
2013-03-23
1
Farmakoterapia przewlekłej
białaczki szpikowej
Katarzyna Sobańska
Katedra i Zakład Farmacji Klinicznej
Uniwersytetu Medycznego im. K.Marcinkowskiego
w Poznaniu
Kierownik: Prof. dr hab. Edmund Grześkowiak
Białaczka (ang. leukaemia)
• zespół chorób nowotworowych układu krwiotwórczego, które
charakteryzują się:
– nieprawidłową proliferacją, dojrzewaniem i uwalnianiem
krwinek białych ze szpiku kostnego i innych tkanek
hematopoetycznych (śledziona, węzły chłonne)
– naciekami narządowymi
– obecnością niedojrzałych postaci tych
komórek we krwi obwodowej
Ryc. 1. Dojrzałe elementy morfotyczne krwi
Białaczka jest chorobą klonalną
• transformacja komórki macierzystej lub komórek wywodzących
sie z wczesnych stadiów hematopoezy
• klon komórek białaczkowych:
– zmiany cytokinetyczne, metaboliczne, zmiany struktury
antygenowej oraz zmiany wrażliwości na czynniki regulujące
proliferację, różnicowanie i dojrzewanie komórek
• niewydolność prawidłowej hematopoezy
• nacieki narządowe kliniczne objawy choroby
Typy białaczek
W zależności od komórek ulegających transformacji:
• białaczki szpikowe – zmiany dotyczą linii mieloidalnych szpiku
• białaczki limfatyczne – zmiany dotyczą linii limfoidalnych szpiku
Ze względu na przebieg choroby wyróżniamy:
• białaczki ostre – charakteryzują się szybkim namnażaniem
niedojrzałych komórek krwi, które wypierają inne rodzaje komórek i
uniemożliwiają tworzenie prawidłowych krwinek;
nagły początek z niespecyficznymi objawami
• białaczki przewlekłe – charakteryzują się nadmiernym rozrostem
względnie dojrzałych, jednak nieprawidłowych krwinek;
powolny przebieg, początkowo bezobjawowy
Przewlekła białaczka szpikowa
(CML ang. chronic myeloid leukaemia)
• to nowotwór, którego istotą jest klonalny rozrost zmienionej
nowotworowo wielopotencjalnej komórki macierzystej szpiku, z
nadmierną proliferacją jednej lub więcej linii mieloidalnych
Ryc. 4. Przewlekła białaczka szpikowa –
rozmaz krwi
Ryc. 5. Przewlekła białaczka szpikowa –
rozmaz szpiku kostnego
Patogeneza CML
• pierwsza choroba nowotworowa, w przypadku której,
udowodniono związek z obecnością mutacji chromosomalnej
• wskaźnikiem cytogenetycznym jest chromosom Philadelphia (Ph)
• obecność chromosomu Philadelphia stwierdza się
u 90-95% pacjentów z CML , a także:
– u 10-20% dorosłych oraz 2-5% dzieci
z ostrą białaczką limfoblastyczną
– wyjątkowo rzadko (2%) u chorych
z ostrą białaczką szpikową,
szpiczakiem plazmocytowym
i chłoniakami immunoblastycznymi
2013-03-23
2
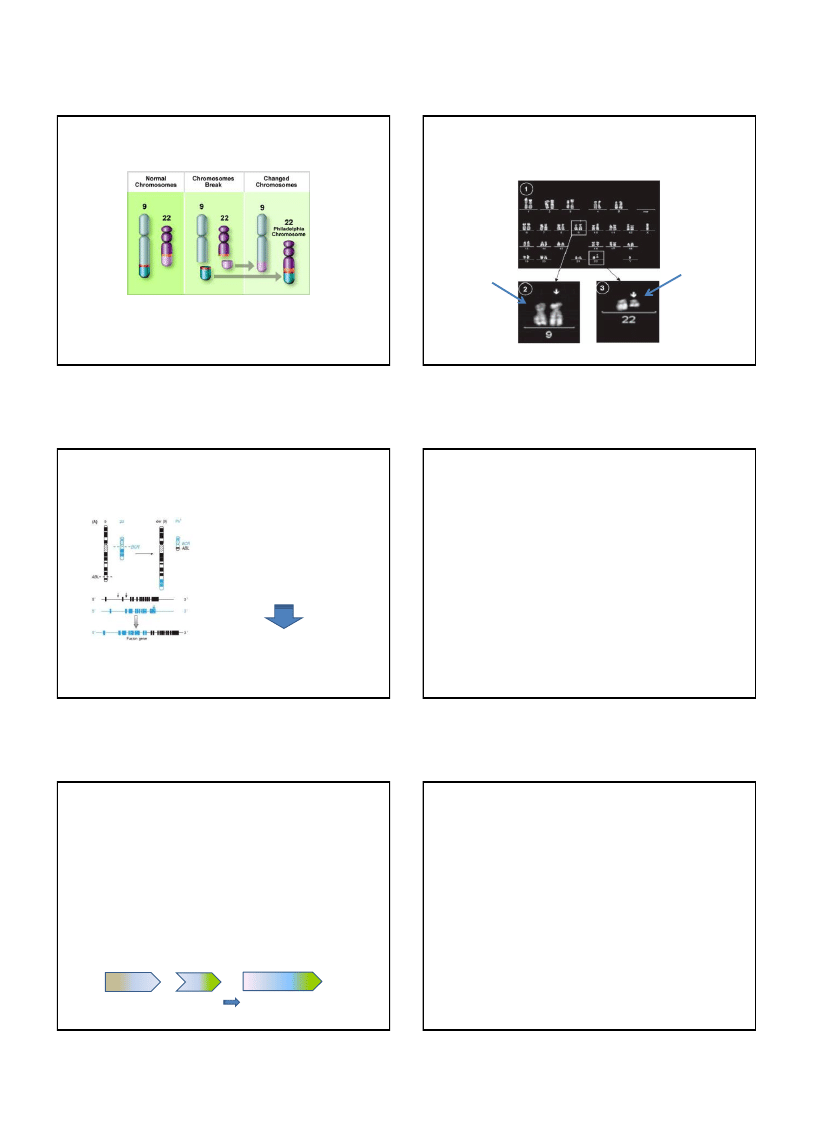
Chromosom Philadelphia
Ryc. 6. Schemat powstawania chromosomu Philadelphia
W następstwie wzajemnej translokacji fragmentów długich ramion
chromosomów 9 i 22 powstaje wydłużony chromosom 9 oraz krótki
chromosom 22 – tzw. chromosom Philadelphia.
Kariotyp pacjenta z przewlekłą białaczką szpikową [46XY,
t(9;22)(q34;q11)]
Obraz klasycznej analizy prążkowej
powstały w wyniku
translokacji pochodny
chromosom 9
chromosom
Phialdelphia
Efektem translokacji jest przeniesienie onkogenu abl (zlokalizowanego w chr.
9) w ściśle określone miejsce złamań (breakpoint cluster region – bcr) na chr.
22 i powstanie fuzyjnego genu bcr-abl
Ekspresja kinazy Bcr-Abl jest podstawowym czynnikiem
transformacji nowotworowej w CML.
gen bcr-abl koduje fuzyjne białko Bcr-Abl
o zwiększonej aktywności kinazy tyrozynowej
Ryc. 8. Schemat powstawania genu Bcr-Abl
Typowe miejsca pęknięć:
w ramieniu długim chromosomu 9 prążek
q34.1 – gen c-abl (ang. Abelson murine
leukemia cellular oncogene)
w ramieniu długim chromosomu 22 prążek
q11.21 - odcinek BCR (ang. Breakpoint Cluster
Region – region skupiska punktów pęknięć),
będący częścią genu bcr
Funkcje genów abl i bcr
gen abl
– ludzki homolog wirusowego onkogenu v-abl przenoszonego przez wirusa
Abelsona, który powoduje białaczkę u ptaków
– koduje białko o masie około 145 kDa, należące do rodziny
niereceptorowych kinaz tyrozyny
– białko to pełni funkcję regulacyjną w cyklu komórkowym
gen bcr
– koduje kilka różnych białek Bcr o masie 83-190 kDa, głównie 2 białka o
masie 160 i 130 kDa
– funkcja tych białek w komórce jest nieznana – przypuszcza się, że podobnie
jak Abl biorą one udział w regulacji cyklu komórkowego
W efekcie translokacji powstają dwa chimeryczne geny fuzji:
bcr-abl na chromosomie 22 oraz
abl-bcr na chromosomie 9
Gen abl-bcr
•
ekspresja genu abl-bcr – u około 60% pacjentów z CML
•
białko będące produktem translacji tego genu prawdopodobnie nie wykazuje
aktywności biologicznej
Gen bcr-abl
•
wykrywany metodami biologii molekularnej w 100% chorych na CML oraz
u 60% zdrowych ludzi
•
koduje białko o nieprawidłowej, zwiększonej aktywności kinazy tyrozynowej
+
=
gen bcr
gen abl
gen bcr-abl
kinaza
tyrozynowa
kinaza tyrozynowa
konstytutywnie aktywna
* Tyrozynowo-swoiste kinazy białkowe
(PTKs, ang. protein tyrosine kinases)
•
enzymy selektywnie fosforylujące tyrozynę w cząsteczkach białek
•
fosforylacja Tyr odgrywa istotna role w procesach, takich jak:
– regulacja wzrostu (również patologicznego), różnicowania
– kontrola cyklu komórkowego
– regulacja kształtu i adhezji
– sygnalizacja transbłonowa i wewnątrzkomórkowa
– kontrola wielu szlaków metabolicznych
•
precyzyjna regulacja aktywności kinaz tyrozynowych jest warunkiem
prawidłowego funkcjonowania wszystkich komórek
wielu chorobach, zwłaszcza nowotworowych, dochodzi do zmian w strukturze
i konstytutywnej aktywacji tych enzymów, czego efektem jest
niekontrolowany wzrost komórkowy

2013-03-23
3
Funkcje kinazy Bcr-Abl
białko o zwiększonej aktywności kinazy tyrozynowej
kinaza Bcr-Abl upośledza funkcje integryn osłabienie zdolności
przylegania komórek hematopoezy do podścieliska i macierzy
pozakomórkowej szpiku
liczne eksperymenty przeprowadzone na liniach komórkowych
wykazały, że BCR-ABL hamuje apoptozę komórek
wzrost procesów proliferacji oraz zmniejszenie odpowiedzi na
czynniki aktywujace apoptoze komórki nowotworowe żyją dłużej
i mogą opuszczać szpik w postaci niedojrzałej
Mechanizm działania kinazy Bcr-Abl
• kinaza Bcr-Abl nie wymaga aktywacji
– jest konstytutywnie aktywna
–
dimeryzacja cząsteczki białka Bcr-Abl
wzajemna fosforylacja reszt
tyrozynowych przyłączenie wielu
enzymów i białek adaptorowych
aktywacja szlaków przekaźnictwa
sygnałów
Kinaza tyrozynowa Bcr-Abl
przyłącza cząsteczkę ATP, następuje
przeniesienie reszty fosforanowej z ATP na tyrozynę białka
substratowego, przez co sygnał proliferacyjny jest nieustannie
przekazywany do jądra komórkowego.
Przewlekła białaczka szpikowa Ph-negatywna
• u 5-10% chorych na CML badania cytogenetyczne
nie pozwalają na wykrycie chromosomu Ph
– u połowy z nich badania molekularne wykazują obecność
translokacji bcr-abl
– przebieg choroby oraz odpowiedź na leczenie u pacjentów
Ph(-)/ bcr-abl(+) są typowe
• u pozostałych 5% pacjentów z CML żadne znane metody
nie pozwalają na wykrycie translokacji bcr-abl
– w przypadku CML Ph(-)/bcr-abl(-), kluczową rolę w
transformacji białaczkowej odgrywają mutacje onkogenu ras
– przebieg jest bardziej agresywny, choroba jest bardziej
oporna na leczenie, szybciej przechodzi w fazę akceleracji i
kryzy blastycznej
Etiologia przewlekłej białaczki szpikowej
w większości przypadków jej przyczyna jest nieznana
jedynym poznanym czynnikiem etiologicznym jest promieniowanie
jonizujące
wzrost zachorowań u pacjentów poddawanych rentgenoterapii z
powodu zesztywniającego zapalenia stawów kręgosłupa
znacząco większy odsetek zachorowań u ludzi, którzy przeżyli wybuch
bomby atomowej w Hiroszimie i Nagasaki – pierwsze przypadki po
2 latach, szczyt występowania po 5-7 latach
rozwój CML w następstwie późnego popromiennego
powikłania radioterapii (w okresie od roku do 25 lat po radioterapii raka
piersi, szyjki macicy oraz drobnokomórkowego raka płuca)
Epidemiologia
CML stanowi około 15% białaczek występujących u dorosłych
roczna zapadalność wynosi 1-1,5 przypadków na 100 000 osób
szczyt zachorowań przypada na 4. i 5. dekadę życia, jednak może wystąpić
w każdym wieku
występuje nieco częściej u mężczyzn niż u kobiet (w stosunku 1,3:1)
stanowi mniej niż 5% białaczek u dzieci; może wystąpić u niemowląt,
ale większość rozpoznań stawianych jest po 6. roku życia
Struktura zachorowań i zgonów na przewlekłą białaczkę szpikowa w Polsce w roku
2009, wg Krajowej Bazy Danych Nowotworowych
2009 r.
Liczba
zachorowań
2009 r.
Liczba zgonów
Kobiety
482
Kobiety
593
Kobiety 55-84 r.ż.
321
Kobiety 50- >85 r.ż.
522
Mężczyźni
540
Mężczyźni
630
Mężczyźni 50-79 r.ż.
321
Mężczyźni 50- >85 r.ż.
523
Obraz kliniczny
u ok. 40% chorych CML zostaje wykryta przypadkowo,
podczas okresowych badan morfologii krwi
objawy kliniczne są często niespecyficzne:
– gorączka
– wzmożona potliwość
– ubytek wagi
– osłabienie spowodowane niedokrwistością
– zawroty głowy
w miarę postępu choroby pojawiają się:
– bóle kostne, związane z proliferacją komórek nowotworowych w
kościach długich i płaskich
– powiększenie śledziony i wątroby powoduje ból i uczucie pełności
w jamie brzusznej (późny objaw)
– wybroczyny do skóry i śluzówek (rozwój małopłytkowości ze skazą
krwotoczną)

2013-03-23
4
Objawy przedmiotowe i podmiotowe
• objawy związane z dużą leukocytozą > 200-300 tys./μl (u 10% chorych):
– utrata masy ciała spowodowana przyspieszonym metabolizmem
– leukostaza – zaburzenia przepływu krwi w mikrokrążeniu
wynikające z gromadzenia się w świetle naczyń dużej liczby
leukocytów, powodujące:
• zaburzenia czynności ośrodkowego układu nerwowego
• zaburzenia widzenia
• bóle głowy
• objawy hipoksemii związane z zaburzeniami przepływu krwi w
naczyniach płucnych
• splenomegalia i/lub hepatomegalia (u 30-40% w chwili rozpoznania)
ból w lewym podżebrzu, uczucie pełności w jamie brzusznej
Nieprawidłowości w badaniach pomocniczych
Morfologia krwi obwodowej
zawsze leukocytoza neutrofilowa, zwykle >
20 tys./μl, nawet 500-700 tys./μl
w rozmazie obecne komórki blastyczne
(odsetek tym większy im większa jest
leukocytoza); zazwyczaj do 10%
obecność we krwi obwodowej komórek linii
neutrofilopoetycznej oraz bazofilia stanowią
charakterystyczne cechy CML (bazofilia oraz
nadpłytkowość, która występuje u około 30%
chorych w chwili rozpoznania, mogą
poprzedzać leukocytozę o kilka lat)
wartości stężenia hemoglobiny i hematokrytu
prawidłowe lub obniżone
1– neutrofil segmentowany,
2– neutrofil pałeczkowaty,
3– metamielocyt,
4– mielocyt,
5– promielocyt,
6– mielobast
Obraz rozmazu krwi obwodowej
Nieprawidłowości w badaniach pomocniczych
Morfologia szpiku
• biopsja aspiracyjna
– szpik bogatokomórkowy ze znaczną
przewaga komórek linii
neutrofilopoetycznej i
megakariopoetycznej
– linia erytropoetyczna jest stłumiona
• trepanobiopsja
– badanie histopatologiczne może ujawnić
zwiększone włóknienie retikulinowe szpiku
i tworzenie nowych naczyń
1– granulocyt segmentowany neutrofilowy,
2– granulocyt pałeczkowaty,
3– metamielocyt neutrofilowy,
4– mielocyt neutrofilowy,
5– promielocyt,
6– mielobast
Obraz rozmazu szpiku kostnego
Badanie cytogenetyczne
•
wykazuje translokację t(9;22)(q34,q11), czyli chr. Philadelphia
•
inne aberracje cytogenetyczne występują z narastającą częstością się w
bardziej zaawansowanych fazach choroby jako marker progresji
•
ocena kariotypu klasyczną metodą prążkową jest konieczna w celu
wykazania obecności Ph; alternatywnie, w przypadku braku metafaz w
hodowli wykonuje się badanie metodą FISH (ang. fluorescent in situ
hybridization – fluorescencyjna hybrydyzacja in situ)
Badanie molekularne
•
wykazuje obecność genu bcr-abl w badaniu RT PCR lub RQ-PCR
•
metoda RT-PCR służy do identyfikacji rodzaju transkryptu bcr- abl,
natomiast badanie RQ-PCR pozwala określić jego ilość z czułością 10
-5
Kariotyp chorego z CML
Inne badania laboratoryjne
• zmniejszona aktywność fosfatazy alkalicznej granulocytów (FAG)
• wzrost stężenia witaminy B12 i białek transportujących
(transkobalamin wytwarzanych przez granulocyty)
• wzrost stężenia kwasu moczowego i dehydrogenazy kwasu
mlekowego (LDH)

2013-03-23
5
Przebieg kliniczny
• 3 (rzadziej 2) fazy: przewlekła, akceleracji oraz przełomu blastycznego
– faza przewlekła faza kryzy blastycznej (przebieg
dwufazowy)
– faza przewlekła faza akceleracji faza kryzy blastycznej
(przebieg trójfazowy)
– u 20-25 % chorych występuje przebieg dwufazowy
• ok. 85% przypadków rozpoznaje sie w fazie przewlekłej choroby
• u około 10% chorych rozpoznanie stawiane jest w zaawansowanych
stadiach: fazie akceleracji (około 6%) lub kryzy blastycznej (około 4%)
Faza przewlekła (chronic phase)
• determinuje długość przeżycia chorego
• trwa zwykle 3-4 lata
• początkowo może przebiegać bezobjawowo (20-30% pacjentów)
• stopniowy wzrost klonu białaczkowego
– hiperleukocytoza (do kilkuset tysięcy), odmłodzenie obrazu
granulocytarnego
– blasty w szpiku i krwi obwodowej <10%
– blasty i promielocyty w szpiku i krwi obwodowej <30%
– bazofile we krwi <20%
– trombocyty >100 tys.
– brak ognisk pozaszpikowego naciekania
• CML zdiagnozowana w tej fazie (większość przypadków) wykazuje dużą
odpowiedź na leczenie i najlepiej rokuje
Faza akceleracji (accelerated phase)
• komórki białaczkowe tracą zdolność różnicowania
• wyraźne objawy: powiększenie śledziony, gorączka niewyjaśniona
innymi przyczynami
• nasilenie objawów ogólnych, utrata masy ciała, bóle kostne, stany
gorączkowe, wzmożona potliwość, bóle śledziony
• rozpoznanie w tej fazie stawiane jest w ok. 6% przypadków
Mechanizm progresji choroby nie jest do końca poznany.
Jedną z przyczyn mogą być dodatkowe zmiany chromosomalne w
komórkach macierzystych szpiku (tzw. ewolucja klonalna).
Ewolucja klonalna
• w miarę rozwoju choroby 50 do 80% pacjentów
nabywa wtórne zmiany chromosomalne w obrębie
klonu komórek Ph(+), które poprzedzają wystąpienie
zmian hematologicznych oraz klinicznych objawów
kryzy blastycznej
– dodatkowy chromosom Ph(+Ph),
– trisomia 8 (+8), rzadziej 19 (+19)
– izochromosom długich ramion 17 [i(17q)]
• mutacje genu p53 pojawiają się u 20-30% pacjentów
w przełomie blastycznym – utrata aktywności
proapoptotycznej białka p53 zahamowanie
apoptozy i wejście w fazę kryzy blastycznej
Faza przełomu blastycznego
• znaczny wzrost ilości komórek blastycznych (obrazem przypomina
ostrą białaczkę)
– w ok. 50% komórki blastyczne maja fenotyp mieloblastów, w 30%
limfoblastów, natomiast w 10% – megakarioblastów
• związana z objawami takimi jak: utrata masy ciała, nadmierne
pocenie, stany gorączkowe oraz bóle w obrębie jamy brzusznej
• złe rokowanie (wszystkie dostępne metody leczenia wykazują
mniejszą skuteczność niż w fazie przewlekłej)
• krótki czas przeżycia od momentu wystąpienia (ok. 2-3 miesiące)
• leczenie wymaga prowadzenia chemioterapii skojarzonej,
w zależności od charakteru transformacji (remisja polega na
cofnięciu sie choroby do fazy przewlekłej)
• rozpoznanie w tej fazie następuje w ok. 4% przypadków
Rozpoznanie
• jedynym kryterium jest stwierdzenie obecności chromosomu Ph
w badaniu cytogenetycznym lub genu bcr-abl metodą FISH lub PCR
Kryteria rozpoznania fazy akceleracji i przełomu blastycznego (wg WHO):
Fazy akceleracji: (obecność 1 lub więcej czynników)
– blasty we krwi obwodowej lub szpiku – 10-19%
– bazofilia ≥ 20%
– małopłytkowość < 100 000/μl (niezwiązana z leczeniem)
– nadpłytkowość > 1 000 000/ μl (oporna na leczenie)
– klonalna ewolucja cytogenetyczna (dodatkowe aberracje
chromosomowe, które nie były obecne przy rozpoznaniu)
– powiększenie śledziony lub wzrost leukocytozy oporne na leczenie
Fazy przełomu blastycznego: (obecność 1 lub więcej czynników)
– odsetek blastów ≥ 20%
– pozaszpikowe nacieki białaczkowe

2013-03-23
6
Czynniki prognostyczne
• pozwalają zakwalifikować chorych do odpowiednich grup rokowniczych,
określających prawdopodobieństwo przeżycia bez progresji choroby
• wskaźnik Sokala
•
trzy grupy ryzyka szybkiej progresji choroby w zależności od:
»
wieku pacjenta
»
wielkości śledziony
»
liczby płytek krwi
»
komórek blastycznych we krwi
• wskaźnik Hasforda
– uwzględnia te same czynniki co model
Sokala, a ponadto liczbę eozynofili i
bazofili we krwi obwodowej
Wskaźnik
Sokala
Mediana przeżycia
(miesiące)
< 0,8
60
0,8-1,2
46
> 1,2
32
Odpowiedź hematologiczna
Odpowiedź hematologiczna (HR ang. hematological response) –
normalizacja parametrów krwi obwodowej:
całkowita remisja hematologiczna CHR – prawidłowy obraz krwi
obwodowej z prawidłowym obrazem rozmazu krwinek białych:
• leukocyty <10x 10
9
/l
• trombocyty <450x 10
9
/l
• mielocyty + metamielocyty <5%
• brak promielocytów i mieloblastów we krwi obwodowej
• granulocyty zasadochłonne <20%
+ brak pozaszpikowych ognisk białaczki
Odpowiedź cytogenetyczna
Odpowiedź cytogenetyczna (CyR ang. cytogenetic response) –
zmniejszenie liczby Ph-pozytywnych metafaz w aspiracie szpiku
kostnego, wykazane w badaniu cytogenetycznym:
minimalna MinimalCyR – 65-95% (Ph+) metafaz
mniejsza MiCyR – 35-65% Ph(+) metafaz
częściowa PCyR – 1-35% Ph(+) metafaz
całkowita CCyr – 0% Ph(+) metafaz
większa MCyR – 0-35% Ph(+) metafaz – stanowi
połączenie odpowiedzi całkowitej i częściowej
Odpowiedź molekularna
Odpowiedź molekularna (MR ang. molecular response) –
zmniejszenie ilości chimerycznego Bcr-Abl transkryptu mRNA,
mierzone metodą RQ-PCR (ang. real-time quantitative
polymerase
chain reaction – reakcja łańcuchowa polimerazy DNA z analizą
ilości
produktu w czasie rzeczywistym):
większa odpowiedź molekularna MMR – zmniejszenie ilości
transkryptu Bcr-Abl we krwi obwodowej ≥ 3 logarytmy w
stosunku do wystandaryzowanych wartości wyjściowych
całkowita remisja molekularna CMR – negatywny wynik
badania RQ-PCR
W praktyce klinicznej obserwuje się trzy poziomy odpowiedzi:
• odpowiedź optymalna
• odpowiedź suboptymalna
• niepowodzenie terapii
Kryteria odpowiedzi optymalnej to uzyskanie:
o
w 3. miesiącu CHR i przynajmniej MiCyR
o
w 6. miesiącu co najmniej PCyR
o
w 12. miesiącu CCyR
o
w 18. miesiącu MMR
Progresja choroby:
o
przejście w fazę akceleracji lub przełom blastyczny
o
śmierć
o
utrata CHR lub MCyR
o
u pacjentów, którzy nie osiągnęli CHR – zwiększenie liczby białych
krwinek (WBC ang. white blood cells) pomimo leczenia
Monitorowanie odpowiedzi na leczenie
• badanie morfologii krwi
– co 2 tyg. do czasu uzyskania CHR, następnie co 2-3 miesiące
• badanie cytogenetyczne (klasyczne badanie metodą prążkową lub FISH)
– po 3 i 6 miesiącach od rozpoczęcia leczenia TKIs,
– następnie co 6 miesięcy do czasu uzyskania CCyR,
– potem co 12 miesięcy, tylko gdy nie ma możliwości wykonywania
badań molekularnych
• badanie molekularne RQ-PCR
– co 3 miesiące do uzyskania MMR, następnie przynajmniej co 6
miesięcy
• badania mutacji genu Bcr-Abl
– w przypadku odpowiedzi suboptymalnej lub niepowodzenia leczenia
– zawsze przed planowaną zmianą TKI lub zastosowaniem innego
leczenia

2013-03-23
7
Leczenie CML
1.
Leczenie cytoredukcyjne – chemioterapia konwencjonalna
nie zmienia istotnie naturalnego przebiegu choroby
celem jest doprowadzenie do zmniejszenia masy kom. białaczkowych
(redukcja leukocytozy do wartości ok. 50 tys./μl)
stosowane są:
hydroksymocznik
busulfan
2.
Leczenie właściwe:
celem jest eliminacja komórek Ph dodatnich – uzyskanie odpowiedzi
hematologicznej, cytogenetycznej i molekularnej
stosowane są:
inhibitory kinazy tyrozynowej Bcr-Abl
interferon α
transplantacja komórek macierzystych szpiku kostnego
od dawcy rodzinnego lub niespokrwenionego (allo-HSCT, ang.
hematopoietic stem cell transplantation)
Klasyczna chemioterapia
Busulfan
•
lek alkilujący DNA
•
ciężkie działania niepożądane
– aplazja szpiku,
– zwłóknienie płuc,
– niepłodność
– zaburzenia miesiączkowania u kobiet
– objawy podobne do zespołu Addisona
•
obecnie stosowany wyłącznie w leczeniu mieloablacyjnym przed
przeszczepieniem macierzystych komórek krwiotwórczych (chemioterapia
BuCy2 (busulfan 4 × 4 mg/kg doustnie; cyklofosfamid 2 × 60 mg/kg dożylnie)
Busulfan i hydroksymocznik
• u 50-80% pacjentów remisja hematologiczna, rzadko remisja
cytogenetyczna
• ich stosowanie wydłuża nieco czas trwania fazy przewlekłej, ale nie
ma
wpływu na naturalny przebieg białaczki
Hydroksymocznik
• inhibitor syntezy DNA hamujący aktywność
reduktazy rybonukleotydowej
• dawka początkowa leku wynosi 40 mg/kg/d.
i jest zmniejszana o 50% przy spadku leukocytozy
poniżej 20 × 10
9
/l
• działania niepożądane:
nudności
brak apetytu
zmiany skórne, zapalenie jamy ustnej
niekiedy niedokrwistość megaloblastyczna
• daje dłuższy czas trwania fazy przewlekłej w porównaniu z Bu
• stosowany cytoredukcyjnie u chorych przygotowywanych do
transplantacji lub przed u chorych z poważnymi chorobami
współistniejącymi, głównie chorobami nowotworowymi, gdzie
przewidywany czas przeżycia < 3 lat
Interferon-α
•
glikoproteina o działaniu antyproliferacyjnym,
immunomodulacyjnym i przeciwwirusowym
•
powoduje zmiany w mikrośrodowisku szpiku,
zwiększa adhezję komórek białaczkowych do podścieliska,
wpływa na miejscowe wytwarzanie cytokin stymulujących przeciwnowotworową
odpowiedź komórkową
•
skuteczność leczenia IFN-a z terapią konwencjonalną:
– odsetek uzyskiwanych remisji hematologicznych (CHR i PHR) wynosił 60–90%,
odpowiedź cytogenetyczna osiągana była u 20–55% chorych, w tym CCyR u
6–30%
– czas trwania fazy przewlekłej wydłuża się średnio o 2 lata
•
INF-α vs. INF-α + małę dawki arabinozydu cytozyny ara-C
– 3-letnie przeżycie u 79% chorych leczonych IFN-a w porównaniu z 86% w
grupie leczenia skojarzonego
– odsetek CHR wynosił odpowiednio 55% i 66%, a MCyR — 24% i 41%
Interferon-α
•
działania niepożądane:
– immunosupresja
– objawy rzekomo grypowe (gorączka, bóle kostno-mięśniowe, złe
samopoczucie)
– spadek masy ciała, uczucie zmęczenia, depresja, bezsenność, łysienie,
– hipoplazja szpiku
– w rzadkich przypadkach powikłania o charakterze autoimmunologicznym, w
tym niedokrwistość hemolityczna, trombocytopenia, zapalenie naczyń
krwionośnych, zespół nerczycowy, niedoczynność tarczycy
•
połączenie interferonu z glikolem polietylenowym (peg-INFα)
– dłuższy jest okres wchłaniania
– stosowany jeden raz w tygodniu)
– mniej toksyczny
•
interferon-α obecnie znajduje zastosowanie
w leczeniu chorych z CML w okresie ciąży
i karmienia piersią
Inhibitory kinaz tyrozynowych –
nowa era leczenia CML
Imatynib
•
lek celowany molekularnie
– kompetytywny inhibitor kinazy Bcr-Abl
– selektywnie hamuje proliferację oraz indukuje apoptozę w komórkach Bcr-Abl +
zawierających chromosom Philadelphia
– hamuje kinazy receptorów: płytkopochodnego czynnika wzrostu PDGF-R (alfa i
beta) oraz czynnika wzrostu komórek macierzystych (SCF) kodowanego przez
protoonkogen c-Kit
•
wskazany w leczeniu dorosłych, dzieci i młodzieży:
– z nowo rozpoznaną CML z chr. Philadelphia (bcr-abl,Ph+),
którzy nie kwalifikują się do zabiegu transplantacji szpiku
– z CML Ph+ w fazie przewlekłej, gdy leczenie INFα
jest nieskuteczne lub w fazie akceleracji choroby, lub
w przebiegu przełomu blastycznego

2013-03-23
8
Mechanizm działania imatynibu
•
blokowanie wiązania ATP w centrum aktywnym kinazy
tyrozynowej
– cząsteczka imatynibu przyłącza sie do miejsca
wiążącego ATP, pomiędzy pętlę aktywacyjna i alfa-C
helisę fragmentu N-końcowego, uniemożliwiając
przeniesienie grupy fosforanowej z ATP na tyrozynę
białka substratowego
– połączenie to zapobiega aktywacji kinazy i stabilizuje
enzym w konformacji nieaktywnej
(zdefosforylowanej)
zablokowanie aktywacji kaskady białek
przekazujących sygnał proliferacyjny do jądra
komórkowego
•
imatynib łączy się tylko z nieaktywną formą kinazy
•
hamuje proliferacje oraz indukuje apoptozę niemal we
wszystkich liniach komórkowych Bcr-Abl pozytywnych
Skuteczność imatynibu
•
w badaniach klinicznych II fazy imatynib stosowano u chorych w fazie przewlekłej,
którzy nie odpowiedzieli na leczenie IFN-a, w fazie akceleracji, przełomu
mieloblastycznego oraz u chorych na Ph (+) ALL
•
w randomizowanym badaniu klinicznym III fazy porównano skuteczność leczenia
imatynibem (dawka 400 mg/d.) oraz IFN-a (5 mln j./m2/d.) w połączeniu z małymi
dawkami ara-C (20 mg/m2/d. przez 10 dni) u chorych z nowo rozpoznaną CML w
fazie przewlekłej
II faza badań
klinicznych
Faza
przewlekła
(n=454)
Faza
akceleracji
(n=181)
Faza kryzy
blastycznej
(n=229)
CHR
95%
34%
8%
MCyR
60%
24%
16%
CCyR
44%
17%
7%
Po 42 mies.
leczenia
Imatinib
(n=553)
INF-α + ara-C
(n=553)
CHR
98%
55%
MCyR
91%
20%
CCyR
84%
7,5%
• wyniki badania IRIS po 6 latach leczenia imatynibem
– 97% pacjentów uzyskało CHR
– 89% pacjentów uzyskało MCyR
– 82% pacjentów uzyskało CCyR
• 6-letni czas życia bez progresji choroby (PFS, progression free survival) do
fazy akceleracji lub kryzy blastycznej wynosił 93%
– żaden pacjent nie wykazał progresji między 5 a 6 rokiem leczenia
• 6-letni czas życia bez nawrotu choroby (EFS, event-free survival)
[nawrót choroby = utrata CHR, MCyR, progresja do fazy AP/BC, śmierć]
wynosił 83%
• 6-letni całkowity czas przeżycia (OS, overall survival) pacjentów leczonych
imatynibem wynosił 88% (zgony związane wyłącznie z CML – 95%)
Skuteczność imatynibu
Oporność na leczenie IM
• pierwotnie istniejąca lub rozwijająca się w trakcie leczenia
(wtórna)
• dotyczy ok. 20-30% przypadków leczenia pierwszorzutowego
• występuje głównie u pacjentów w zaawansowanych stadiach
CML, szczególnie w fazie przełomu blastycznego (u 70%
chorych)
Typ oporności
pierwotna
wtórna
brak CHR po 3 miesiącach
utrata CHR lub CyR (nabyta
hematologiczna i/lub
cytogenetyczna oporność na lek
w odniesieniu do utraty wcześniej
uzyskanej odpowiedzi)
brak CyR po 6 miesiącach
brak co najmniej PCyR po 12
miesiącach
brak CCyR po 18 miesiącach
Mechanizmy oporności :
mutacje punktowe w obrębie domeny kinazy tyrozynowej
Bcr-Abl (ponad 90 typów mutacji, które uniemożliwiają
wiązanie lub zmniejszają wrażliwość na ten lek)
amplifikacja i nadekspresja genu bcr-abl
ewolucja klonalna – wykształcenie alternatywnych,
onkogennych dróg przewodzenia sygnału w komórkach
nowotworowych
zaburzenia w transporcie leku do komórek – zwiększona
aktywność białek transportowych, które usuwają lek z
komórek (glikoproteina P) lub zmniejszony dokomórkowy
transport leku (hOCT1)
Inhibitory kinaz tyrozynowych II generacji
Dazatynib
inhibitor kinazy tyrozynowej Bcr-Abl
przyłącza się zarówno do formy aktywnej jak i nieaktywnej enzymu)
– 325-krotnie silniejszy inhibitor niezmutowanego genu bcr-abl niż imatynib
– zachowuje skuteczność w przypadkach mutacji genu bcr-abl, z wyjątkiem
mutacji T315I/A, F317L i V299L
hamuje kinazy z rodziny Src, PDGFR i c-Kit
– poprzez działanie na kinazę Src pokonuje oporność
w zaawansowanych stadiach CML
• stosowany:
– noworozpoznaną CML Ph+ w fazie przewlekłej,
– CML w fazie przewlekłej, w fazie akceleracji lub w fazie przełomu
blastycznego w przypadku oporności lub nietolerancji na
uprzednie leczenie, w tym leczenie imatynibem
2013-03-23
9
Inhibitory kinaz tyrozynowych II generacji
Nilotynib
pochodna imatynibu, charakteryzuje się lepszym topograficznym
dopasowaniem do struktury kinazy Bcr-Abl
wiąże się z nieaktywną formą kinazy, blokując ją 30-krotnie silniej niż
imatynib
zachowuje skuteczność we wszystkich typach oporności na imatynib z
wyjątkiem mutacji T315I, Y253H/F, E255V/K i F359V
wykazuje porównywalną do imatynibu aktywność wobec kinaz c-Kit i
PDGFR, nie działa na kinazę Src
wskazany w leczeniu dorosłych pacjentów
z nowo rozpoznaną CML Ph+ w fazie przewlekłej
w fazie przewlekłej lub fazie akceleracji, w przypadku
oporności lub nietolerancji na uprzednie leczenie,
w tym leczenie imatynibem
brak danych dotyczących skuteczności u pacjentów
w przełomie blastycznym
Skuteczność dazatynibu i nilotynibu
• wyniki II fazy badań klinicznych
– 52 pacjentów otrzymało dasatynib w dawce 100 mg/dobę lub 50 mg/2x
dziennie
– 53 pacjentów otrzymywało nilotynib w dawce 400 mg/2x dziennie
• skuteczność obu leków jest podobna
• odsetek osiąganych CCyR u chorych
po niepowodzeniu terapii imatynibem
– 44% dla dazatynibu
– 42% dla nilotynibu
II faza badań
klinicznych
Dasatynib
(n=52)
Nilotynib
(n=53)
CCyR
po 3 mies.
79%
90%
po 6 mies.
93%
95%
po 12 mies.
95%
93%
po 18 mies.
88%
95%
MMR
po 12 mies.
34%
47%
po 18 mies.
48%
65%
INHIBITOR KINAZY
TYROZYNOWEJ
DZIAŁANIA NIEPOŻĄDANE
CZĘSTOŚĆ
WYSTĘPOWANIA [%]
Imatynib
(400mg/dobę)
Niehematologiczne:
obrzęki (obwodowe i wokół oczu)
60
nudności
50
skurcze mięśni
49
bóle mięśniowo-szkieletowe
47
biegunka
45
wysypka i inne odczyny skórne
40
zmęczenie
39
bóle głowy
37
bóle w obrębie jamy brzusznej
37
bóle stawów
31
Hematologiczne 3-4 stopnia:
neutropenia
17
trombocytopenia
9
Dazatynib
(100 mg/dobę)
Niehematologiczne:
retencja płynów (w tym wysięk opłucnowy)
34
biegunka
27
bóle głowy
12
wysypka
11
bóle mieśniowo-szkieletowe
11
Hematologiczne 3-4 stopnia :
neutropenia
21
trombocytopenia
19
Nilotinib
(2 x 300mg/dobę)
Niehematologiczne:
wysypka skórna
32
świąd
16
bóle głowy
14
nudności
14
zmęczenie
11
bóle mięśni
10
Hematologiczne 3-4 stopnia:
neutropenia
12
trombocytopenia
10
Allogeniczna transplantacja komórek
macierzystych szpiku (allo-HSCT)
allo-HSCT od dawcy rodzinnego lub
niespokrewnionego jest jedyną znaną metodą, która
daje szanse na całkowite wyleczenie
możliwa do przeprowadzenia u mniej niż połowy
pacjentów
istotnym ograniczeniem dla allo-SCT jest:
– brak dawcy zgodnego w antygenach układu HLA
– wysoka toksyczność, obserwowana
zwłaszcza u chorych w bardziej
zaawansowanym wieku
(powyżej 40–50 r.ż.)
Allogeniczna transplantacja komórek macierzystych
szpiku (allo-HSCT)
• w odniesieniu do CML udokumentowano istnienie efektu
„przeszczep przeciwko białaczce” (graft versus leukemia – GVL)
zależnego od immunokompetentnych komórek pochodzących od
dawcy
• odpowiedzialne za nie są limfocyty T i komórki NK dawcy, które na
przestrzeni czasu całkowicie eliminują wszystkie rezydualne komórki
białaczkowe biorcy lub utrzymują je w stanie „uśpienia”
• na powierzchni komórek białaczkowych CML zidentyfikowano
antygeny, które mogą stanowić cel dla limfocytów T. Należą do nich:
– antygeny specyficzne, takie jak peptydy Bcr-Abl
– antygeny towarzyszące (proteinaza 3, WT1, elastaza,
telomeraza, surwiwina, PRAME, katepsyna, mieloperoksydaza)
oraz antygeny zgodności tkankowej mniejsze
Skala prognostyczna wg Gratwohla
dla chorych na CML, pozwalającą
określić ryzyko wystąpienia powikłań
potransplantacyjnych
Czynniki
Punkty
Dawca
Spokrewniony zgodny w HLA
0
Niespokrewniony/nie w pełni
zgodny
1
Zaawansowanie choroby
Faza przewlekła
0
Faza akceleracji
1
Faza przełomu blastycznego
2
Wiek chorego
<20 lat
0
20-40 lat
1
> 40lat
2
Płeć dawcy/biorcy
inne
0
mężczyzna biorca- kobieta
dawca
1
Czas od rozpoznania do
przeszczepu
< 12 miesięcy
0
> 12 miesięcy
1
Punkty
MRT* (%)
5-letnie
przeżycie
0
21
76
1
21
73
2
35
59
3
47
49
4
53
38
5
45
39
6
81
19
* śmiertelność związana z allo-HSCT
(treatment related mortality )

2013-03-23
10
Allo-HSCT
• od dawcy spokrewnionego
– optymalnym dawcą jest zgodne w antygenach układu HLA
rodzeństwo
– u 60–80% chorych uzyskuje się 5-letnie przeżycie
• od dawcy niespokrewnionego zgodnego w antygenach HLA lub dawcy
rodzinnego nie w pełni zgodnego ryzyko powikłań z powodu nasilonej
choroby „przeszczep przeciwko gospodarzowi” (GVHD, graft versus host
disease)
– przeżycia 5-letnie są nieco mniej liczne (50–70%) i w głównej mierze
zależą od wieku pacjenta
• niektóre badania wskazują podobne wyniki transplantacji od dawcy
rodzinnego i niespokrewnionego, głównie dzięki bardziej doskonałym
metodom doboru w antygenach HLA
Skuteczność allo-HSCT
• wypadkowa intensywności leczenia przygotowującego (kondycjonowania) i
reakcji „przeszczep przewiwko białaczce” GVL (graft versus leukemia)
• standardowe leczenie polega na zastosowaniu terapii ablacyjnej:
o chemioterapii BuCy2 (busulfan 4 × 4 mg/kg doustnie; cyklofosfamid 2 ×
60 mg/kg dożylnie) lub radiochemioterapii TBI/Cy (total body
irradiation; naświetlanie całego ciała 12 Gy, cyklofosfamid 2 × 60 mg/kg)
o chemioterapia BuCy2 – lepiej tolerowana, mniej toksyczna i bardziej
skuteczna w zapobieganiu wznowie w porównaniu z TBI/Cy
• transplantacje o zredukowanym kondycjonowaniu (mini-allo-SCT)
o u osób w zaawansowanym wieku (>50 r.ż.) i ze współistniejącymi
chorobami
o cytostatyki podaje się w dawkach niepowodujących mieloablacji
o eradykację komórek nowotworowych uzyskuje się, stosując w
późniejszym okresie infuzję limfocytów T dawcy (DLI, donor lymphocytes
infusion), która wzmaga reakcję GVL
Powikłania allo-HSCT
•
GvHD (graft-versus-host disease) – choroba przeszczep przeciwko
gospodarzowi – reakcja w organizmie biorcy wywołana obecnością obcych
antygenowo limfocytów dawcy
•
przewlekła GVHD występuje u ok. 50% chorych po allo-HSCT
•
u chorych z postacią ciężką GVHD dochodzi często do rozległego zajęcia skóry,
błon śluzowych, oczu, przewodu pokarmowego i innych narządów
•
wystąpienie objawów GvHD wpływa na przeżycie pacjentów – 80% chorych z
postacią łagodną przeżywa 10 lat, z postacią ciężką tylko 5%
Wskazania do allo-HSCT
• allo-HSCT nie jest obecnie rekomendowany w pierwszej linii leczenia
chorych w fazie przewlekłej CML
• można rozważyć jako terapię I rzutu u chorych dzieci oraz dorosłych do 40.
r.ż., z wysokim ryzykiem progresji choroby (wg Sokala lub Hasforda) i
niskim stopniem powikłań transplantacyjnych wg Grathwola (0-2)
• jako terapia II rzutu:
w przypadku mutacji opornych na dostępne kinazy tyrozynowe
(mutacja T315I) lub
w przypadku progresji do kryzy lub akceleracji w trakcie terapii
imatynibem po osiągnięciu fazy przewlekłej przy pomocy TKI II
generacji
• jako terapia III rzutu:
w przypadku oporności, nietolerancji TKI II generacji lub
w przypadku progresji do kryzy blastycznej lub fazy akceleracji w
trakcie terapii TKI II
Strategie postępowania terapeutycznego
• leczenie pierwszoliniowe
– inhibitory kinaz tyrozynowych
• imatynib (400 mg/dobę)
– u chorych w wieku > 75lat – możliwość zmniejszenia dawki
– skutecznie minimum to 300 mg/dobę
• dazatynib (100 mg/dobę)
• nilotynib (600 mg/dobę)
– interferon α – kobiety w ciąży i w okresie laktacji
– u chorych, u których przewidywanych czas przeżycia < 3 lat – hydroksymocznik
• leczenie drugoliniowe
– oporność pierwotna lub wtórna na imatynib – inhibitory kinaz tyrozynowych II
generacji – dazatynib (100mg/dobę) lub nilotynib (800mg/dobę)
• uwzględnienie następujących czynników: profil toksyczności, choroby
współistniejące, interakcje, analiza mutacji genu Bcr-Abl
» w przypadku mutacji Y253H, E255W i F359C – dazatynib
» w przypadku mutacji V299L, Q252H i F317L – nilotynib
» w przypadku mutacji T315I – alloHSCT
– odpowiedź suboptymalna – imatynib (600-800 mg/dobę) lub inhibitory kinaz
tyrozynowych II generacji
• niepowodzenie terapii II liniowej (brak przynajmniej odpowiedzi
suboptymalnej)
– allo-HSCT lub
– leczenie eksperymentalne z wykorzystaniem prowadzonych badań
klinicznych lub
– leczenie cytoredukcyjne za pomocą hydroksymocznika
• leczenie fazy akceleracji i przełomu blastycznego
– we wszystkich wypadkach dążenie do allo-HSCT
– I liniowo – imatynib 600-800 mg/dobę (z wyjątkiem mutacji T315I)
– leczenie kryzy blastycznej opornej na imatynib – polichemioterapią
przewidziana dla ostrych białaczek mielo- lub limfoblastycznych
– II liniowo
• leczenie fazy akceleracji opornej na imiatinib – dazatynib 140
mg/dobę lub nilotynib 800 mg/dobę
• leczenie fazy kryzy blastycznej – dazatynib 140 mg/dobę
Wyszukiwarka
Podobne podstrony:
Przewlekła białaczka szpikowa jest chorobą nowotworową szpiku kostnego i krwi
Przewlekła białaczka szpikowa(1), Pierwsza Pomoc Przedmedyczna, Medycyna, Onkologia
Przewlekła białaczka szpikowa
Współczesne zasady leczenia przewlekłej białaczki szpikowej
13 Przewlekła białaczka szpikowa
Bialaczka szpikowa przewlekla
Przewlekła białaczka limfatyczna
przewlekłe białaczki limfocytowe
2 Patomechanizm objawów klinicznych w ostrej białaczce szpikowejid 19599 ppt
Ostrsa białaczka szpikowa
przewlekła białaczka limfocytowa
Ostra Bialaczka Szpikowa Proces Pielegnowania id 341705
przewlekła białaczka limfocytowa 2
Bialaczki szpikowe ostre
więcej podobnych podstron