2012-03-28
1
Wykład 6
Koloidy
1
CHEMIA FIZYCZNA
Szkoła Główna Gospodarstwa Wiejskiego
Wydział Nauk o Żywności
Wykład 6.
Koloidy
dr inż. Bożena Parczewska-Plesnar
Katedra Chemii
WNoŻ, bud. 32, p. 2035
E-mail: bparczewska@wp.pl
2
Do opisu stanu równowagi adsorpcyjnej stosuje się najczęściej tzw.
równania izoterm adsorpcji określające charakter zależności ilości
adsorbatu zaadsorbowanego na 1 g adsorbentu od ciśnienia (jeśli
adsorbat jest gazem) lub stężenia adsorbatu (w przypadku roztworu)
przy zachowaniu stałości temperatury.
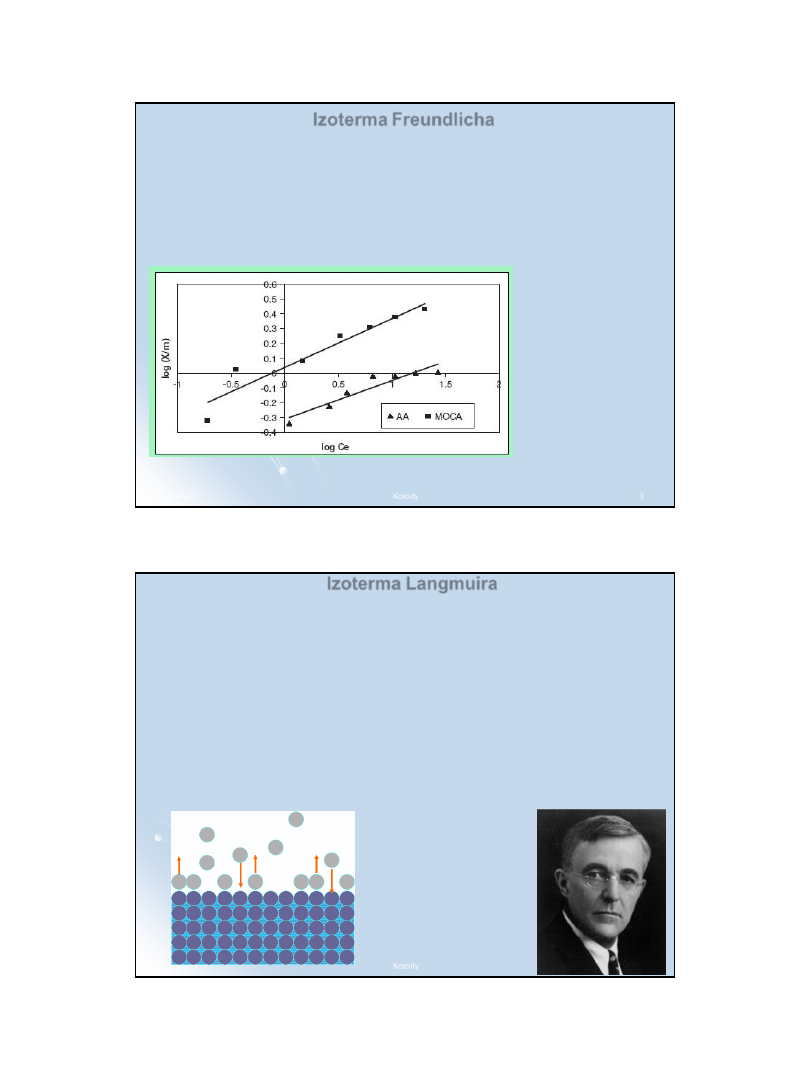
Izoterma Freundlicha
W 1895 r. Boedeker
podał po raz pierwszy empiryczne równanie
izotermy adsorpcji w postaci:
gdzie: k i n są stałymi, p to ciśnienie gazowego adsorbatu.
Równanie to zwane jest równaniem izotermy adsorpcji Freundlicha,
gdyż to Freundlich przypisał temu równaniu wielkie znaczenie i
rozpowszechnił jego stosowanie.
W przypadku adsorpcji z roztworów ciekłych, ciśnienie zastępuje się
stężeniem adsorbatu w roztworze:
n
p
k
a
Często przedstawia się równanie tej izotermy w postaci
logarytmicznej:
n
c
k
a
k
log
c
log
n
a
log
Wykład 6
Koloidy
2012-03-28
2
3
W równaniu:
loga
jest liniową funkcją logc, dlatego wartość n można wyliczyć ze
współczynnika kierunkowego prostej, którą wyznacza się
eksperymentalnie na podstawie serii punktów adsorpcji zmierzonych
dla roztworów o różnym stężeniu adsorbatu.
Izoterma Freundlicha
k
log
c
log
n
a
log
Równanie
izotermy
Freundlicha
stosuje się do
opisu adsorpcji
fizycznej gazów i
składników
roztworów
ciekłych.
Wykład 6
Koloidy
y
a
log
x
c
log
Wykład 6
Koloidy
4
Adsorpcję chemiczną opisuje izoterma Langmuira opierająca się na
następujących założeniach:
powierzchnia stałych adsorbentów ma na swej powierzchni
szczególnie aktywne miejsca zwane
centrami aktywnymi
,
proces adsorpcji zachodzi na centrach aktywnych,
każde centrum może zaadsorbować tylko jedną cząstkę, czyli
adsorbent pokrywa się
warstwą monomolekularną
,
cząsteczki zaadsorbowane na centrach adsorpcji nie oddziałują
wzajemnie na siebie,
proces adsorpcji ma charakter dynamicznej równowagi pomiędzy
adsorpcją i procesem odwrotnym do adsorpcji – desorpcją:
cząsteczka w fazie gazowej
⇄ cząsteczka w adsorbencie
Izoterma Langmuira
USA, 1881
–1957
1932 Nagroda
Nobla za odkrycie i
badania nad
chemią powierzchni
2012-03-28
3
Stopień pokrycia
q
definiujemy jako stosunek ilości moli
zaadsorbowanej substancji (n) do ilości moli tej substancji potrzebnej
do pokrycia całej powierzchni adsorbentu warstwą monomolekularną
(n
o
):
Wartość
q
zawiera się między 0 gdy adsorpcja nie występuje a 1 gdy
adsorpcja jest równa a
max
, czyli cała powierzchnia jest pokryta warstwą
monomolekularną.
Izotermę adsorpcji Langmuira
przedstawia równanie:
lub
gdzie: p -
ciśnienie gazu,
b -
współczynnik adsorpcji.
5
Izoterma Langmuira
max
o
a
a
n
n
q
Izoterma adsorpcji Langmuira dobrze opisuje przypadki chemisorpcji,
natomiast w przypadku adsorpcji fizycznej na ogół zawodzi.
p
b
1
p
b
a
a
max
a
1
2
3
p
max
a
a
p
b
a
a
max
p
b
p
b
a
a
max
1
1
– gdy p małe, to 1+b
.
p
≈ 1
3
– gdy p duże, to 1+b
.
p
≈ b
.
p
Wykład 6
Koloidy
p
b
1
p
b
q
6
W przypadku adsorpcji fizycznej dochodzi do tworzenia się warstw
wielomolekularnych
, ponieważ ten sam rodzaj sił, jakie odpowiedzialne
są za adsorpcję fizyczną pierwszej warstwy adsorbatu, działa też
pomiędzy nią a dalszymi, zbliżającymi się do powierzchni adsorbentu
cząsteczkami.
Izoterma BET (Brunauer, Emmet i Teller)
Wykład 6
Koloidy
2012-03-28
4
Wykład 6
Koloidy
7
Równanie opisujące ilość
substancji zaadsorbowanej w
takich warunkach przez
jednostkową masę adsorbentu,
jako funkcję ciśnienia gazu
znane pod nazwą BET ma
postać:
p
p
1)
-
(C
+
1
p)
-
p
(
p
C
a
=
a
o
o
max
C jest pewną funkcją temperatury,
określoną przez różnicę
pomiędzy ciepłem adsorpcji w
pierwszej monomolekularnej
warstwie i ciepłem skraplania:
Izoterma BET
)RT
E
(E
k
m
e
C
gdzie: p
o
-
jest prężnością pary nasyconej adsorbatu w temperaturze, w
której odbywa się adsorpcja,
C
– stała.
8
Adsorpcja elektrolitów
Opisane tu izotermy adsorpcji dobrze opisują adsorpcję w prostych
układach gazowych lub ciekłych. W przypadku mieszanin
wieloskładnikowych opis jest bardziej złożony, ponieważ ustalająca się
równowaga adsorpcji to wynik konkurowania składników układu w
oddziaływaniu z adsorbentem. Wówczas stosuje się bardziej
skomplikowane zależności.
W przyrodzie i technice często spotykana jest adsorpcja elektrolitów.
Adsorpcji ulega najczęściej tylko jeden rodzaj jonów zawartych w
roztworze, wskutek czego na powierzchni adsorbentu gromadzi się
ładunek elektryczny. Ładunek ten zostaje skompensowany albo przez
jony przeciwnego znaku gromadzące się w cieczy tuż przy powierzchni
adsorbentu, albo też przez przejście z powierzchni adsorbentu do
roztworu równoważnej ilości innych jonów tego samego znaku.
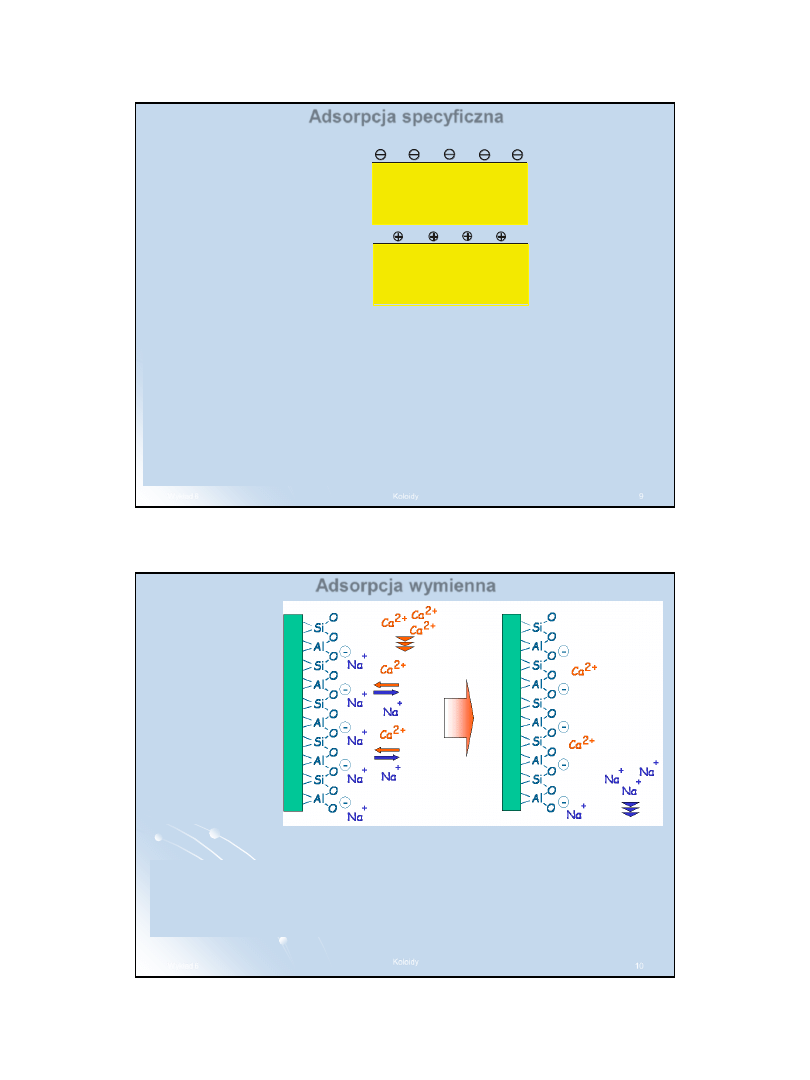
Pierwszy rodzaj adsorpcji nazwiemy
adsorpcją specyficzną
, drugi
rodzaj -
adsorpcją wymienną
.
Wykład 6
Koloidy
2012-03-28
5
9
Adsorpcja specyficzna
W przypadku adsorpcji
specyficznej, substancja o
budowie jonowej na ogół
najłatwiej adsorbuje te jony,
które wchodzą w skład jej
sieci przestrzennej.
Osad AgI
adsorbuje więc z
roztworu AgN0
3
jony srebra,
natomiast z roztworu KI jony
jodkowe.
sieć AgI
zaadsorbowane jony Ag
+
ciecz (jony NO
3
-
)
+
+
+
+
+
+
+
+
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
+
+
+
+
+
+
+
+
-
-
-
-
-
+
+
+
+
+
+
+
+
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
+
+
+
+
sieć AgI
zaadsorbowane jony I
-
ciecz (jony K
+
)
W przypadku adsorpcji jonów srebra powierzchnia adsorbentu ładuje
się dodatnio i zaczyna przyciągać w pobliże granicy faz jony znaku
przeciwnego, tj. jony NO
3
-
.
W ten sposób
na granicy faz tworzy się elektryczna warstwa podwójna
złożona z zaadsorbowanych jonów Ag
+
i z zobojętniających je, lecz
znajdujących się w cieczy, jonów NO
3
-
.
Gdy adsorpcji ulegają jony l
-
, adsorbent ładuje się ujemnie, w cieczy
natomiast na granicy faz gromadzą się jony dodatnie, K
+.
Wykład 6
Koloidy
10
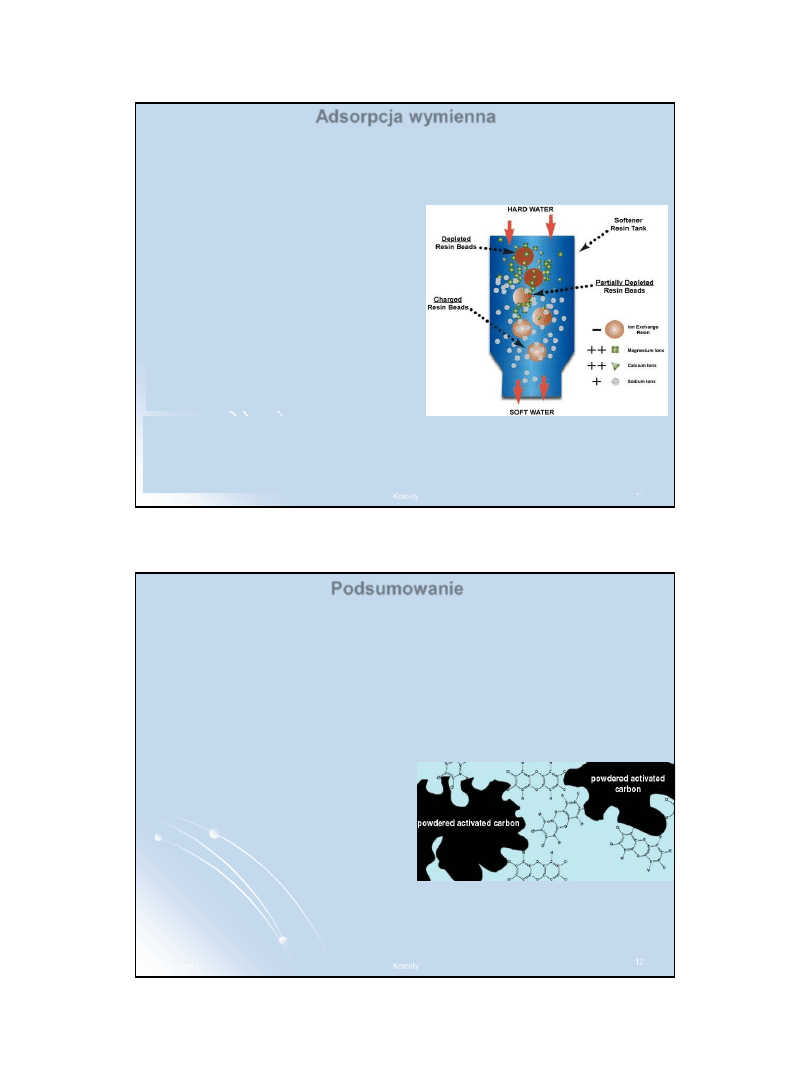
Adsorpcja wymienna
Typowym
przykładem
adsorbentów
wykazujących
adsorpcję
wymienną są
występujące w
przyrodzie oraz
otrzymywane
sztucznie zeolity
(krystaliczne
glinokrzemiany).
Materiały te adsorbują z wody np. jony Ca
2+
i Mg
2+
równocześnie
oddając do roztworu w równoważnej ilości jony sodu lub potasu,
dzięki czemu adsorbent pozostaje obojętny pod względem
elektrycznym.
Wykład 6
Koloidy
2012-03-28
6
11
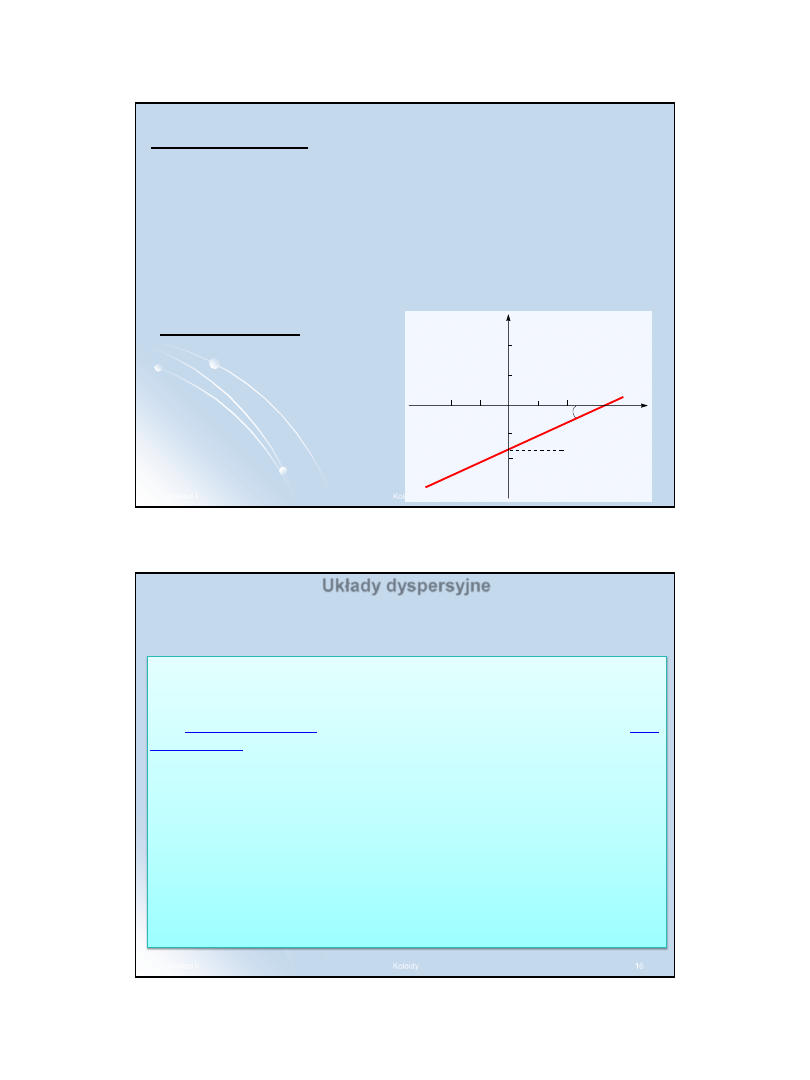
Adsorpcja wymienna
Szerokie zastosowanie znalazły syntetyczne żywice organiczne zwane
jonitami. Zależnie od swego składu mogą one wymieniać kationy
(kationity) lub aniony (anionity). Posługując się jonitami, możemy
przeprowadzić praktycznie całkowitą demineralizację wody.
Wodę przepuszcza się najpierw
przez warstwę kationitu
zawierającego jony wodorowe.
Usuwamy z niej wówczas jony metali
(Na
+
, Ca
2+,
Mg
2+
), a wprowadzamy na
ich miejsce jony H
+
. Następnie na
anionicie wymieniamy aniony (SO
4
2-
,
Cl
-
) na jony OH
-
. Zużyte jonity można
łatwo zregenerować, przemywając
roztworem kwasu lub wodorotlenku
litowca.
Jonity stosuje się także w procesach wydzielania kationów metali
szlachetnych: złota, srebra, platyny, z rozcieńczonych roztworów, do
rozdzielania pierwiastków promieniotwórczych, rozdzielania metali ziem
rzadkich itd.
Wykład 6
Koloidy
12
Charakter adsorpcji
można orientacyjnie określić na podstawie
przebiegu izotermy adsorpcji
•
Jeżeli wykres zależności a=f(c) lub a=f(p) asymptotycznie dąży do
wartości maksymalnej, to adsorpcja monomolekularna powinna być
opisana równaniem Langmuira
•
Jeżeli wartość a wzrasta nieograniczenie ze wzrostem wartości c lub
p, to adsorpcja wielowarstwowa
– polimolekularna, opisana izotermą
Freundlicha lub BET.
Podsumowanie
Rodzaje adsorbentów
węgle aktywowane (tzw. bezpostaciowa odmiana węgla o bardzo
rozwiniętej powierzchni otrzymywana z drewna lub torfu, odpowiednio
przygotowana).
związki glinu (głównie tlenki),
związki krzemu (żele krzemionkowe
wąsko- i szeroko porowate, ziemie
okrzemkowe),
związki glinu i krzemu (żele
glinokrzemianowe, krystaliczne
glinokrzemiany
– zeolity)
Wykład 6
Koloidy

2012-03-28
7
węgiel aktywowany szeroko stosowany jest w chemii
spożywczej, przemyśle farmaceutycznym, obronnym (w
pochłaniaczach masek gazowych), w gospodarce komunalnej,
do oczyszczania wód;
w procesach odzyskiwania cynku, niklu, siarki z rud poprzez
flotację i metodą pianową;
reakcje katalityczne
–
zachodzące w obecności adsorbentów
zwanych
katalizatorami
–
tzw.
kataliza wielofazowa
(heterogeniczna):
katalizator tworzy z reagentami związki pośrednie (obniża barierę
energetyczną reakcji, przyspiesza osiągnięcie stanu równowagi
reakcji) i zwiększa szybkość reakcji;
efekty katalityczne występują najsilniej w przypadku adsorpcji
chemicznej (chemisorpcji);
adsorpcja fizyczna powoduje zwiększenie lokalnego stężenia
reagentów i jest dla nich źródłem energii cieplnej.
13
Zastosowanie procesów adsorpcji
Wykład 6
Koloidy
14
Katalizator
Proces
Żel krzemionkowo–glinowy
Kraking ciężkich frakcji ropy
naftowej
Żel tlenku chromu (III) Cr
2
O
3
na
Al
2
O
3
, tlenek niklowo
–glinowy
Uwodornianie, odwodornienie
węglowodorów
H
3
PO
4
na ziemi okrzemkowej
Polimeryzacja alkenów
Żelazo
Synteza amoniaku
Miedź
Utlenianie alkoholi do
aldehydów
Co, ThO
2
, Mg na ziemi
okrzemkowej
Synteza węglowodorów z H
2
i
CO
Przykłady procesów i reakcji katalitycznych
Wykład 6
Koloidy
2012-03-28
8
Koloidy
15
a
1
, a
2
– wartości adsorpcji rzeczywistej wyznaczone
doświadczalnie w obu roztworach
c
r1
, c
r2
– wyznaczone doświadczalnie stężenia końcowe
adsorbatu w roztworach w stanie równowagi.
Metoda analityczna:
r
nlogc
+
logk
=
loga
Rozwiązanie układu dwóch równań z dwiema niewiadomymi
dla dwu roztworów adsorbatu różniących się stężeniem:
r1
1
nlogc
+
logk
=
loga
r2
2
nlogc
+
logk
=
loga
Metody wyznaczania współczynników izotermy Freundlicha
Metoda graficzna:
r
nlogc
+
logk
=
loga
lg k
lg a
-2
2
lg c
r
-1
1
-1
1
2
-2
n = tg
loga
y
r
logc
x
b
ax
y
k
log
b
n
a
Wykład 6
16
Układy dyspersyjne
W układzie wielofazowym zjawiska powierzchniowe odgrywają tym
większą rolę, im bardziej rozwinięta jest powierzchnia stanowiąca
granicę faz.
Układy, w których uzyskano znaczne rozwinięcie powierzchni wskutek
rozdrobnienia jednej z
faz, nazywamy układami dyspersyjnymi.
W
każdym układzie dyspersyjnym możemy wyróżnić fazę rozproszoną
oraz
fazę rozpraszającą, otaczającą i oddzielającą od siebie cząstki fazy
rozproszonej.
Obie fazy mogą występować w różnych stanach skupienia: stałym,
ciekłym i gazowym. A więc:
ciało stałe może być rozproszone w cieczy, gazie lub innym ciele
stałym;
ciecz może być rozproszona w gazie, ciele stałym lub w innej cieczy
nie mieszającej się z nią;
gaz może być rozproszony w cieczy lub w ciele stałym. Nie jest
oczywiście możliwe rozproszenie fazy gazowej w innej fazie gazowej,
gdyż, jakiekolwiek dwa gazy zawsze mieszają się ze sobą dając roztwór
homogeniczny.
Wykład 6
Koloidy
2012-03-28
9
17
Układy dyspersyjne
Podział układów dyspersyjnych ze względu na stopień rozdrobnienia
fazy zdyspergowanej (f
z
)
Układ dyspersyjny
Układ
jednorodny
(homogeniczny)
f
z
< 1 nm
•
rozdrobnienie
molekularne
•
jednakowe
właściwości
fizykochemiczne
w każdym
elemencie
układu
Układ niejednorodny (heterogeniczny)
•
w obrębie układu występują obszary o
odmiennych właściwościach
rozdrobnienie
koloidalne
(roztwór koloidalny,
zol)
1nm
≤ f
z
< 200 nm
Rozdrobnienie
grubodyspersyjne
(makroskopowe)
f
z
≥ 200 nm
Mieszaniny takie można
stosunkowo łatwo rozdzielić
metodami fizycznymi.
Koloidy
Wykład 6
18
Układy dyspersyjne
Koloidy
Układy o rozdrobnieniu molekularnym – roztwory rzeczywiste, zostały
włączone do klasyfikacji dla pełniejszego obrazu. W tym przypadku nie
można mówić o istnieniu fazy rozproszonej i istnieniu granicy faz.
Granica pomiędzy rozproszeniem koloidalnym a rozproszeniem
molekularnym jest
granicą wybraną umownie. W rzeczywistości istnieje
raczej stopniowe przejście od układów o rozdrobnieniu molekularnym
do
układów o rozdrobnieniu koloidalnym. Podobnie przejście od
układów rozdrobnionych koloidalnie do układów grubodyspersyjnych,
np. zawiesin, ma
charakter ciągły. Granicę 200nm wybrano jako granicę,
poniżej której cząstek rozproszonych nie można obserwować za
pomocą zwykłego mikroskopu optycznego.
1nm
200nm
<1nm
>200nm
Wykład 6

2012-03-28
10
19
Układy koloidalne - podział
Faza
rozpraszająca
f
d
Faza
rozproszona
f
z
Rodzaj
koloidu
Przykłady
Nazwa
szczegółowa
koloidu
Gaz
Gaz
Nie istnieje
Ciecz
Aerozole
(gazozole)
Mgła, chmury Aerozole ciekłe
(mgły)
Ciało stałe
Dym, kurz
Aerozole stałe
(dymy)
Ciecz
Gaz
Zole,
roztwory
koloidalne
Piana
Piany
Ciecz
Mleko, białko Lizole, emulsje
Ciało stałe
Zole tlenków
metali,
wodorotlenków
Zawiesiny
koloidalne
Ciało stałe
Gaz
Dirozole
Pumeks,
okluzje gazowe
Piany stałe
Ciecz
Kwarc mleczny
Emulsje stałe
(piany stałe)
Ciała stałe
Szkło
rubinowe,
perły
fosforowe
Zole stałe
Wykład 6
Koloidy
Wykład 6
Koloidy
20
20
Przykłady układów koloidalnych
Naturalne:
mleko
–
kropelki tłuszczu i białka w wodzie
mgła, chmury
–
kropelki wody rozproszone w powietrzu
krew
–
krwinki, płytki, białe ciałka w osoczu
błoto
–
zawiesina gleby w wodzie
Wytworzone sztucznie:
farby
– barwniki, wypełniacze i substancje pokrywające
rozproszone w rozpuszczalniku
ciekłe kryształy
– uporządkowane struktury drobnych cząstek
będących długimi cząsteczkami
kosmetyki
– tłuszcze, witaminy, i to co wymienione w reklamie,
wszystko zawieszone w wodzie lub tłuszczu
lekarstwa
– czynnik aktywny rozproszony w obojętnej matrycy
Zjawiska powierzchniowe
Roztwory koloidalne, zole, zależnie od środka rozpraszającego, określa
się bardziej szczegółowymi nazwami, np. alkozole, w przypadku gdy
fazą rozpraszającą jest alkohol, lub aerozole, gdy funkcję tę spełnia
powietrze. Najczęściej jednak mamy do czynienia z hydrozolami, w
których ośrodkiem rozpraszającym jest woda. W dalszym ciągu
będziemy mówić tylko o hydrozolach.
Wykład 13

2012-03-28
11
21
Układy płynne i spoiste
Koloidy
W
układach dyspersyjnych, nazywanych
układami spoistymi, cząstki rozproszone
stykają się z sobą bezpośrednio i tworzą
układ mniej lub bardziej sztywny.
Przykładem tego rodzaju układów
spoistych są żele, a także liczne ciała stałe
otrzymywane w
stanie dużego
rozdrobnienia, np. węgiel aktywny.
Koloidalny
roztwór złota
Wykład 6
W
układach dyspersyjnych
cząstki fazy rozproszonej
mogą być całkowicie od
siebie izolowane przez fazę
rozpraszającą. W
przypadku cząstek
koloidalnych mówimy
wtedy o roztworze
koloidalnym, czyli zolu.
Roztwory koloidalne, w
których faza rozpraszająca
jest
cieczą lub gazem,
zachowują swoją płynność.
Wykład 6
22
Właściwości optyczne układów koloidowych
Koloidy
Zole pod
względem swego wyglądu
pozornie nie różnią się od roztworów
rzeczywistych. Jedne i drugie są
przezroczyste i w zasadzie klarowne.
Jeżeli jednak będziemy obserwować w
ciemnym pokoju wiązkę światła
przechodzącą przez ciecz, wystąpią
wyraźne różnice w zachowaniu się obu
roztworów.
W
przypadku roztworu rzeczywistego wiązka będzie niewidoczna
dla
obserwatora spoglądającego w kierunku prostopadłym do biegu
promieni.
W przypadku roztworu koloidalnego natomiast obserwator
dostrzeże smugę światła w cieczy
, analogiczną do smugi światła
widocznej w powietrzu, gdy do
mrocznego pokoju wpada wąski
promień słoneczny. Powstawanie takiej smugi polega na rozpraszaniu
światła przez cząstki koloidalne, podobnie jak smuga widoczna w
pokoju jest
rezultatem rozpraszania światła przez cząstki kurzu
unoszące się w powietrzu.
Zjawisko rozpraszania światła przez roztwory koloidalne nazywa się
efektem Tyndalla.
2012-03-28
12
23
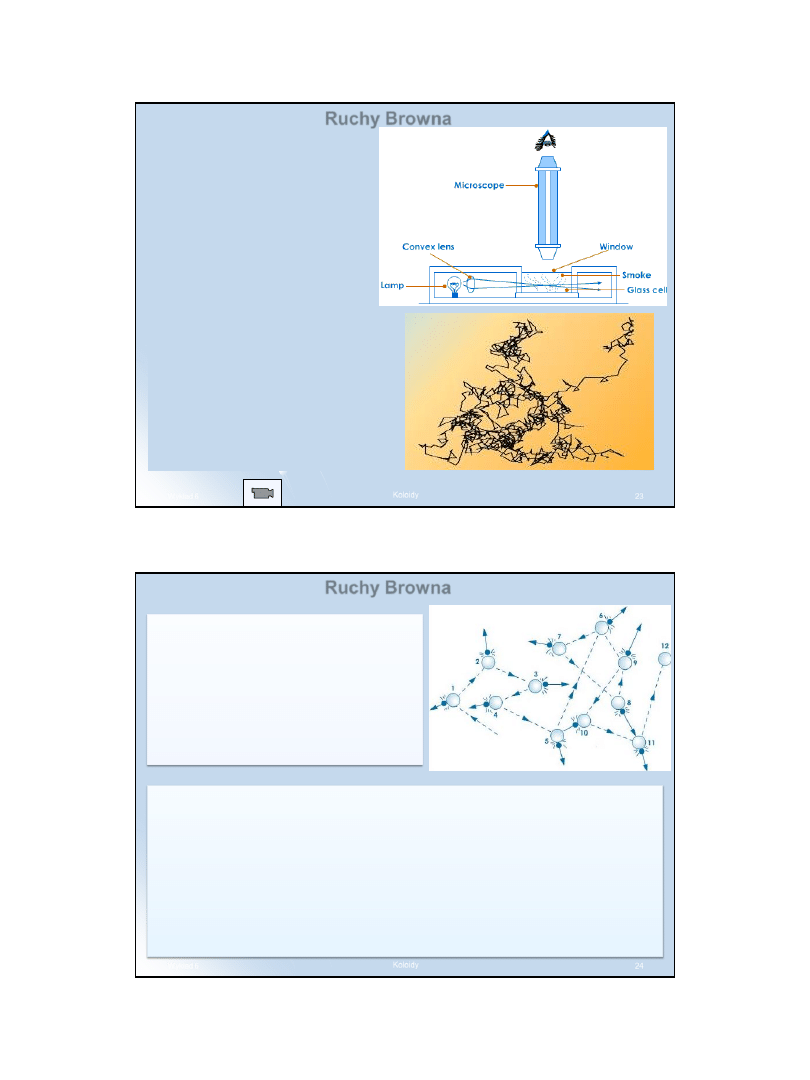
Ruchy Browna
Koloidy
Cząstki koloidalne są zbyt
małe, by można je było
dostrzec pod
zwykłym
mikroskopem optycznym.
Oświetlając roztwór koloidalny
z
boku, zamiast od dołu,
możemy jednak obserwować
pod
mikroskopem świetlne
plamki powstałe wskutek
rozpraszania światła przez
pojedyncze cząstki koloidalne.
W
urządzeniu tego rodzaju,
zwanym ultramikroskopem, nie
udaje się rozróżnić kształtu, barwy
ani rozmiarów cząstek
koloidalnych, można natomiast
śledzić ich bezładne ruchy, zwane
ruchami Browna
.
Wykład 6
24
Ruchy Browna
Koloidy
Bezładne ruchy cząstek koloidowych
zostały odkryte przez botanika
angielskiego Roberta Browna w
1827r.
Stwierdził on na podstawie
obserwacji mikroskopowych, że
drobne pyłki kwiatowe zawieszone w
wodzie znajdują się w ustawicznym,
bezładnym, zygzakowatym ruchu.
Podobnym ruchem poruszają się cząstki koloidalne. Szybkość tych
ruchów wzrasta gdy maleje masa cząstki. Naturę tego zjawiska wyjaśnił
Marian Smoluchowski i Albert Einstein (1905r.) na gruncie teorii
kinetyczno-molekularnej.
Cząsteczki cieczy wykonują ustawiczne ruchy drgające i zderzają się z
cząstkami koloidalnymi. Prawdopodobieństwo dokładnego
zrównoważenia uderzeń nadchodzących z jednej strony cząstki przez
uderzenia nadchodzące z drugiej strony jest małe i w rezultacie cząstka
się przesuwa w sposób zupełnie nieregularny, chaotyczny.
Wykład 6
2012-03-28
13
25
Właściwości kinetyczne układów koloidowych
Sedymentacja
–
opadanie cząstek koloidowych na dno naczynia pod
wpływem siły ciężkości. Prędkość opadania cząstek koloidowych
v
określa wzór:
9
g
)
d
(d
r
2
=
v
0
2
r
– promień cząstki koloidowej, d – gęstość
ośrodka rozpraszającego, d
o
– gęstość cząstki
koloidowej, g
– przyspieszenie ziemskie,
–
lepkość
Zastosowania
: pomiar wielkości cząstek ważny w produkcji: artykułów
spożywczych, kosmetyków, farmaceutyków, papieru, cementu, farb
i barwników, przeróbki kopalin
Wykład 6
r
N
3
t
T
R
=
x
A
2
Δx – średnia wartość przesunięcia w
czasie
Δt wyznaczona na podstawie
dużej liczby pomiarów dla pojedynczej
cząstki koloidowej, r – promień cząstki
koloidowej, η – współczynnik lepkości
ośrodka, R – stała gazowa, T –
temperatura w K, N
A
– liczba Avogadra.
Wykład 6
26
Otrzymywanie koloidów
Koloidy
Są dwie grupy metod otrzymywania koloidów:
metody dyspersyjne
(zmniejszanie rozmiarów
cząstek o charakterze grubodyspersyjnym do
rozmiarów koloidalnych) i
metody kondensacyjne
(łączenie cząstek znajdujących się w stanie
rozproszenia molekularnego).
Metody dyspersyjne:
Najprostsza metoda dyspersyjna polega na
mieleniu
rozdrabnianych
substancji w
młynach koloidowych o bardzo dużej liczbie obrotów.
Metoda
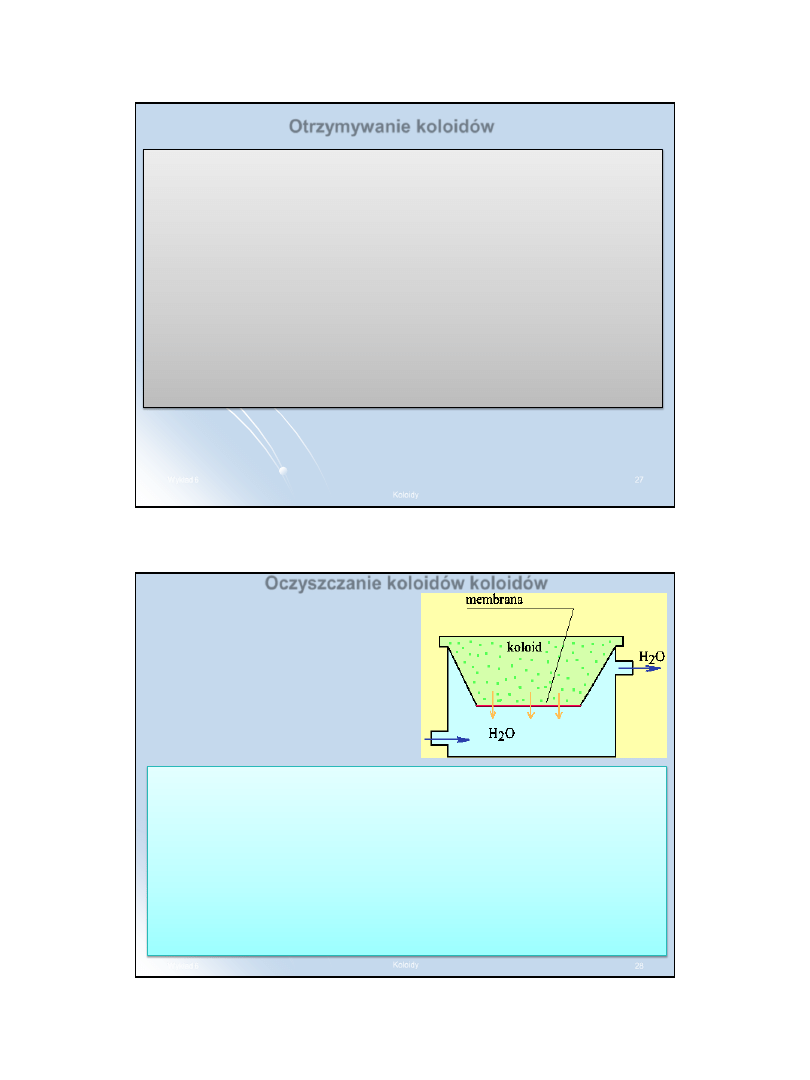
rozpylania katodowego
(metoda Brediga)
– polega na
wytworzeniu, za pomocą prądu stałego, łuku elektrycznego pomiędzy
dwiema elektrodami metalowymi zanurzonymi w wodzie, z
których
jedną - katodę, stanowi metal poddawany rozdrobnieniu. Pary metalu
wydzielające się w tych warunkach są gwałtownie chłodzone w wodzie i
zestalają się tworząc cząstki koloidalne.
Zawiesiny można niekiedy także przeprowadzić w stan koloidalny,
działając
falami ultradźwiękowymi
.
Wiele substancji o
bardzo dużej masie cząsteczkowej -
substancje
makromolekularne
, w
których pojedyncze cząsteczki chemiczne mają
rozmiary cząstek koloidalnych np. żelatyna lub niektóre białka,
wykazuje zdolność
samorzutnego tworzenia roztworów koloidalnych
w
zetknięciu z wodą.
2012-03-28
14
27
Otrzymywanie koloidów
Koloidy
Metody kondensacyjne
polegają na
wydzieleniu trudno rozpuszczalnej
substancji z
roztworu silnie rozcieńczonego w trakcie różnego rodzaju
reakcji chemicznych
, np. podwójnej wymiany, utleniania, redukcji,
hydrolizy itp.
Warunki panujące w roztworze są przy tym tak dobrane, że
nierozpuszczalny produkt reakcji nie może wytworzyć agregatów
większych niż te o rozmiarach cząstek koloidalnych.
Nie wydziela się on
wówczas w formie osadu, lecz tworzy roztwór koloidalny.
Na przykład:
hydroliza rozcieńczonego roztworu FeCl
3
w temperaturze wrzenia
prowadzi do
powstania roztworu koloidalnego wodorotlenku żelaza(III),
redukcja chlorku złota taniną lub formaldehydem powoduje powstanie
koloidalnego roztworu złota metalicznego.
Wykład 6
28
Oczyszczanie koloidów koloidów
Koloidy
Roztwór koloidalny otrzymany
którymkolwiek z opisanych sposobów
jest
silnie zanieczyszczony różnymi
elektrolitami.
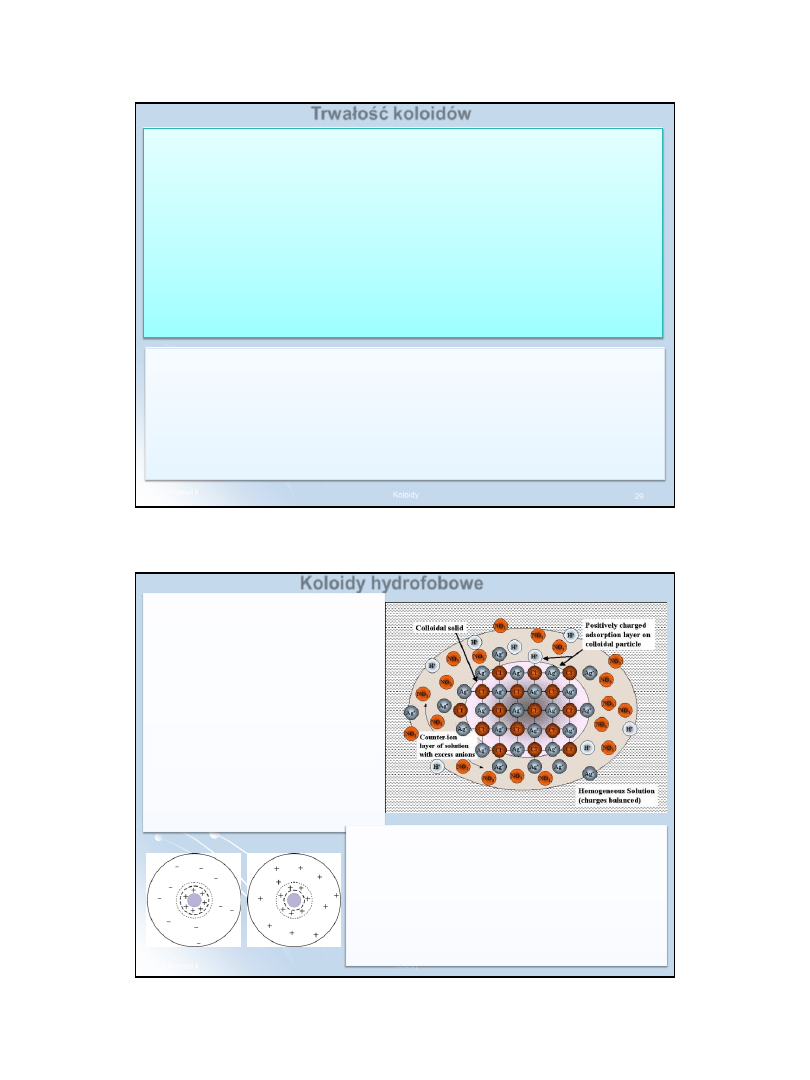
Oczyszczanie roztworów koloidalnych
przeprowadzać można za pomocą
dializy.
Urządzenie służące do tego
celu, dializator, przedstawiono na
rysunku.
Wykład 6
Istotną jego część stanowi błona z papieru pergaminowego, kolodium
lub innego materiału zawierającego mikropory na tyle wąskie, że nie
mogą przez nie przechodzić cząstki o rozmiarach koloidalnych, łatwo
natomiast mogą się przedostawać cząstki o rozmiarach molekularnych.
Błona ta stanowi dno dializatora.
Dializator po wypełnieniu roztworem koloidalnym zostaje częściowo
zanurzony w
zbiorniku, przez który przepływa czysta woda.
Zanieczyszczenia roztworu koloidalnego dyfundują stopniowo przez
błonę na zewnątrz i zostają uniesione przez prąd wody. Wewnątrz
dializatora pozostaje roztwór koloidalny pozbawiony elektrolitów.
2012-03-28
15
29
Trwałość koloidów
Koloidy
Silnie rozdrobniona substancja, tj. substancja o bardzo znacznie
rozwiniętej powierzchni, jest bogatsza w energię od substancji słabo
rozdrobnionej, wskutek czego wykazuje dążność do zmniejszania swej
powierzchni.
Można oczekiwać, że roztwory koloidalne będą nietrwałe i że cząstki
koloidalne będą miały tendencję do skupiania się w większe agregaty,
czyli do
koagulacji
.
Efektem obserwowanym jest wówczas
sedymentacja (strącanie się) agregatów cząstek w roztworze lub
powstanie żelu.
W
rzeczywistości jednak mamy często do czynienia z roztworami
koloidalnymi o
praktycznie nieograniczonej trwałości.
Wykład 6
Cząstki zawdzięczają swą trwałość albo elektrostatycznemu
odpychaniu się cząstek, które zyskują jednoimienny ładunek
elektryczny wskutek selektywnej adsorpcji jednego z
jonów zawartych
w roztworze
–
koloidy liofobowe
, albo utworzeniu się na powierzchni
cząstek otoczek z cząsteczek rozpuszczalnika –
koloidy liofilowe
. Jeżeli
fazą rozpraszającą jest woda, układy koloidalne obu rodzajów noszą
nazwy: koloidów
hydrofobowych
i
hydrofilowych
.
30
Koloidy hydrofobowe
Koloidy
Cząstki koloidalne odznaczają
się dużą zdolnością adsorpcji.
Jeżeli adsorpcja ma charakter
selektywny, czyli ulega jej tylko
jeden rodzaj jonów zawartych w
roztworze, to
wszystkie cząstki
zyskują ten sam ładunek
elektryczny, a ich wzajemne
odpychanie przeciwstawia się
tendencji do koagulacji.
Adsorpcji na cząstkach
koloidalnych mogą ulegać
zarówno jony H
+
i OH
-
, jak i inne
jony obecne w roztworze.
Wykład 6
Cząstkę złożoną z trudno rozpuszczalnego
agregatu i związanych z nim warstewek
jonowych nazywamy micelą.
Znak ładunku elektrycznego miceli nie jest
cechą charakterystyczną danej substancji.
W zależności od środowiska ta sama
cząstka koloidalna może mieć ładunek
dodatni lub ujemny.

2012-03-28
16
31
Koloidy hydrofobowe
Koloidy
Wykład 6
Znak ładunku elektrycznego cząstek koloidalnych możemy określić,
jeżeli w roztworze koloidalnym umieścimy dwie elektrody, pomiędzy
którymi istnieje różnica potencjałów. Cząstki naładowane dodatnio
wędrują do bieguna ujemnego, cząstki naładowane ujemnie - do
dodatniego
.
Zjawisko wędrówki cząstek koloidalnych pod wpływem pola
elektrycznego nosi nazwę elektroforezy
.
Za pomocą badań
elektroforetycznych stwierdzono, że
zole wodorotlenków metali, takich jak
Fe(OH)
3
, Cd(OH)
2
, Al(OH)
3
, Cr(OH)
3
, i
zole tlenków metali, np. TiO
2
, ZrO
2
,
ładują się na ogół dodatnio,
natomiast zole metali, np. zole Au, Pt,
Ag, oraz zole siarczków, np. As
2
S
3
,
Sb
2
S
3
– ujemnie.
32
Punkt izoelektryczny
Koloidy
Jeżeli chcemy doprowadzić do koagulacji koloidu hydrofobowego, to
musimy zobojętnić ładunek elektryczny nagromadzony na powierzchni
cząstek: najłatwiej to osiągnąć przez dodanie do roztworu pewnej ilości
elektrolitu zawierającego dobrze adsorbujące się jony przeciwnego
znaku.
Wykład 6
Im większy jest ładunek
dodanych jonów, tym
silniejsze jest
ich działanie
koagulujące.
Koagulacja
rozpoczyna się w chwili, gdy
zostanie osiągnięte niemal
całkowite zobojętnienie
elektryczne cząstek
, czyli
punkt izoelektryczny
.
2012-03-28
17
33
Punkt izoelektryczny
Koloidy
Koagulacja może być odwracalna i nieodwracalna.
Wykład 6
Proces koagulacji nieodwracalnej nazywany jest denaturacją.
Peptyzacja polega na usunięciu z osadu zaadsorbowanych jonów
koagulujących. Cząstki koloidalne odzyskują swój pierwotny ładunek i
ponownie zaczynają się odpychać. Peptyzację taką obserwujemy na
przykład podczas przemywania czystą wodą świeżo strąconych
siarczków metali ciężkich.
Procesem odwrotnym do koagulacji jest peptyzacja - zjawisko
przechodzenia skoagulowanego osadu lub żelu z powrotem w stan
roztworu koloidalnego.
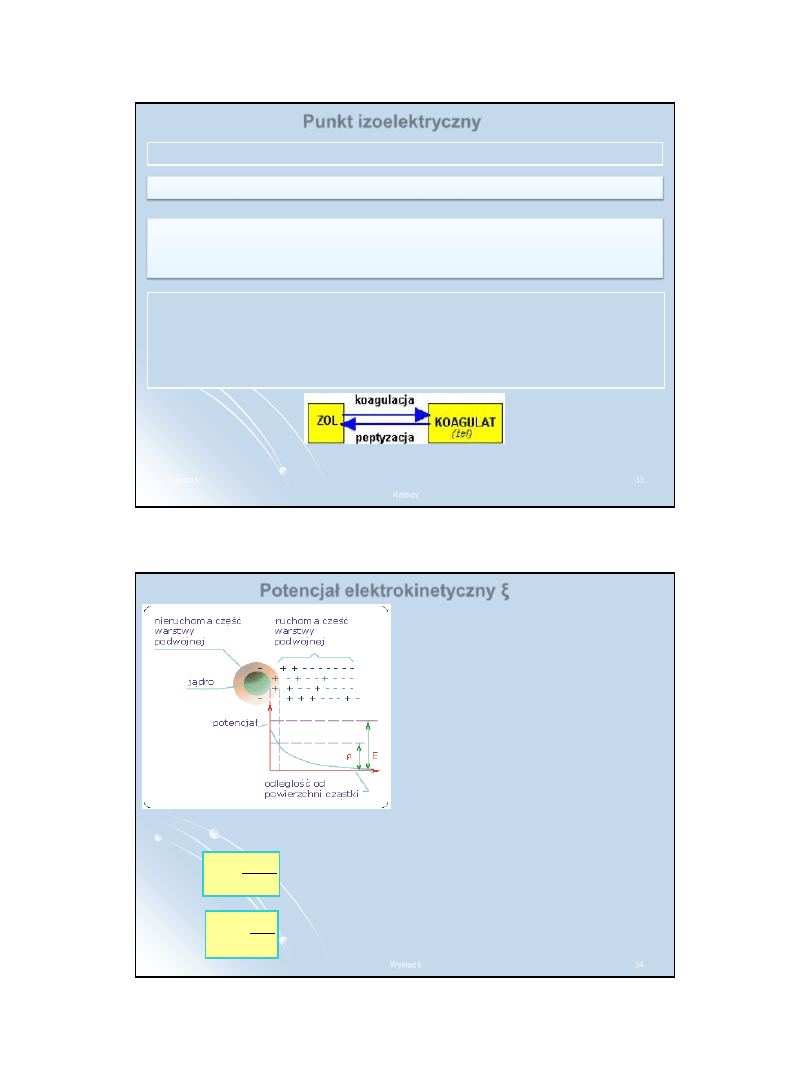
Potencjał elektrokinetyczny ξ
Na granicy warstwy adsorpcyjnej i
dyfuzyjnej powstaje różnica
potencjałów zwana
potencjałem
elektrokinetycznym ξ
(czyt. dzeta)
.
Elektroosmoza
– przyłożenie pola
elektrycznego powoduje ruch
warstwy dyfuzyjnej jonów i
cząsteczek rozpuszczalnika.
Jest to
jednokierunkowy ruch ośrodka
dyspersyjnego przez błonę
półprzepuszczalną
Ruchliwość elektroosmotyczna
czyli prędkość przesuwania się
warstwy rozpuszczalnika podczas elektroosmozy:
η
ξ
ε
E
=
v
E
– natężenie pola elektrycznego, równe
ilorazowi
U/l
(napięcia przez odległość między
elektrodami podłączonymi do źródła prądu),
ε
– stała dielektryczna ośrodka,
η
– lepkość ośrodka.
stąd:
ε
E
η
v
ξ
34
Wykład 6

2012-03-28
18
35
Równowaga membranowa Donnana
1. Koloid R-Na
(dysocjujący) oddzielony od rozpuszczalnika
(wody) błoną półprzepuszczalną
H
2
O
R Na
+
-
H
2
O
+
Na
+
Na
błona
półprzepuszczalna
Jony Na
+
wędrują na drugą stronę błony. Aby
nie powodowało to różnicy potencjałów
pomiędzy jedną a drugą stroną błony –
zobojętnienie ładunku następuje w wyniku
hydrolizy membranowej:
R
–
+ H
2
O RH + OH
–
Jony OH
–
wędrują w prawo i równoważą
ładunek jonów Na
+
2. Koloid R-Na
o stężeniu c
1
oddzielony błoną
półprzepuszczalną od roztworu wodnego NaCl o stężeniu c
2
c
1
c
1
c
2
c
2
błona
półprzepuszczalna
+
Na
H
2
O
R Na
+
-
H
2
O
Cl-
Dążąc do stanu równowagi jony Na
+
i Cl
–
przechodzą przez błonę, a jony R
–
pozostają po jednej stronie membrany.
Błona zachowuje się tak, jakby
przepuszczała jony tylko w jednym
kierunku, a przeszkadzała ich
przepływowi w kierunku przeciwnym.
Wykład 6
36
błona
półprzepuszczalna
+
Na
H
2
O
R Na
+
-
H
2
O
Cl-
Cl-
c
1
c
1
+ x x
c
2
- x
c
2
- x
W stanie
równowagi:
C
ząstki koloidowe nie mogąc
przechodzić przez błonę
półprzepuszczalną powodują, że po
osiągnięciu równowagi
obserwujemy nierównomierne
rozmieszczenie jonów elektrolitu po
obu stronach błony.
Donnan
na podstawie praw termodynamiki stwierdził, że iloczyny
stężeń jonów Cl
–
i Na
+
po obu stronach membrany są sobie równe
Stąd:
x)
-
x)(c
-
(c
x
x)
(c
2
2
1
x
– stężenie jonów Cl
–
i jonów, Na
+
, które przedyfundowały przez błonę
2
1
2
2
2c
+
c
c
=
x
Znając c
1
i c
2
, po obliczeniu x, można
obliczyć stosunek stężeń jonów po obu
stronach membrany
Równowaga membranowa:
•
w
komórkach oraz tkankach roślin i zwierząt
•
w surowcach i produktach
spożywczych
•
koloidy dysocjujące na jony wywierają wpływ na wędrówkę soli
wbrew ciśnieniu osmotycznemu
Wykład 6

2012-03-28
19
37
Koloidy hydrofilowe
Koloidy
Hydrofilowe roztwory koloidalne są najczęściej roztworami substancji
wielkocząsteczkowych: białka, skrobi, dekstryn, niektórych barwników
itd. W swojej strukturze mają grupy funkcyjne mogące tworzyć wiązanie
wodorowe z cząsteczkami wody. Roztwory te zawdzięczają swoją
trwałość zdolności cząstek do tworzenia na swej powierzchni
wielowarstwowej otoczki złożonej z cząsteczek wody. Otoczka
hydratacyjna chroni cząstki koloidalne od bezpośredniego zetknięcia
się i w ten sposób uniemożliwia ich koagulację.
Wykład 6
38
Koloidy białek
Koloidy
Cząstki niektórych zoli białek wykazują pewien ładunek elektryczny,
którego obecność przyczynia się dodatkowo do zwiększenia trwałości
zolu.
Ładunek ten nie powstaje jednak wskutek adsorpcji jonów z roztworu
przez cząstki koloidalne, jak to ma miejsce w przypadku koloidów
hydrofobowych.
Jest on natomiast wynikiem dysocjacji grup
kwasowych lub zasadowych wchodzących w skład białka.
Grupa kwasowa COOH ulega dysocjacji w
myśl równania:
R-COOH + H
2
O
⇌ R-ROO
-
+ H
3
O
+
grupa zasadowa NH
2
natomiast:
R-NH
2
+ H
2
O
⇌ R-NH
3
+
+ OH
-
W
pierwszym przypadku cząstka koloidalna ładuje się ujemnie, w
drugim dodatnio.
Obydwie równowagi kwasowo-zasadowe zależą od stężenia jonów
H
3
O
+
w
roztworze. Im roztwór bardziej kwaśny, tym silniejsza
dysocjacja zasadowa, im bardziej zasadowy, tym silniejsza dysocjacja
kwasowa.
Wykład 6
2012-03-28
20
39
Koloidy hydrofilowe
Koloidy
Koloidy hydrofilowe różnią się od koloidów hydrofobowych także
swoim zachowaniem się wobec dodatków elektrolitów. Ulegają one
mianowicie koagulacji dopiero pod
wpływem dużej ilości dodanych soli.
Sądzi się, że jony obecne w elektrolicie, ulegając silnej hydratacji,
powodują równocześnie zmniejszenie liczby cząsteczek wody
tworzących otoczki cząstek koloidalnych, a co za tym idzie zmniejszenie
trwałości układu koloidalnego i jego koagulację.
Wykład 6
Koloidy hydrofilowe odznaczają się na ogół większą trwałością niż
koloidy hydrofobowe, a ich dodatek do
zolów hydrofobowych może
znacznie zwiększyć trwałość tych ostatnich (koloidy ochronne).
Koagulacja koloidów liofilowych (np. białek) pod wpływem czynników
bardziej drastycznych jak wysoka temperatura, stężone kwasy, zasady
lub sole metali ciężkich jest nieodwracalna -
denaturacja
. Proces ten
jest nieodwracalny, gdyż produkt (żel) nie da się przeprowadzić w
pierwotną formę zolu. Podczas denaturacji dochodzi do uszkodzenia
struktury koloidu.
40
Żele
{kind=link}
Liczne zole koloidów hydrofilowych, np. zol żelatyny, przygotowane
na gorąco, zastygają podczas chłodzenia w jednolitą, galaretowatą
masę zwaną żelem.
Żele o podobnych właściwościach można otrzymać również przez
dodanie do
niektórych zoli hydrofobowych odpowiednich, ani za
małych, ani za dużych, ilości elektrolitów. W ten sposób powstaje na
przykład żel wodorotlenku żelaza(III) i żel kwasu krzemowego.
Przemiana zolu w
żel następuje pod warunkiem, że stężenie koloidu
nie jest
mniejsze od pewnej granicznej wartości.
Żelatyna tworzy w temperaturze pokojowej galaretę w przypadku
zawartości ponad 1-1,5%, natomiast kwas krzemowy w przypadku
zawartości, zależnie od warunków, ponad 3-6%.
Wiele żeli podczas ogrzewania przechodzi z powrotem w zole. Tego
rodzaju odwracalne układy koloidalne tworzy z wodą żelatyna.
Wykład 6
Wyszukiwarka
Podobne podstrony:
chf wykład 6, Studia, Chemia, fizyczna, wykłady
chf wykład 3, Studia, Chemia, fizyczna, wykłady
chf wykład 8, Studia, Chemia, fizyczna, wykłady
chf wykład 1, Studia, Chemia, fizyczna, wykłady
ChF Wyklad 1p(1)
chf wykład 5, Studia, Chemia, fizyczna, wykłady
chf wykład 4, Studia, Chemia, fizyczna, wykłady
chf wykład 7, Studia, Chemia, fizyczna, wykłady
chf wykład 2, Studia, Chemia, fizyczna, wykłady
chf wykład 6, Studia, Chemia, fizyczna, wykłady
Napęd Elektryczny wykład
wykład5
Psychologia wykład 1 Stres i radzenie sobie z nim zjazd B
Wykład 04
geriatria p pokarmowy wyklad materialy
ostre stany w alergologii wyklad 2003
WYKŁAD VII
Wykład 1, WPŁYW ŻYWIENIA NA ZDROWIE W RÓŻNYCH ETAPACH ŻYCIA CZŁOWIEKA
Zaburzenia nerwicowe wyklad
więcej podobnych podstron