-
si³kami zasadniczymi. Porcje nie po-
winny byæ zbyt du¿e, aby nie zniechê-
caæ dziecka. Najlepiej, jeœli czas trwa-
nia posi³ku nie przekracza 30 min.
Nale¿y ograniczyæ podawanie produk-
tów o ma³ej wartoœci od¿ywczej (np.
lepiej soki ni¿ woda mineralna czy her-
bata) i nie u¿ywaæ produktów o obni-
¿onej wartoœci energetycznej w miej-
sce pe³nowartoœciowych (np. mleko
pe³ne a nie odt³uszczone). Z naszego
doœwiadczenia wynika, ¿e jednorazo-
wa rozmowa nie zapewnia najczêœciej
oczekiwanego skutku i dla uzyskania
prawid³owej diety niezbêdne jest wie-
lokrotne omawianie zagadnieñ zwi¹-
zanych z ¿ywieniem. Jednoczeœnie pa-
miêtaæ nale¿y, ¿e zbytnie koncentrowa-
defekt
choroba
utrata energii
utrata energii
ograniczenia dietetyczne
depresja
komórkowy
p³uc
przez przewód
przez nerki
spowodowane
pokarmowy
dolegliwoœciami
bólowymi
wzrost zapotrzebowania
spadek poda¿y
na energiê
energii
niedobór energii
os³abienie miêœni
os³abienie uk³adu
zanik mi¹¿szu
oddechowych
odpornoœciowego
p³ucnego
pogorszenie funkcji p³uc
nasilenie objawów

p r a k t y k a m e d y c z n a
Przewodnik
Lekarza
89
nie siê na problemie diety i masy cia³a
wywo³ywaæ mo¿e negatywne reakcje
zwi¹zane z tematem ¿ywienia, a nawet
trudnoœci
natury
behawioralnej
w przyjmowaniu pokarmu.
W razie braku skutecznoœci zacho-
wawczych metod leczenia rozwa¿yæ
nale¿y zastosowanie metod inwazyj-
nych. Prowadzenie ¿ywienia enteralne-
go (przez sondê albo te¿ przezskórn¹
gastrostomiê za³o¿on¹ przy u¿yciu me-
tod endoskopowych lub ultrasonogra-
ficznych) lub parenteralnego – stanowi
bardzo istotny element leczniczy w za-
pobieganiu niedo¿ywienia, utrzymaniu
prawid³owego rozwoju fizycznego pa-
cjenta, jak równie¿ w zapobieganiu za-
ostrzeñ choroby oskrzelowo-p³ucnej
w CF. Przy znacznych zaburzeniach
stanu od¿ywienia pomoc¹ dla oceny
potrzeb energetycznych mog¹ byæ wzo-
ry do obliczania podstawowego wydat-
ku energetycznego (PWE) opracowa-
ne przez Schofielda [3]. ¯ywienie po-
zajelitowe poleca siê stosowaæ
u chorych z niedoborami bia³kowo-
-energetycznymi wystêpuj¹cymi w po-
³¹czeniu z niedoborami pierwiastków
œladowych i witamin.
Niekiedy stosowaæ nale¿y dietê uzu-
pe³niaj¹c¹, zaœ w jej wyborze kierowaæ
siê powinno: wiekiem dziecka, stadium
rozwoju (wiêksze zapotrzebowanie
w okresie skoku pokwitaniowego)
i drog¹ poda¿y. Dodatkowa poda¿ ener-
gii drog¹ doustn¹ winna byæ oparta na
produktach akceptowanych przede
wszystkim ze wzglêdu na ich smak
(przy poda¿y przez zg³êbnik lub gastro-
stomiê ten problem nie wystêpuje).
W razie potrzeby wzbogacenia diety
w energiê zalecaæ nale¿y preparaty,
w których wêglowodany wystêpuj¹
w postaci polimerów glukozy. Gdy za-
lecamy diety zawieraj¹ce t³uszcze œred-
nio³añcuchowe (MCT) pamiêtaæ trze-
ba, ¿e ich gêstoœæ energetyczna jest
mniejsza ni¿ t³uszczów opartych na
kwasach d³ugo³añcuchowych (LCT)
o ok. 0,6 Kcal/g, a ich poda¿ zmniejsza
dostêpnoœæ niezbêdnych nienasyconych
kwasów t³uszczowych. W leczeniu ¿y-
wieniowym dietami dodatkowymi na-
le¿y preferowaæ takie, które cechuj¹ siê
wysok¹ gêstoœci¹ energetyczn¹ (>1
Kcal/ml). W wyborze diety peptydowej
lub elementarnej trzeba kierowaæ siê
wydolnoœci¹ uk³adu pokarmowego
i braæ pod uwagê mo¿liwoœæ wspó³ist-
nienia innych chorób, np. alergii pokar-
mowej czy nietolerancji laktozy. Jedno-
czeœnie nale¿y pamiêtaæ, ¿e powik³a-
niem stosowania diety elementarnej
mo¿e byæ wyst¹pienie biegunki osmo-
tycznej.
Istotnym elementem prawid³owego
sposobu ¿ywienia jest suplementacja
witamin rozpuszczalnych w t³uszczach
(tab. 2.). Dzieci, u których wystêpuj¹
straty t³uszczów w kale, wymagaj¹ pre-
paratów wodnych roztworów tych wi-
tamin.
Podstaw¹ leczenia wystêpuj¹cych
w CF zaburzeñ trawienia i wch³ania-
nia (nastêpstwo zewn¹trzwydzielni-
czej niewydolnoœci trzustki i upoœle-
dzonej czynnoœci jelit) jest – obok le-
czenia ¿ywieniowego – w³aœciwe
stosowanie preparatów enzymów
trzustkowych. Wskazaniem do rozpo-
czêcia suplementacji enzymatycznej
u chorych na mukowiscydozê jest wy-
st¹pienie klinicznych objawów niewy-
dolnoœci zewn¹trzwydzielniczej trzust-
ki (biegunka t³uszczowa, brak/s³aby
przyrost masy cia³a u dzieci oraz spa-
dek masy cia³a u doros³ych) potwier-
dzonych w badaniach laboratoryjnych
(elastaza-1 w stolcu, dobowe wydala-
nie t³uszczów lub test oddechowy,
ewentualnie steatokryt). Preparaty en-
zymów trzustkowych powinny byæ
podawane w trakcie posi³ków, najle-
piej w 2 porcjach (na pocz¹tku i w po-
³owie karmienia) po to, by mo¿liwie
najlepiej wymieszane zosta³y z treœci¹
pokarmow¹. U niemowl¹t i ma³ych
dzieci dopuszcza siê mo¿liwoœæ otwie-
rania kapsu³ek preparatów enzymów
trzustkowych z zachowaniem warun-
ków podawania mikrogranulek z so-
kiem, herbat¹ lub wod¹, zaœ unikaniem
ich podawania, np. z mlekiem lub
w preparatach mlekozastêpczych (ze
wzglêdu na wczesn¹ ich inaktywacjê
w ¿o³¹dku). Empirycznie zalecane
dawki enzymów wynosz¹:
3 dla niemowl¹t 2 000–4 000 j. FIP li-
pazy/120 ml mieszanki mlecznej
(lub 1 karmienie piersi¹),
3 dla dzieci poni¿ej 4. roku ¿ycia
1 000 j. FIP lipazy/kg m.c./posi³ek,
3 dla dzieci powy¿ej 4. roku ¿ycia
2 500 j. FIP lipazy/kg m.c./posi³ek.
Nie nale¿y przekraczaæ dobowej
dawki 10 000 j. FIP lipazy/kg m.c./do-
bê. Ogólnie nale¿y przyj¹æ, ¿e dobo-
wa dawka enzymów powinna byæ do-
biera indywidualnie na podstawie
oznaczenia bilansu wydalanych t³usz-
czów w kale, a tak¿e oceny ustêpowa-
nia patologicznych objawów klinicz-
nych i dostosowania do zalecanej wiel-
koœci dobowego spo¿ycia pokarmów.
Za wystarczaj¹ce w tym wzglêdzie po-
krycie zapotrzebowania enzymatycz-
nego nale¿y przyj¹æ nastêpuj¹ce kry-
teria:
3 ustêpowanie lub brak wzdêæ i bó-
lów brzucha,
3 zmniejszenie liczby stolców (mniej
ni¿ 3/dobê) oraz ich normalizacja
morfologiczna (uformowane, bez
oleistego wygl¹du, bez niestrawio-
nych resztek pokarmowych),
3 dobowa utrata t³uszczu mniejsza ni¿
10–15 g/dobê (optymalnie poni¿ej
7 g/dobê),
3 ustêpowanie objawów os³uchowych
wzmo¿onej perystaltyki jelit,
3 tendencja do przyrostu masy i wyso-
koœci cia³a.
TTaabb.. 22..
DDoobboow
wee
ddaaw
wkkii
ssuupplleem
meennttaaccyyjjnnee
w
wiittaam
miinn
rroozzppuusszzcczzaallnnyycchh
w
w tt³³uusszzcczzaacchh
Witamina A
5 000–10 000 j.m.
do 1. roku ¿ycia 2 000–4 000 j.m.
od 2. do 7. roku ¿ycia 4 000–5 000 j.m.
powy¿ej 7. roku ¿ycia 5 000–10 000 j.m.
Witamina D*
400–800 j.m.
Witamina E
25–400 mg
0.–6. mies. ¿ycia 25 mg
7.–12. mies. ¿ycia 50 mg
od 2. do 4. roku ¿ycia 100 mg
powy¿ej 4. roku ¿ycia 200–400 mg
Witamina K**
2,5–10 mg/tydz.
0.–12. mies. ¿ycia 2,5 mg/tydz.
powy¿ej 1. roku ¿ycia 5 mg/tydz. i wiêcej
Beta-karoten*** 1 mg/kg m.c. (do 20 mg)
* w przypadku chorych z cholestaz¹, suplementacja witamin¹ D powinna byæ prowadzona preparatem 25 0H-D
3
,
** wskazaniem do leczniczego stosowania witaminy K s¹ zaburzenia krzepniêcia, hepatosplenomegalia,
kr wioplucie, d³ugotr wa³a i intensywna antybiotykoterapia oraz zabiegi operacyjne,
*** brak jednolitej opinii w kwestii wielkoœci dawki

p r a k t y k a m e d y c z n a
Przewodnik
Lekarza
90
W przypadku braku skutecznoœci po-
dawanych preparatów enzymów trzust-
kowych nale¿y rozwa¿yæ celowoœæ w³¹-
czenia H
2
-blokerów, inhibitorów pom-
py protonowej i/lub preparatów kwasów
¿ó³ciowych (brak zgodnoœci co do sku-
tecznoœci powy¿szych metod oraz mo¿-
liwoœci wyst¹pienia powik³añ w czasie
ich d³ugotrwa³ego stosowania). Poda-
wanie ³¹cznie z preparatami enzymów
trzustkowych leków przyspieszaj¹cych
opró¿nianie ¿o³¹dka i skracaj¹cych czas
pasa¿u jelitowego mo¿e zmniejszyæ sku-
tecznoϾ suplementacji enzymatycznej.
Przekroczenie maksymalnej zalecanej
dobowej dawki preparatów enzymów
trzustkowych (tj. 10 000 j. FIP lipazy/kg
m.c.) niesie ze sob¹ mo¿liwoœæ wyst¹-
pienia niezwykle groŸnego powik³ania
w postaci kolonopatii w³ókniej¹cej.
W celu zmniejszenia lub unikniêcia ry-
zyka wyst¹pienie tego powik³ania (za-
równo u dzieci, jak i u doros³ych pacjen-
tów z CF) pacjentom nale¿y podawaæ
preparaty enzymów trzustkowych nie
zawieraj¹ce w swoim sk³adzie polime-
rów kwasu metakrylowego. Aktualnie
wœród preparatów dostêpnych na pol-
skim rynku warunki takie spe³nia jedy-
nie Kreon 10 000 i Kreon 25 000.
W okresie upa³ów i przy wyst¹pie-
niu gor¹czki u pacjentów z CF koniecz-
ne jest uzupe³nianie diety sol¹ (NaCl) –
u niemowl¹t: 100 mg/kg m.c./dobê,
w wieku 1–5 lat: 500 mg/kg m.c./dobê,
w wieku 6–10 lat: 1 000 mg/kg m.c./do-
bê, powy¿ej 11. roku ¿ycia: 1 500 mg/kg
m.c./dobê.
W przypadku stwierdzenia zaburzeñ
dotycz¹cych w¹troby lub dróg ¿ó³cio-
wych (¿ó³taczka w wywiadzie, wystê-
powanie cholestazy), a tak¿e w celu za-
pobiegania powik³aniom w¹trobowym
w przebiegu CF, wskazane jest podawa-
nie preparatu kwasu ursodezoksycholo-
wego w dawce dobowej 10–30 mg/kg
m.c. W przypadku podejrzenia nadci-
œnienia wrotnego (szczególnie u chorych
ze splenomegali¹) wymagana jest dia-
gnostyka (USG jamy brzusznej z bada-
niem dopplerowskim, badanie endosko-
powe prze³yku) i leczenie w oœrodku
specjalistycznym.
Ze wzglêdu na czêste wystêpowanie
odp³ywu ¿o³¹dkowo-prze³ykowego
u chorych na mukowiscydozê (zazwy-
czaj bez typowych objawów ze strony
przewodu pokarmowego) wskazane jest
okresowe wykonywanie specjalistycz-
nych badañ diagnostycznych (g³ównie
24-godzinne badanie pH-metryczne
prze³yku).
Najwa¿niejszym zagadnieniem w te-
rapii mukowiscydozy – obok leczenia
¿ywieniowego – jest leczenie zmian
oskrzelowo-p³ucnych. Wymaga ono
skojarzonego stosowania leków muko-
litycznych i rozszerzaj¹cych oskrzela,
antybiotykoterapii i fizjoterapii oraz le-
czenia przeciwzapalnego. Leki mukoli-
tyczne stosowane s¹ w celu up³ynnienia
wydzieliny drzewa oskrzelowego. Naj-
skuteczniejsz¹ metod¹ ich poda¿y jest
wykonanie inhalacji. Leki mukolitycz-
ne mog¹ byæ równie¿ podawane doust-
nie i do¿ylnie. Do inhalacji u¿ywa siê
preparatów ambroksolu lub N-acetyloc-
ysteiny albo hipertonicznych roztworów
NaCl. Jednak¿e najskuteczniejszym
obecnie lekiem mukolitycznym jest
ludzka rekombinowana DN-aza. Leki
rozszerzaj¹ce oskrzela podawane s¹ dro-
g¹ wziewn¹ i znajduj¹ zastosowanie
u pacjentów z cechami odwracalnej ob-
turacji. Ich zastosowanie powinno po-
przedzaæ wziewn¹ poda¿ leków muko-
litycznych i antybiotyków. Do¿ylne an-
tybiotyki stosuje siê w przypadkach
nowych zaka¿eñ oskrzelowo-p³ucnych
oraz w zaostrzeniu przewlek³ych zmian
zapalnych. Wskazanym jest wtedy prze-
prowadzenie co najmniej 14-dniowej
kuracji do¿ylnej (wyj¹tkowo doustnej),
je¿eli to mo¿liwe w oparciu o lekowra¿-
liwoϾ flory bakteryjnej drzewa oskrze-
lowego. Ze wzglêdu na zwiêkszon¹ eli-
minacjê leków oraz s³ab¹ penetracjê do
gêstej, lepkiej wydzieliny oskrzelowej
w mukowiscydozie stosuje siê wiêksze
dawki antybiotyków. Przy braku wyni-
ków badañ bakteriologicznych najczê-
œciej prowadzi siê terapiê skojarzon¹
aminoglikozydem i ceftazydymem. Po
uzyskaniu œwie¿ych wyników dokonu-
je siê ewentualnej modyfikacji leczenia.
W razie koniecznoœci czêstego, do¿yl-
nego stosowania antybiotyków nale¿y
rozwa¿yæ za³o¿enie cewnika do ¿y³y
centralnej (Vascuport, Port-a-cath, Cuf-
f-cath), który mo¿na tak¿e wykorzystaæ
dla celów ¿ywienia pozajelitowego.
W przypadku przewlek³ego zaka¿enia
Pseudomonas aeruginosa wskazana jest
d³ugotrwa³a antybiotykoterapia wziew-
na (kolistyna, gentamycyna lub tobra-
mycyna). W przewlek³ym leczeniu prze-
ciwzapalnym zastosowanie znajduj¹ za-
równo glikokortykosteroidy (systemowo
b¹dŸ wziewnie), jak i leki niesterydowe
(systemowo – ibuprofen, wziewnie – np.
nedokromil sodu). Bardzo wa¿nym za-
gadnieniem jest fizjoterapia, która
w istotny sposób wp³ywa na skutecz-
noϾ prowadzonego leczenia (metody
bierne – oklepywanie, uciskanie + dre-
na¿; metody czynne – technika natê¿o-
nego wydechu, technika aktywnego cy-
klu oddechowego, drena¿ autogenny,
æwiczenia z zastosowaniem Fluttera lub
maski PEP) [3].
W kompleksowym leczeniu muko-
wiscydozy nie mo¿na zapomnieæ o in-
nych, czêœciej lub rzadziej wystêpuj¹-
cych, objawach klinicznych (np. polipy
nosowe, zapalenia zatok, cukrzyca, oste-
oartropatia, niep³odnoœæ) oraz o mo¿li-
woœci wyst¹pienia ka¿dej choroby wie-
ku dzieciêcego/doros³ego (czêsto zapo-
mina siê o tym, ¿e u chorego na
mukowiscydozê mo¿e pojawiæ siê ka¿-
da inna choroba!). Aby uzyskaæ najwy¿-
sz¹ skutecznoœæ prowadzonego postê-
powania terapeutycznego (z³o¿onoœæ
choroby), leczenie chorego na mukowi-
scydozê powinno byæ prowadzone przez
oœrodek specjalistyczny, przy sta³ej
wspó³pracy lekarza rodzinnego.
Piœmiennictwo
1. Riordan JR, Rommens JM, Kerem B, Alon N,
Rozmahel R, Grzelczak Z, Zieleñski J, Lok S,
Plavsic N, Chou JL, Drumm ML, Ianuzzi ML,
Collins FC, Tsui LC. Identification of the cy-
stic fibrosis gene: cloning and characteriza-
tion of complementary DNA. Science 1989;
245: 1066-73. Published erratum appears in
Science 1989; 245: 1437.
2. Tümmler B, Puchelle E. CFTR: a multiface-
ted molecule. Trends Cell Biol 1997; 7: 250-
1.
3. Bo¿kowa K, Cichy W, Jarosz J, Ksi¹¿yk J, £u-
kasik M, Mazurczak T, Milanowski A, Nowa-
kowska A, Orlik T, Pawlik J, Piotrowski R,
Pogorzelski A, Prusak J, Sands D, Skorupa W,
Stolarczyk A, Teysseire M, Walkowiak J,
Wierzbicka M, Witt M, ¯ebrak J. Zasady roz-
poznawania i leczenia mukowiscydozy. Pol-
ska Grupa Robocza Mukowiscydozy, Warsza-
wa 1999; 5-20.
4. Zieleñski J. Genotype and phenotype in cystic
fibrosis. Respiration 2000; 67: 117-33.
5. Ahmed N, Durie P. Gastrointestinal and pan-
creatic complications of cystic fibrosis. Infan-
cy and childhood. International Seminars in
Pediatric Gastroenterology and Nutrition
2000; 4: 1-7.
dr n. med. Jaros³aw Walkowiak
Klinika Gastroenterologii Dzieciêcej
i Chorób Metabolicznych
Instytutu Pediatrii
Akademii Medycznej w Poznaniu
kierownik Kliniki prof. dr hab. n. med.
Marian Krawczyñski
prof. dr hab. n. med. Wojciech Cichy
Klinika Gastroenterologii Dzieciêcej
i Chorób Metabolicznych
Instytutu Pediatrii
Akademii Medycznej w Poznaniu
e-mail: cichy@sk5.usoms.poznan.pl
Wyszukiwarka
Podobne podstrony:
MUKOWISCYDOZA 4
Mukowiscydoza, Niedokrwistość sierpowao krwinkowa
Mukowiscydoza etiologia, zmiany narzadowe i ich nastepstwa[1]
mukowizcydoza, FIZJOTERAPIA
Objawy gastroenterologiczne w przebiegu mukowiscydozy, Pielęgniarstwo
Mukowiscydoza
Mukowiscydoza
materiały do egz (orto, pulmo, itp), Mukowiscydoza, Mukowiscydoza
mukowiscydoza (2), Fizjoterapia, . fizjoterapia
Pediatria. Mukowiscydoza, Wykłady, PEDIATRIA
Mukowiscydoza itd, KRZYWICA
Mukowiscydoza itd, KRZYWICA
MUKOWISCYDOZA(1)
MUKOWISCYDOZA F3
Rola diety w mukowiscydozie, różności, dietetyka, ciekawostki, diety, normy
więcej podobnych podstron