
PRZEWLEKŁA BIAŁACZKA
SZPIKOWA
Magdalena Nieśpiałowska
Katedra i Zakład Diagnostyki Laboratoryjnej

2

KLASYFIKACJA NOWOTWORÓW
MIELOIDALNYCH WG WHO
1. Ostra białaczka szpikowa
2. Zespoły mielodysplastyczne (MDS)
3. Nowotwory mieloproliferacyjne (MPN)
Przewlekła białaczka szpikowa
Czerwienica prawdziwa
Nadpłytkowość samoistna
Pierwotne zwłóknienie szpiku
Przewlekła białaczka neutrofilowa
Przewlekła białaczka eozynofilowa nieokreślona inaczej
Zespół hipereozynofilowy
Mastocytoza
Niesklasyfikowane nowotwory mieloproliferacyjne.
4. MDS/MPN – nowotwory mielodysplastyczno-mieloproliferacyjne
Przewlekła białaczka mielomonocytowa
Młodzieńcza postać białaczki mielomonocytowej
Atypowa przewlekła białaczka szpikowa
MPN/MDS niesklasyfikowany
5. Nowotwory mieloproliferacyjne przebiegające z eozynofilią.
N szpikowe z rearanżacją PDGFRA
N szpikowe z rearanżacją PDGFRB
N szpikowe z rearanżacją FGF1
3
Od 2008 r. obowiązuje nowa klasyfikacja CMPD (chronic
myeloproliferative diseases) wg WHO.

DEFINICJA
Przewlekła białaczka szpikowa (CML- chronic myeloid
leukemia) jest klonalną chorobą komórki macierzystej
szpiku wywołaną pojawieniem się chimerowego genu
BCR-ABL1
U większości (90 -95%) chorych stwierdza się w
komórkach hematopoetycznych obecność
nieprawidłowego genu Filadelfia. Powstaje on w wyniku
translokacji fragmentu DNA z chromosomu 9 do
chromosomu 22 – t[9;22]. Translokacji ulega gen ABL
kodujący kinazę tyrozynową, zostaje on wbudowany na
chromosomie 22 do genu BCR kodującego kinazę
serynowo-treoninową. W wyniku tego zaburzenia
powstaje białko fuzyjne BCR-ABL o aktywności kinazy
tyrozynowej.
4

CHROMOSOM PHILADELPHIA
Chromosom Philadelphia został odkryty i
opisany w 1960 roku przez:
Petera Nowella z Uniwersytetu w Pensylwanii
Davida Hungerforda z Institute for Cancer
Research
W 1973 roku Janet D. Rowey z Uniwersytetu w
Chicago udowodniła, że translokacja
pomiędzy
chromosomem
9 i 22 (t(9;22)
(q34;q11)) stanowi przyczynę powstania tej
anomalii
5
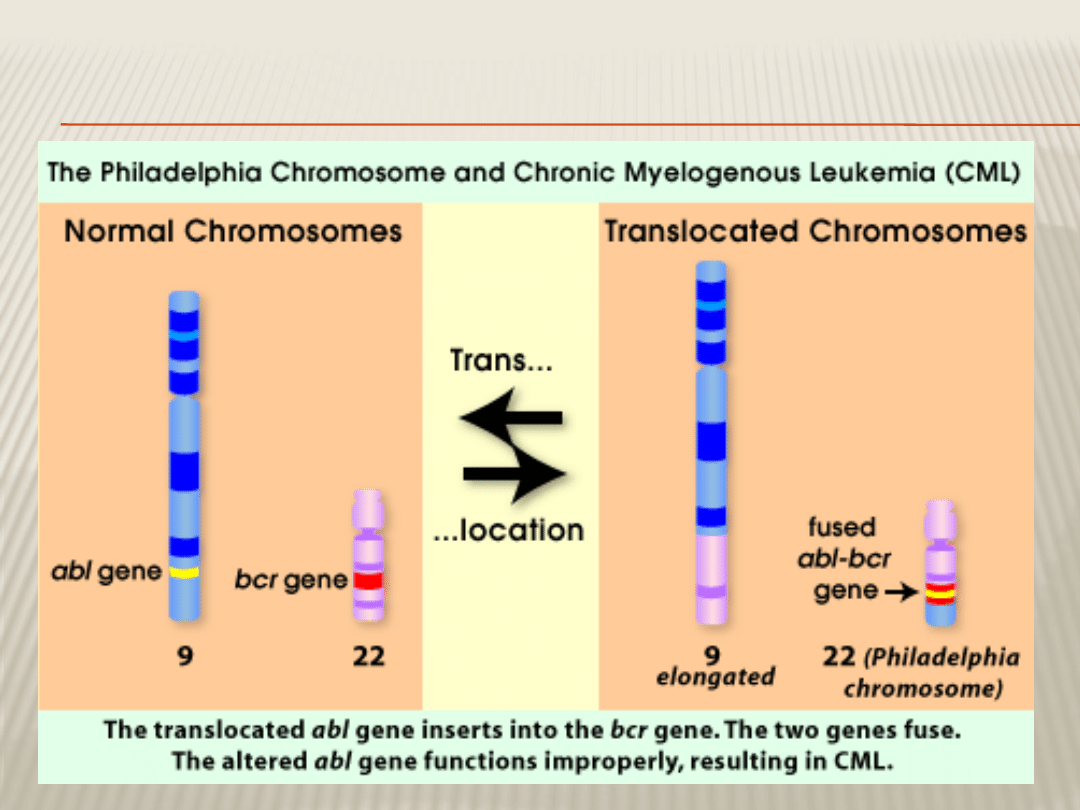
TRANSLOKACJA (9;22) (Q34;Q11)
6

T(9;22)(Q34;Q11)
W wyniku translokacji (9;22) powstają 2 geny fuzyjne:
Na chromosomie 9 - ABL-BCR kodujący powstawanie
białka abl/bcr nieaktywnego biologicznie
Na chromosomie 22 – chimerowy gen BCR-ABL
którego produktem jest białko o aktywności kinazy
tyrozynowej:
zakłócenie adhezji komórkowej
aktywacja lub zakłócone przekazywanie sygnałów
fizjologicznych w wielu szlakach komórkowych
zahamowanie apoptozy
indukcja degradacji wielu białek kluczowych dla procesu
hematopoezy głównie kaspazy 3
7

GEN I BIAŁKO ABL
Gen abl jest protoonkogenem kodującym białko
cytoplazmatyczne i jądrowe z rodziny kinaz
tyrozynowych, które jest odpowiedzialne za
różnicowanie, podział, adhezję i odpowiedź na
uszkodzenia komórek. W warunkach normalnych
ekspresja genu ABL, a zatem produkcja białka, podlega
ścisłej regulacji.
Białko ABL jest obecne w jądrze komórkowym w
połączeniu z F-aktyną i uczestniczy w procesach:
hamowania cyklu komórkowego w odpowiedzi na stres
genotoksyczny.
przekazywanie sygnału komórkowego z receptorów dla
czynnika wzrostu i integryn.
8

GEN BCR/ABL
Białko BCR jest obecne na wczesnych prekursorach
mieloidalnych
Po połączeniu fragmentu abl z fragmentem bcr powstały
gen znajduje się ciągle w pozycji włączonej (staje się
onkogenem) i wymyka się spod kontroli komórki.
Produkowane nowe białko przyczynia się do wzrostu
częstotliwości podziałów komórkowych.
Blokując naprawę DNA, powoduje szybkie gromadzenie
się mutacji w nowych pokoleniach komórek.
Wzmożona produkcja białka bcr/abl upośledza również
zdolność komórek do apoptozy oraz do interakcji i
adhezji do podścieliska. Wszystko to prowadzi do
wzrostu masy nowotworu i w konsekwencji doprowadza
do kryzy blastycznej
9

HISTORIA
Przewlekła białaczka szpikowa została
opisana w 1845 roku przez Rudolfa
Virchowa.
Początkowo Virchow sądził, że ma do
czynienia z ciężką infekcją bakteryjną,
manifestującą się olbrzymią liczbą
leukocytów we krwi obwodowej.
Ponieważ krew opisywanego przez niego
pacjenta miała białawy odcień, nazwał tę
chorobę mianem "białaczki".
10

PATOGENEZA
W większości przypadków przyczyna
rozwoju tej choroby nie jest znana.
Jedynym czynnikiem etiologicznym,
którego znaczenie zostało
bezsprzecznie udowodnione, jest
promieniowanie jonizujące (stąd
gwałtowny wzrost zachorowań na tę
chorobę w Japonii po atomowych
atakach na Hiroszimę i Nagasaki.
11

EPIDEMIOLOGIA
Roczna zapadalność na CML wynosi 1/100
tys. osób populacji ogólnej
Częściej dotyczy mężczyzn niż kobiet (1,3 : 1)
Średni wiek to ok. 50 r.ż.
Stanowi ok. 15 - 25 % białaczek wśród
dorosłych,
Stanowi < 5% białaczek dziecięcych
Rocznie w Polsce pojawia się ponad 350
nowych przypadków zachorowań
12

NATURALNY PRZEBIEG CHOROBY
Wyodrębniamy 3 fazy choroby:
1.
Przewlekła (średnio 4-5 lat)
2.
Akceleracji (9-12 miesięcy)
3.
Przełom blastyczny (3-6 miesięcy)
Obserwowano przypadki, gdzie faza
przewlekła przechodziła bezpośrednio
w fazę przełomu blastycznego.
13

CML - OBRAZ KLINICZNY
U 30% chorych dolegliwości podmiotowe w chwili rozpoznania
choroby.
Zmęczenie, zmniejszona tolerancja wysiłku fizycznego
Utrata apetytu
Dyskomfort w jamie brzusznej
Uczucie
pełności
w
nadbrzuszu,
spowodowane
powiększeniem śledziony
Utrata masy ciała
Nadmierna potliwość
Ból w okolicy podżebrowej, promieniujący do lewego barku
(niedokrwienie lub zawał śledziony)
Objawy moczówki (ustępujące po podaniu wazopresyny)
Pokrzywka ( związana ze wzrostem histaminy)
W miarę trwania choroby objawy nasilają się.
14

CML - OBRAZ KLINICZNY
W badaniu przedmiotowym stwierdza się:
powiększenie śledziony,
tkliwość dolnej części mostka (charakterystyczny objaw),
gorączka z plamisto-grudkowymi, fioletowymi zmianami na skórze
twarzy, ramion i tułowia.
rzadko stwierdza się objawy sugerujące nadczynność tarczycy –
nietolerancja ciepła
zapalenia stawów (związane z podwyższeniem stężenia kwasu
moczowego)
U ok. 5% pacjentów z nowo rozpoznaną białaczką objawy są bardziej
intensywne i przypominają fazę akceleracji:
gorączka
bóle kostne
znaczne powiększenie śledziony
nacieki narządowe.
15

DIAGNOSTYKA CML
Morfologia krwi z rozmazem
Badanie cytomorfologiczne i histologiczne szpiku
(trepanobiopsja).
Kariotyp – odsetek komórek z obecną t(9;22) (q34;q11) oraz
dodatkowe aberracje (chromosom Filadelfia),
W przypadku rozpoznania fazy akceleracji lub przełomu
blastycznego należy wykonać ocenę cytomorfologiczną,
cytometryczną i immunohistochemiczną klonu
odpowiedzialnego za progresję choroby.
60% chorych ma przełom mieloblastyczny
20-30% limfoblastyczny
10% erytroblastyczny
3% megakarioblastyczny
Może mieć to znaczenie w monitorowaniu choroby resztkowej lub
wznowy choroby
16

NORMY WSKAŹNIKÓW UKŁADU
BIAŁOKRWINKOWEGO
Rodzaj
komórek
Wartości
prawidłowe
(10
9
/l)
Wzór
odsetkowy
(%)
Leukocyty
ogółem
4 - 10
100
Neutrofile
1,6 – 7,5
45 - 75
Eozynofile
0,1 – 0,5
0 - 5
Bazofile
0,02 – 0,15
0 - 2
Limfocyty
1,5 – 4,5
16 - 45
Monocyty
0,4 – 0,8
4 - 10
17

ZMIANY W MORFOLOGII KRWI OBWODOWEJ
Niedokrwistość
Liczba leukocytów przekracza
25 x 10
9
/l. U osób
nieleczonych stale wzrasta
liczba białych krwinek oraz
płytek krwi (nawet do 1000 x
10
9
/l)
U 50% chorych WBC > 100 x
10
9
/l
Podwyższona liczba płytek
krwi (50% chorych), lub w
normie
W rozmazie krwi obecne są wszystkie
postaci rozwojowe granulocytów
(mieloblasty, promielocyty, mielocyty,
metamielocyty, pałki, granulocyty
podzielone).
Mieloblasty stanowią 0-10%, komponent
dojrzewający (ok. 40%), postaci
dojrzałe (30%) wszystkich leukocytów.
Obecne są także pojedyncze
erytroblasty. Liczba bazofili lub
eozynofili może wzrastać do 20%.
Cechy zaburzenia dojrzewania
granulcytów
Obniżenie lub brak aktywności FAG
Bezwzględna liczba limfocytów jest
zwiększona (średnio 15 x 10
9
/l) w
wyniku wzrostu limfocytów T
h
oraz T
s
.
Liczba komórek NK jest obniżona.
18
W momencie rozpoznania
choroby
W fazie przewlekłej choroby
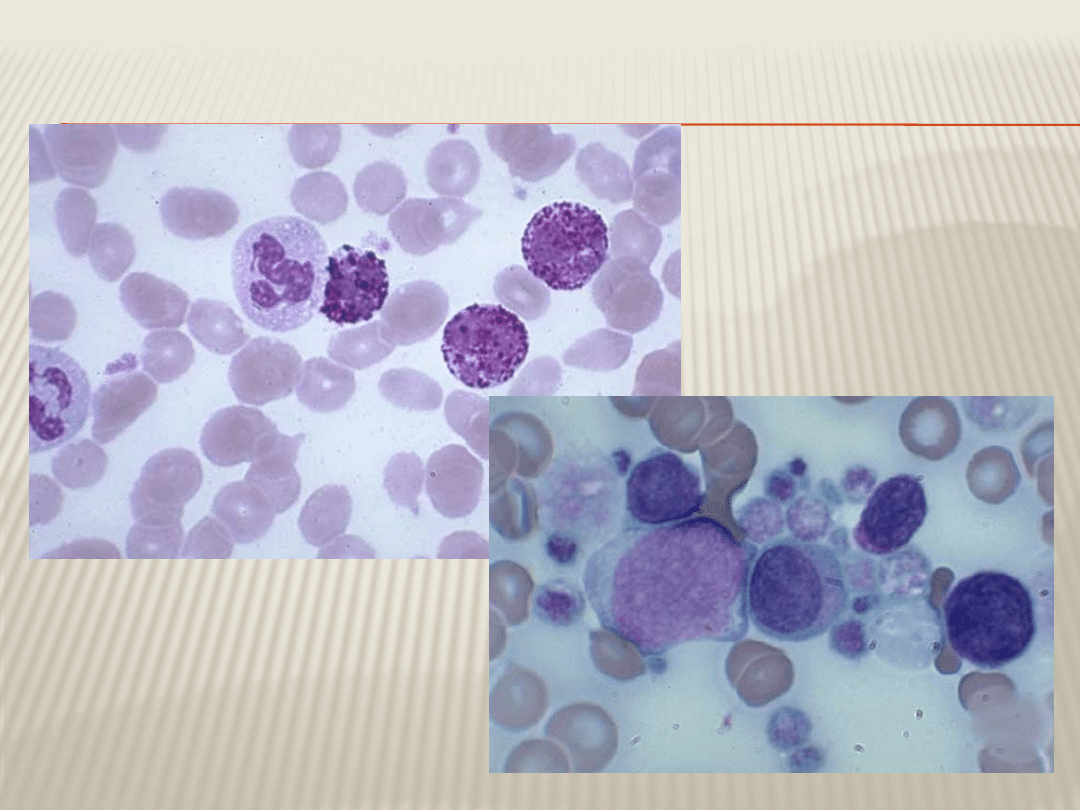
CML – KREW OBWODOWA
19
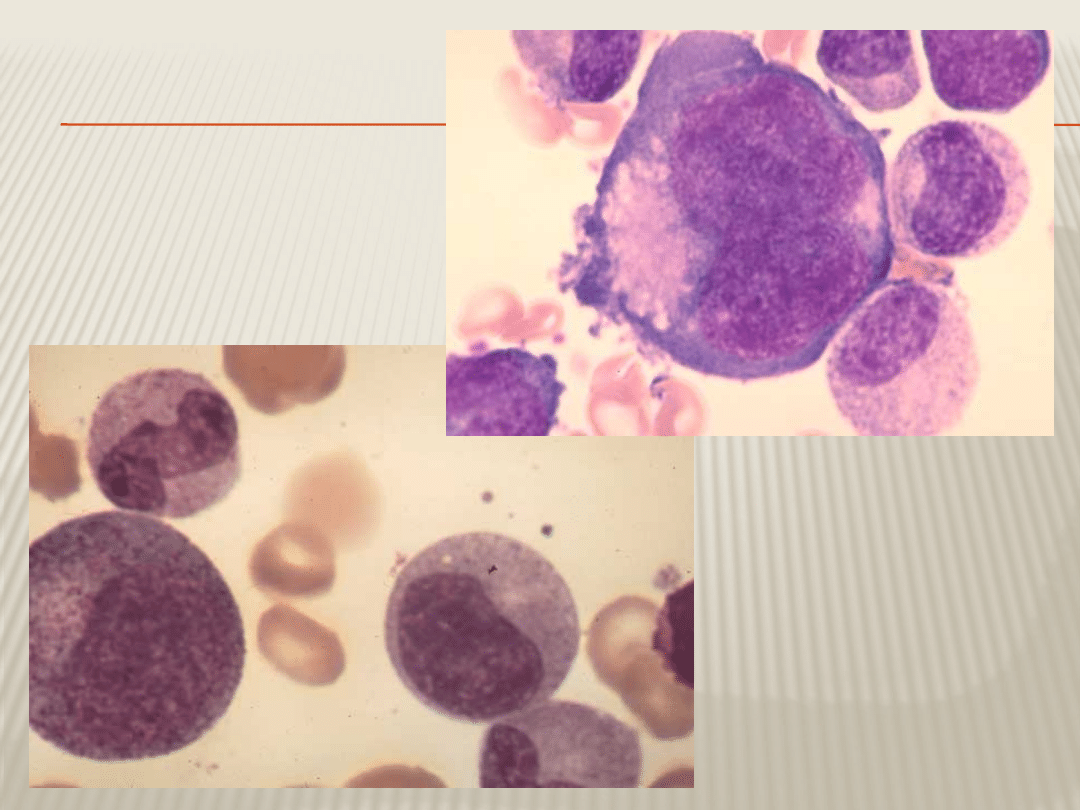
20

OBRAZ SZPIKU KOSTNEGO
Szpik uzyskany metodą biopsji aspiracyjnej jest wybitnie
bogatokomórkowy,
Dominują komórki z linii granulocytarnej (do 95%) ze zwiększeniem
odsetka mielocytów, liczba mieloblastów nie przekracza 15%,
Zawartość granulocytów kwasochłonnych i zasadochłonnych może
być zwiększona (koreluje z odsetkiem tych komórek w rozmazie
krwi obwodowej).
Gorzej reprezentowany układ erytroblastyczny, z zachowaniem
prawidłowości w morfologii i dojrzewaniu komórek.
Liczba megakariocytów prawidłowa lub zwiększona
Trepanobiopsja – w badaniu histopatologicznym szpiku
obserwuje się nasilone włóknienie kolagenowe i retikulinowe,
proporcjonalne do wzrostu liczby megakariocytów w szpiku.
Klinicznie koreluje z wielkością śledziony, nasileniem
niedokrwistości i liczbą blastów we krwi obwodowej.
21

BADANIA DODATKOWE
poziomu witaminy B12 w osoczu
(zwiększona zdolność wiązania
witaminy B12 przez białka osocza
pochodzące z granulocytów)
Prawidłowy lub podwyższony poziom
żelaza
stężenie kwasu moczowego
22

INNE ZABURZENIA
Brak lub niska aktywność fosfatazy
alkalicznej granulocytów (FAG), jej
aktywność może wzrosnąć do normy w
przypadkach ze współistniejącą
infekcją.
W przypadku normalizacji WBC w
wyniku leczenia, FAG również może
osiągnąć poziom w granicach normy.
23

KRYTERIA ROZPOZNANIA FAZY AKCELERACJI
Występuje przynajmniej 1 z poniższych objawów:
Liczba blastów w rozmazie krwi obwodowej lub szpiku – 10-19%
Liczba bazofili w rozmazie krwi obwodowej >20%
Utrzymująca się małopłytkowość <100x10
6
/l niezwiązana z
leczeniem lub utrzymująca się nadpłytkowość >1000x10
6
/l
niereagująca na leczenie
Powiększenie śledziony oraz wzrost liczby leukocytów we krwi,
niereagująca na leczenie
Klonalna ewolucja choroby w badaniu cytogenetycznym
(pojawiające się w trakcie trwania choroby dodatkowe zaburzenia
cytogenetyczne, nie wykrywane w czasie rozpoznania)
Cechy proliferacji w układzie megakariocytarnym (tworzenie
klaserów) powiązane z włóknieniem kolagenowym lub
retikulinowym w obrębie jam szpikowych i cechy ciężkiej dysplazji
w układzie granulocytarnym.
24

KRYTERIA ROZPOZNANIA FAZY BLASTYCZNEJ
Występuje co najmniej 1 z wymienionych
objawów
Wzrost liczby blastów w rozmazie krwi
obwodowej lub szpiku >20%
Objawy pozaszpikowej proliferacji
komórek blastycznych
W badaniu histopatologicznym szpiku
kostnego tworzenie dużych skupisk
komórek blastycznych lub klaserów
25

RÓŻNICOWANIE I LECZENIE
Przewlekłą białaczkę szpikową należy różnicować
z:
innymi nowotworami mieloproliferacyjnymi
odczynem białaczkowym występującym w
innych nowotworach i chorobach infekcyjnych
( FAG)
osteomielofibrozą
przewlekłą białaczką eozynofilową
chorobami przebiegającymi z
nadpłytkowościami
26

LECZENIE
Celem leczenia jest osiągnięcie trzech rodzajów remisji choroby:
remisji hematologicznej
remisji cytogenetycznej (zmniejszenie ilości komórek szpikowych
zawierających chromosom Philadelphia - najlepiej do 0)
remisji molekularnej (zmniejszenie liczby cząsteczek kinazy
tyrozynowej bcr-abl - najlepiej do 0).
Leczenie inhibitorami kinazy tyrozynowej
II generacji – Glivec
Remisja hematologiczna – ustąpienie objawów podmiotowych
związanych z chorobą, zmian w badaniu przedmiotowym typowych
dla CML oraz normalizacja wyników badań laboratoryjnych (morfologii
krwi obwodowej, liczby płytek krwi, rozmazu krwi i szpiku)
27
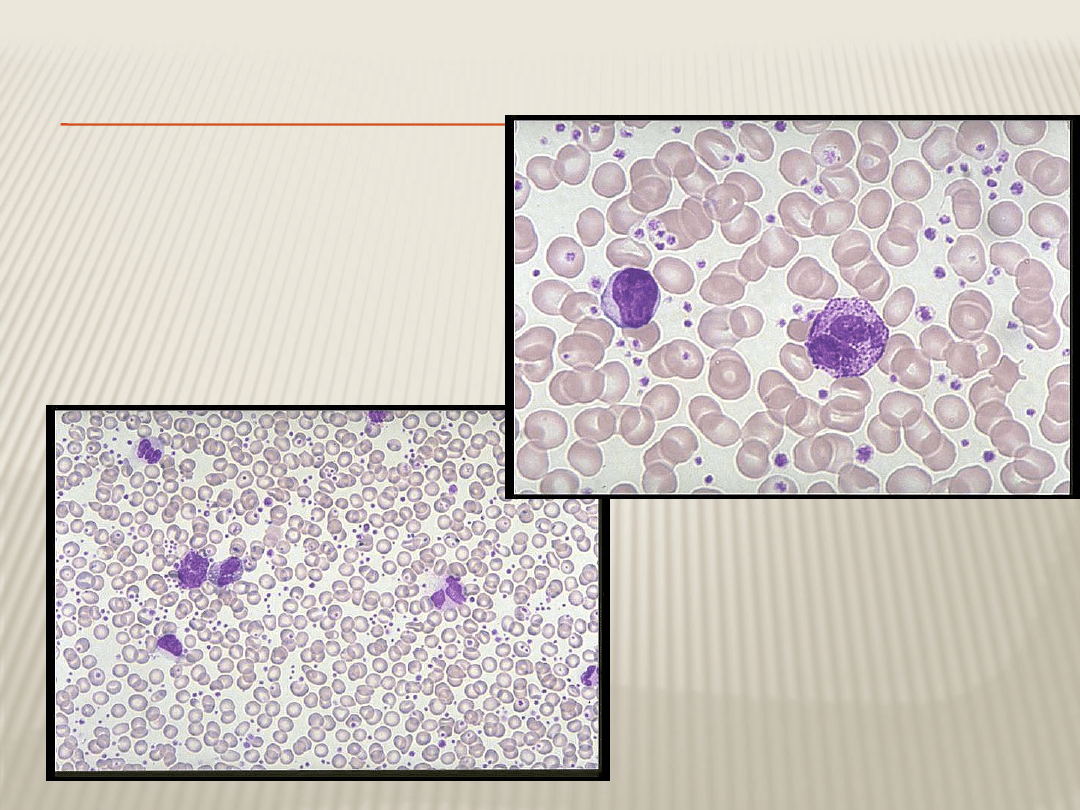
ROZMAZ KRWI CML
28
Pow. x 200
Pow. x 400
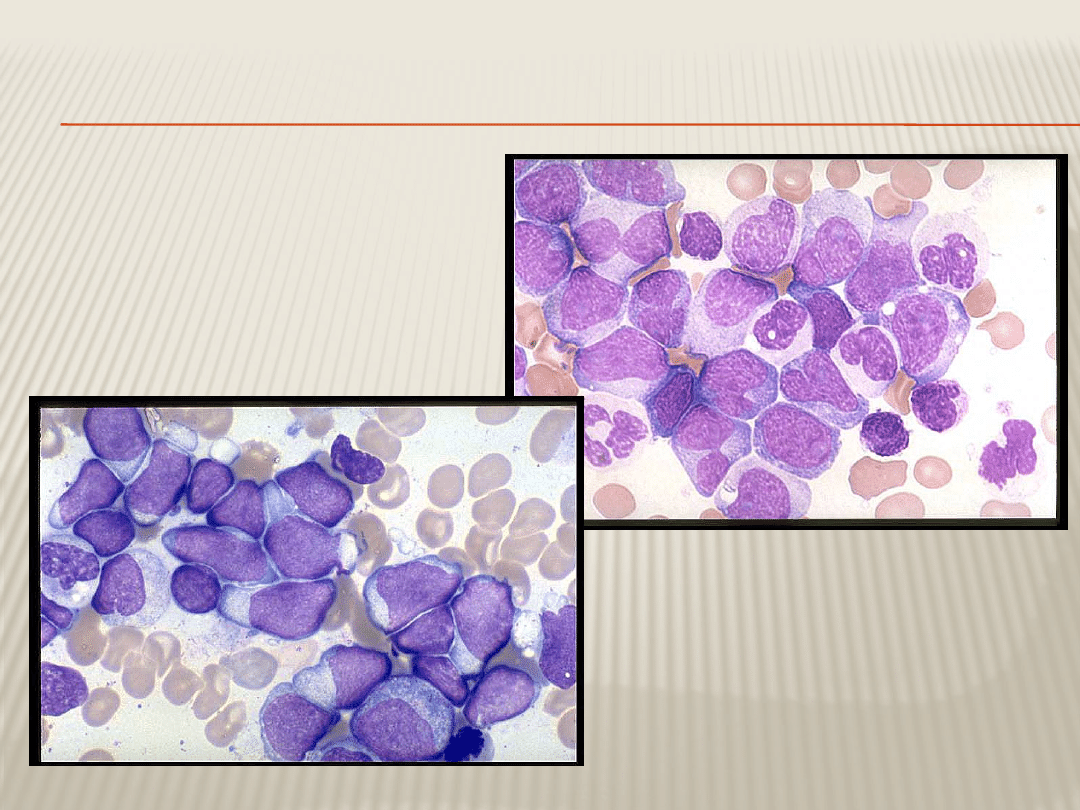
ROZMAZ KRWI – KRYZA BLASTYCZNA
29
Rozplem linii mieloidalej
Rozplem linii
limfoidalnej

DZIĘKUJĘ ZA UWAGĘ
30
Document Outline
- Slide 1
- Slide 2
- Klasyfikacja nowotworów mieloidalnych wg who
- Definicja
- Chromosom Philadelphia
- Translokacja (9;22) (q34;q11)
- T(9;22)(q34;q11)
- Gen i białko ABL
- Gen bcr/abl
- Historia
- Patogeneza
- Epidemiologia
- Naturalny przebieg choroby
- CML - Obraz kliniczny
- CML - Obraz kliniczny
- Diagnostyka CML
- Slide 17
- Zmiany w Morfologii krwi obwodowej
- CML – krew obwodowa
- Slide 20
- obraz szpiku kostnego
- Badania dodatkowe
- Inne zaburzenia
- Kryteria rozpoznania fazy akceleracji
- Kryteria rozpoznania fazy blastycznej
- Różnicowanie i leczenie
- leczenie
- Rozmaz krwi CML
- Rozmaz krwi – kryza blastyczna
- Slide 30
Wyszukiwarka
Podobne podstrony:
Farmakoterapia przewlekłej białaczki szpikowej new
Przewlekła białaczka szpikowa jest chorobą nowotworową szpiku kostnego i krwi
Przewlekła białaczka szpikowa(1), Pierwsza Pomoc Przedmedyczna, Medycyna, Onkologia
Przewlekła białaczka szpikowa
Współczesne zasady leczenia przewlekłej białaczki szpikowej
Bialaczka szpikowa przewlekla
Przewlekła białaczka limfatyczna
przewlekłe białaczki limfocytowe
2 Patomechanizm objawów klinicznych w ostrej białaczce szpikowejid 19599 ppt
Ostrsa białaczka szpikowa
przewlekła białaczka limfocytowa
Ostra Bialaczka Szpikowa Proces Pielegnowania id 341705
przewlekła białaczka limfocytowa 2
Bialaczki szpikowe ostre
więcej podobnych podstron