
Przewlekłe białaczki
Przewlekłe białaczki
limfocytowe (PBL )
limfocytowe (PBL )
Mirosława Pietruczuk
Zakład Diagnostyki
Laboratoryjnej
Katedry Diagnostyki
Laboratoryjnej
Uniwersytetu Medycznego w Łodzi
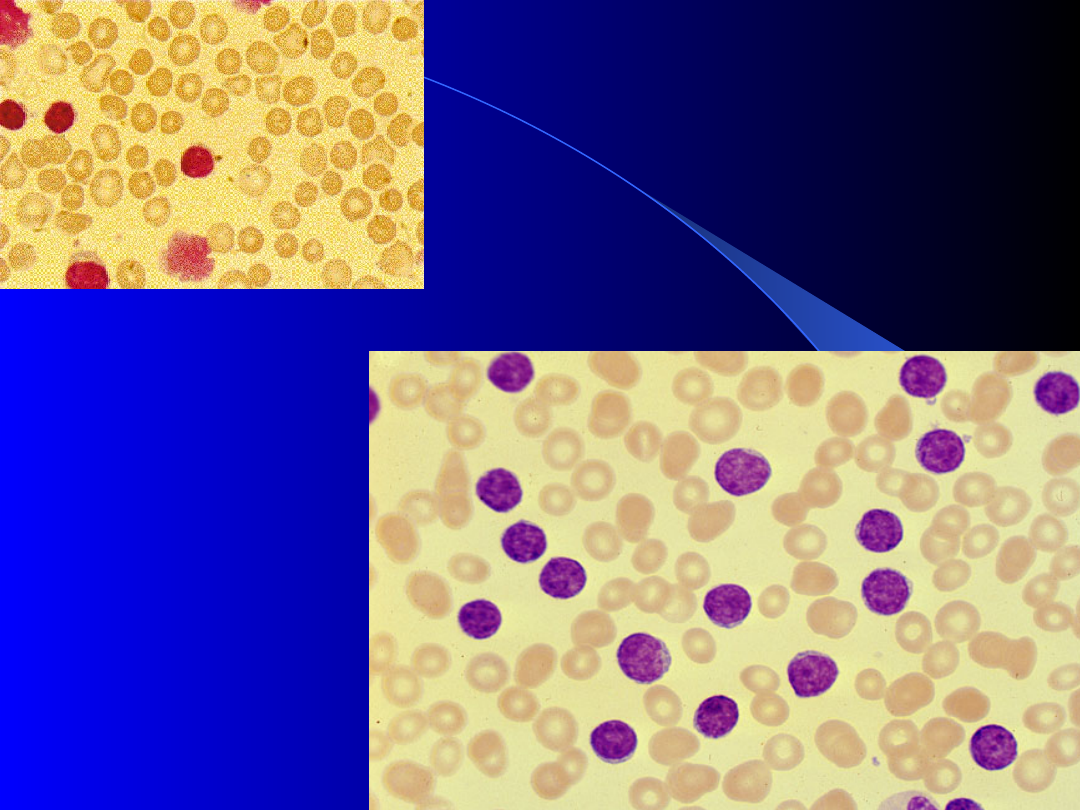
Typ zwykły PBL:
Typ zwykły PBL:
Przewlekła
Przewlekła
białaczka
białaczka
limfocytowa
limfocytowa
B - komórkowa
B - komórkowa
Hiperleukocytoza
Limfocytoza -
>4.0x10
9
/l
we krwi obwodowej
Różnego stopnia
pancytopenia:
(neutropenia
erytrocytopenia
trombocytopenia)
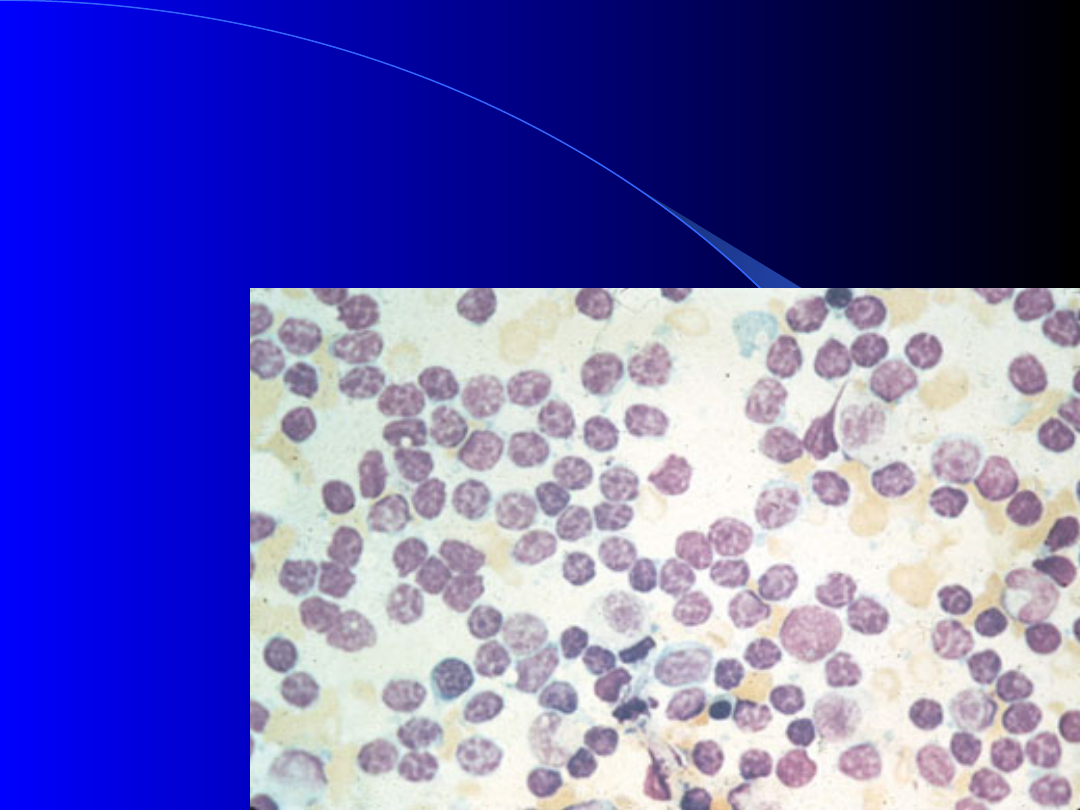
Szpik kostny w PBL –
Szpik kostny w PBL –
B - komórkowej
B - komórkowej
40% komórek szpiku kostnego
stanowią limfocyty
Hypogammaglobulinemia (powikłania
infekcyjne)
NAIH
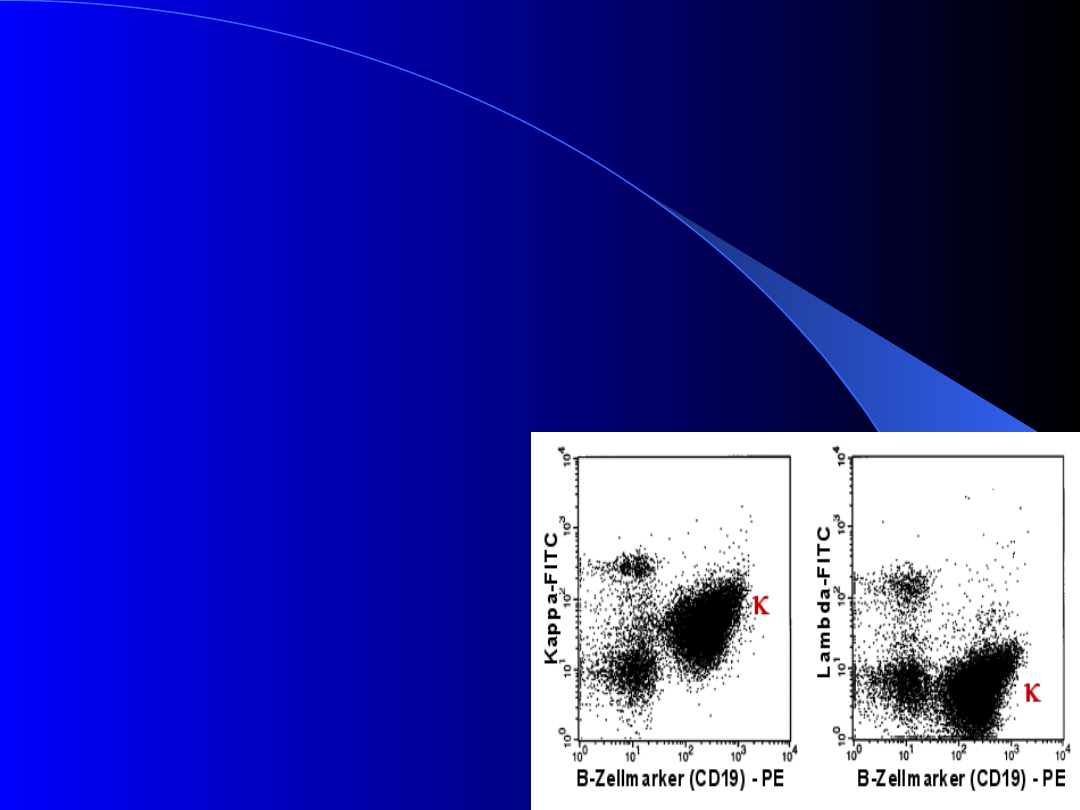
Klonalność limfocytów w
Klonalność limfocytów w
PBL
PBL
Nowotworowy charakter i niedojrzałość
komórek B w PBL wyraża się ich
klonalnością:
1)
występowaniem
powierzchniowych
łańcuchów lekkich typu
wyłącznie kappa
lub wyłącznie lambda
2) mniejszą ekspresją
powierzchniowych
immunoglobulin (sIg),
przy przewadze
fenotypu IgM, IgD.

Co zmieniło się w podejściu
Co zmieniło się w podejściu
do CLL i dlaczego warto o
do CLL i dlaczego warto o
tym wiedzieć.
tym wiedzieć.
1.
CLL stanowią bardzo heterogenną grupę
chorób
, zarówno w aspekcie klinicznym
jak i możliwości progresji choroby
(przeżycie > 10lat u chorych z małym
ryzykiem, 5-7 lat ze średnim ryzykiem i
<2 lat w postaciach agresywnych).
2.
Nowe
możliwości
diagnostyczne:
cytometria
przepływowa
i
badania
genetyczne.
3. Nowe możliwości terapii CLL (terapia
celowana).

CLL , heterogenna
CLL , heterogenna
grupa chorób – aspekt
grupa chorób – aspekt
kliniczny
kliniczny
Klasyfikacja PBL-B wg. Rai:
Okres 0:
Limfocytoza we krwi obwodowej>
4,0x10
9
/l,
limfocyty stanowią >40% utkania
szpikowego
Okres 1:
Objawy
okresu
zerowgo
i
powiększenie węzłów chłonnych

Okres 2:
Objawy okresu 0 lub (i) 1 oraz
powiększenie śledziony lub (i) wątroby.
Okres 3:
Dołączenie się niedokrwistości (Hb<6,2
mmol/l)
do
któregokolwiek
z
poprzednich okresów
Okres 4:
Dołączenie
się
małopłytkowości(<100x10
9
/l
lub (i) zespołu hemolitycznego typu
NAIH, do któregokolwiek z poprzednich
okresów.

CLL jako heterogenna
CLL jako heterogenna
grupa chorób.
grupa chorób.
1. Część pacjentów nie wymaga leczenia
2. Część
chorych
na
CLL
podlega
standardowej chemioterapii i odpowiada na
nią remisją
3. U części chorych szybko następuje progresja
choroby po leczeniu
4. U
części
chorych
występuje
szybka
transformacja CLL w agresywne postaci
chłoniaka
5. U części chorych brak jest odpowiedzi na
leczenie
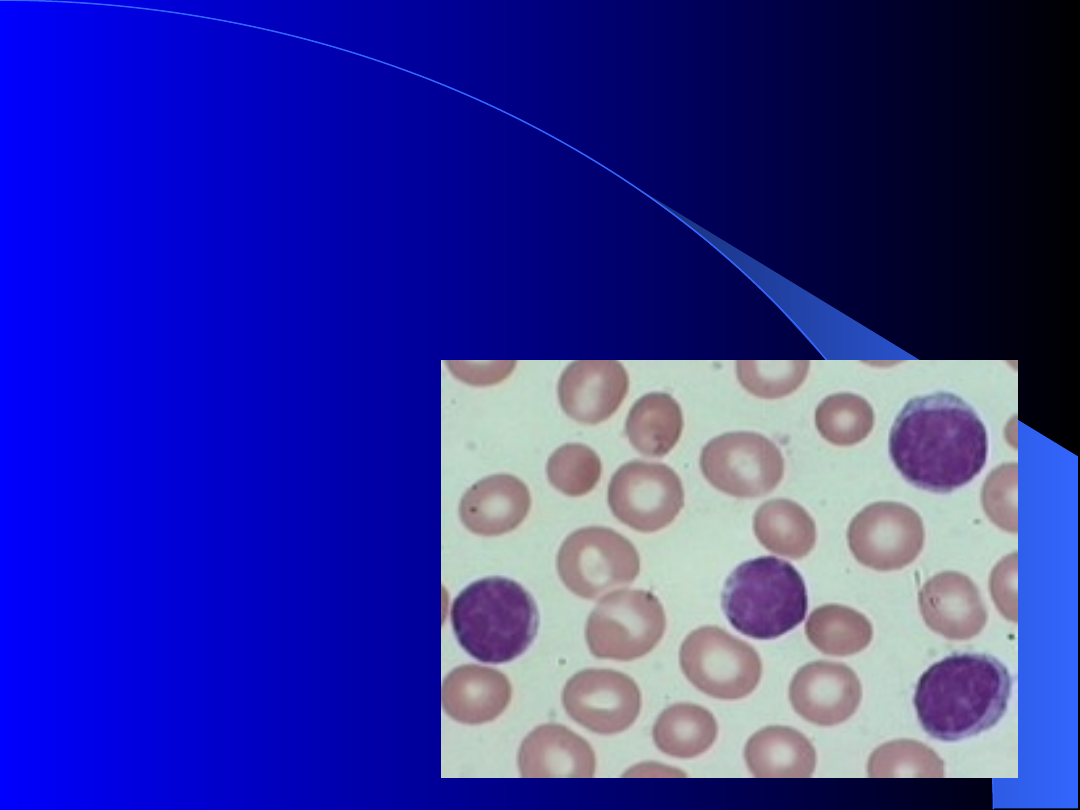
Diagnostyka CLL
Diagnostyka CLL
W oparciu o kryteria IWCLL i NCI
-Working Group Guidelines:
1) liczba limfocytów we krwi obwodowej
> 5.0x 10
9
/l i podstawowy fenotyp
CD19+, CD5+, CD23+, FMC-7 minus.
2)
„pre CLL” –
obecność
monoklonalnej
limfocytozy
<5,0x10
9
/l

Klasyfikacja WHO/ REAL
Klasyfikacja WHO/ REAL
przewlekłych białaczek
przewlekłych białaczek
limfocytowych
limfocytowych
B – komórkowe:
1. Przewlekła białaczka limfocytowa B
– komórkowa (PBL-B)
A) Typ zwykły PBL-B:
CD19/CD5/
CD20/CD37/
CD23
/HLA-DR
Słabsza FMC-7/CD22/sIg
B) PBL-B z prolimfocytami >10% a < 55%
C) Zespół Richtericha

Klasyfikacja CLL
Klasyfikacja CLL
2. Białaczka prolimfocytowa B-
komórkowa
(B-BP) z >55%
prolimfocytów:
CD79b/
CD19/
CD20/CD37/HLA-DR/sIg
ujemny CD5
,
słaba CD23
3. Białaczka włochatokomórkowa:
CD103/CD11c/CD25
/sIg/
CD19/
CD20/
CD37/FMC-7/CD23

CLL- klasyfikacja
CLL- klasyfikacja
4. Białaczka
plazmatycznokomórkowa:
CD38/
słaba CD10,
Ujemne:CD19/CD20/CD37/CD5/CD23/
HLA-DR/sIg
5. Zespoły białaczka/chłoniak:
Śledzionowy (SLVL), z komórek płaszcza
(MCL), komórek olbrzymich, grudkowy
(FL), Makroglobulinemia Waldenstroma

T- komórkowe CLL
T- komórkowe CLL
1. Białaczka z dużych ziarnistych limfocytów
(LGL):
CD2/CD3/CD7/
CD56/CD57/CD8
Ujemne CD5/CD4
2. Białaczka prolimfocytowa T-
komórkowa
CD2/CD7/
CD4/CD25
3. Białaczka chłoniak T-komórkowy
(ATLL):
CD2/CD3/CD5/CD4/CD25
4. Zespół Sezar”ego:
CD2/CD3/CD5/
CD4
/CD26 ujemny CD8

Leczenie CLL
Leczenie CLL
1) chlorambucyl + prednizon
2)
COP
(cyklofosfamid, winkrystyna,
prednison)
CHOP
(+ hydroksyrubicyna)
3) Analogi nukleozydów:
FAMB
(fludarabina),
2CdA
(2 -
chlorodeoksyadenozyna)
4) Bioterapia:
Campath –1H
(anty CD5),
interferon alfa (IFN-α)
5) przeszczepianie komórek
macierzystych (allogeniczne i
autologiczne)
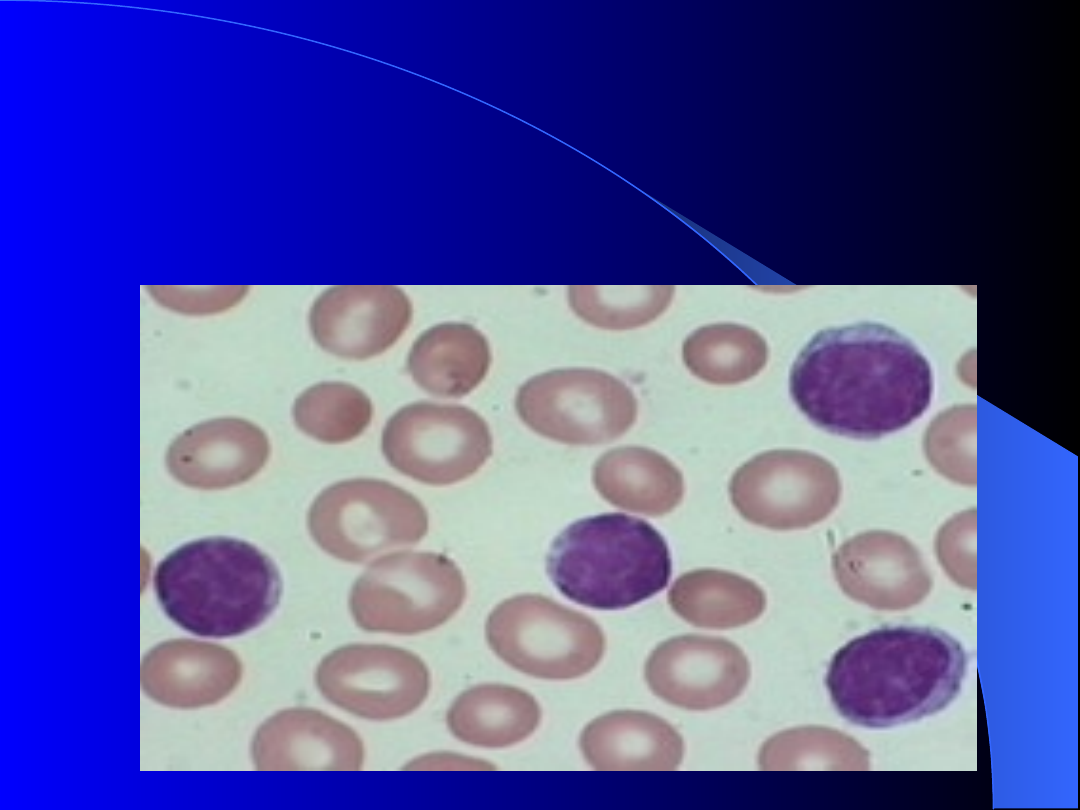
Od czego więc głównie
Od czego więc głównie
zależą - przebieg choroby
zależą - przebieg choroby
- efekty leczenia CLL.
- efekty leczenia CLL.

Rodzinne występowanie CLL
Rodzinne występowanie CLL
1)
Subkliniczne formy CLL
obserwuje się
w młodym wieku. Opisywano występowanie
rodzinne tej postaci z możliwością transmisji
w różne choroby układu chłonnego.
2) Fizjologicznie monoklonalną ekspansję
populacji
limfocytów
CD5+/CD19+
obserwuje się u 13% populacji, u zdrowych
młodych ludzi z prawidłową leukocytozą.
Nosi
to
nazwę
CLUS
–
„clonal
lymphocytosis
of
undetermined
significance.”

3)
Stwierdzono
genetyczny
związek
pomiędzy
występowaniem
„CLUS”
i
wczesnym
CLL
(obecność
delecji: del(13q14).
4) Najczęściej obserwowane
aberracje chromosomalne w
CLL, dotyczą chromosomów:
13, 1, 6, 3 i 17.

Patogeneza CLL-
Patogeneza CLL-
pochodzenie komórek
pochodzenie komórek
1.
W
większości
przypadków
CLL,
nowotworowe limfocyty B pochodzą
z
limfocytów
B
pamięci
z
centrów
postgerminalnych.
2. W przypadku
agresywnych postaci CLL
( bez mutacji IgVH):
A) nowotworowe limfocyty B pochodzą
z
pregerminalnych
centrów
z
„dziewiczych” limfocytów B.
B) nowotworowe limfocyty B pochodzą z
aktywowanych limfocytów B, narażonych na
ciągły kontakt z antygenem.

Hipermutacje genu dla
Hipermutacje genu dla
IgVH
IgVH
1) Najczęściej
hipermutacje genu dla IgVH
dotyczą rearanżacji VH1-69 i VH3-21.
2) W nie zmutowanych CLL, najczęściej brak
mutacji dotyczy rearanżacji VH1-69.
3) Stwierdzenie braku mutacji genu dla IgVH
jest równoznaczne z złym rokowaniem w CLL.
4)
Jednak badania genetyczne nie są
dostępne rutynowo i stąd problem z
wyodrębnieniem „agresywnych postaci”
CLL.
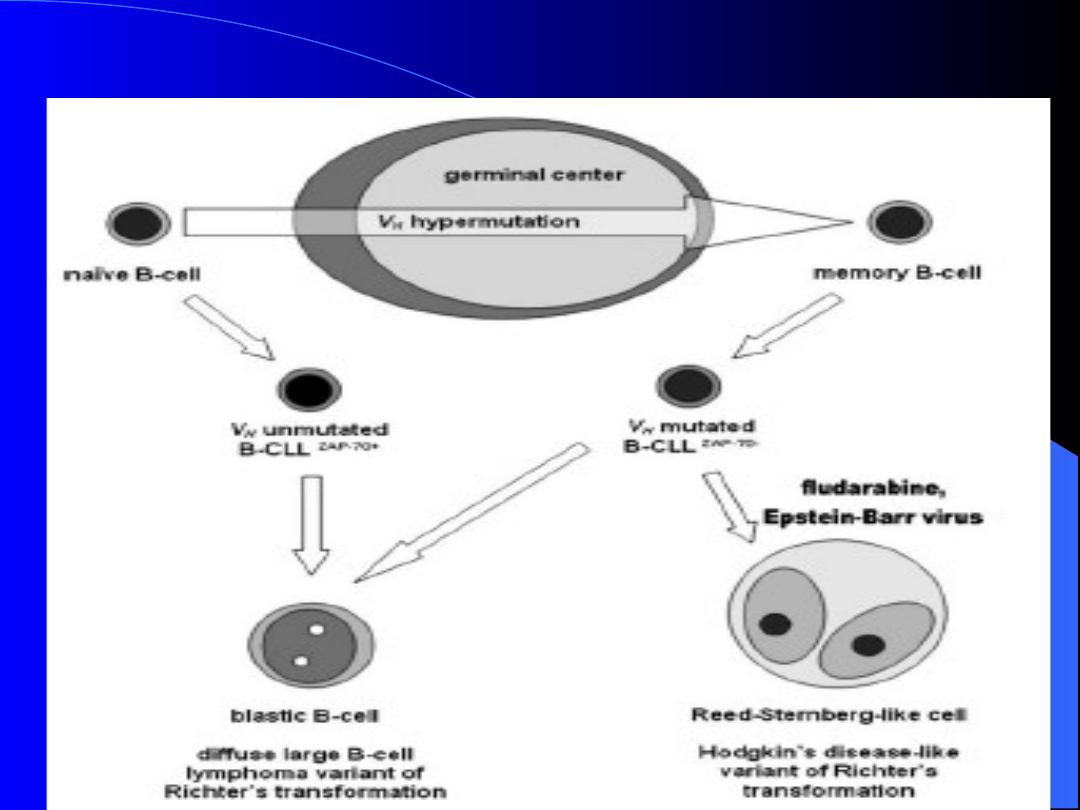
ZAP-70
ZAP-70

Dlatego jedyną
Dlatego jedyną
nadzieją są czynniki
nadzieją są czynniki
prognostyczne
prognostyczne
1)
Najlepszym
czynnikiem
prognostycznym jest:
-
występowanie
(
dobry
czynnik
rokowniczy
)
- lub brak (
zły czynnik rokowniczy
)
mutacji
genu
dla
regionu
zmiennego
łańcucha
ciężkiego
immunoglobuliny (IgVH)

Czynniki prognostyczne
Czynniki prognostyczne
2) występowanie delecji chromosomu 11q23
zły czynnik prognostyczny
3) wyciszenie lub mutacja genu p53
zły czynnik rokowniczy
4) wysoka ekspresja antygenu CD38 i CD44
zły czynnik rokowniczy
5) podwyższone stężenie kinazy tymidynowej
zły czynnik rokowniczy
6) wysokie stężenie β2 mikroglobuliny
zły czynnik rokowniczy
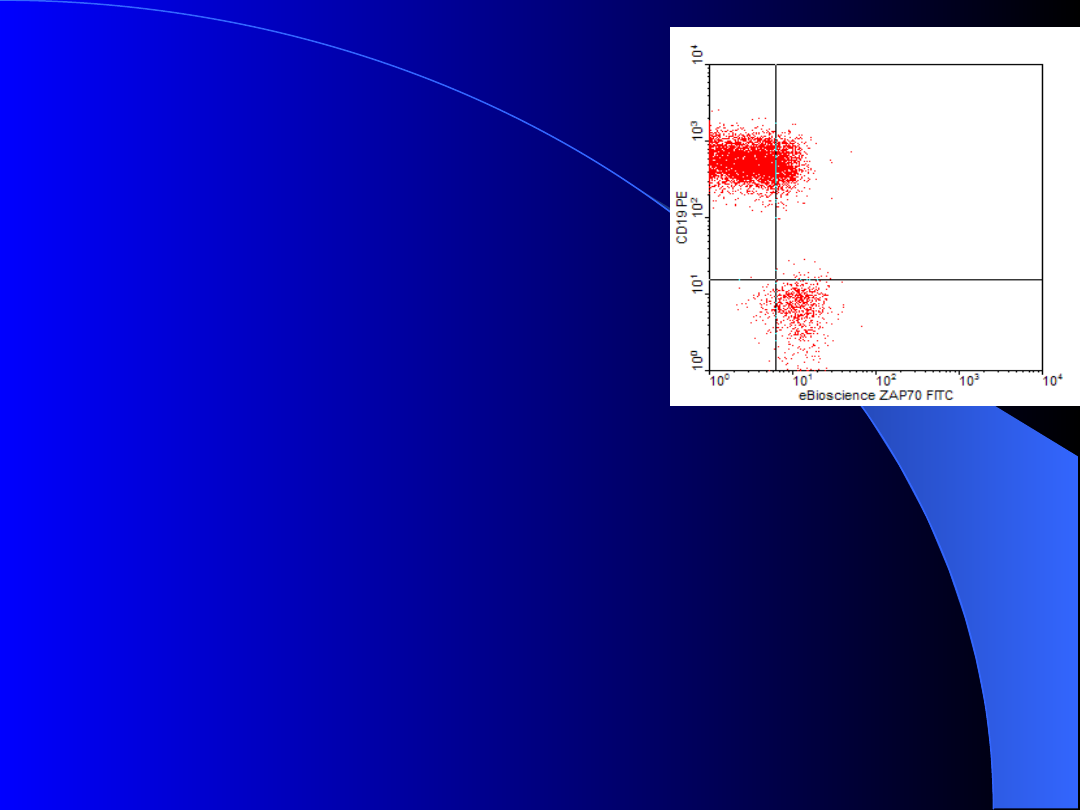
Nowy czynnik
Nowy czynnik
rokowoniczy
rokowoniczy
ZAP-70
ZAP-70
Zalety:
- można oznaczać rutynowo
przy użyciu przeciwciał
monoklonalnych na cytometrze przepływowym
- dobrze koreluje z mutacją genu dla IgVH
obecna mutacja genu dla IgVH brak ekspresji
ZAP-70
brak mutacji genu dla IgVH wysoka ekpresja
ZAP-70)

ZAP -70
ZAP -70
1) Gen kodujący białko
ZAP – 70
fizjologicznie
występuje w limfocytach T
2) Białko
ZAP-70
to kinaza tyrozynowa,
niezbędna
limfocytom
T
do
przewodzenia sygnału.
3) W normalnych limfocytach B, białko
ZAP-70
fizjologicznie nie występuje.
4) Wzrost ekspresji białka
ZAP-70
występuje u chorych z agresywną
postacią CLL.
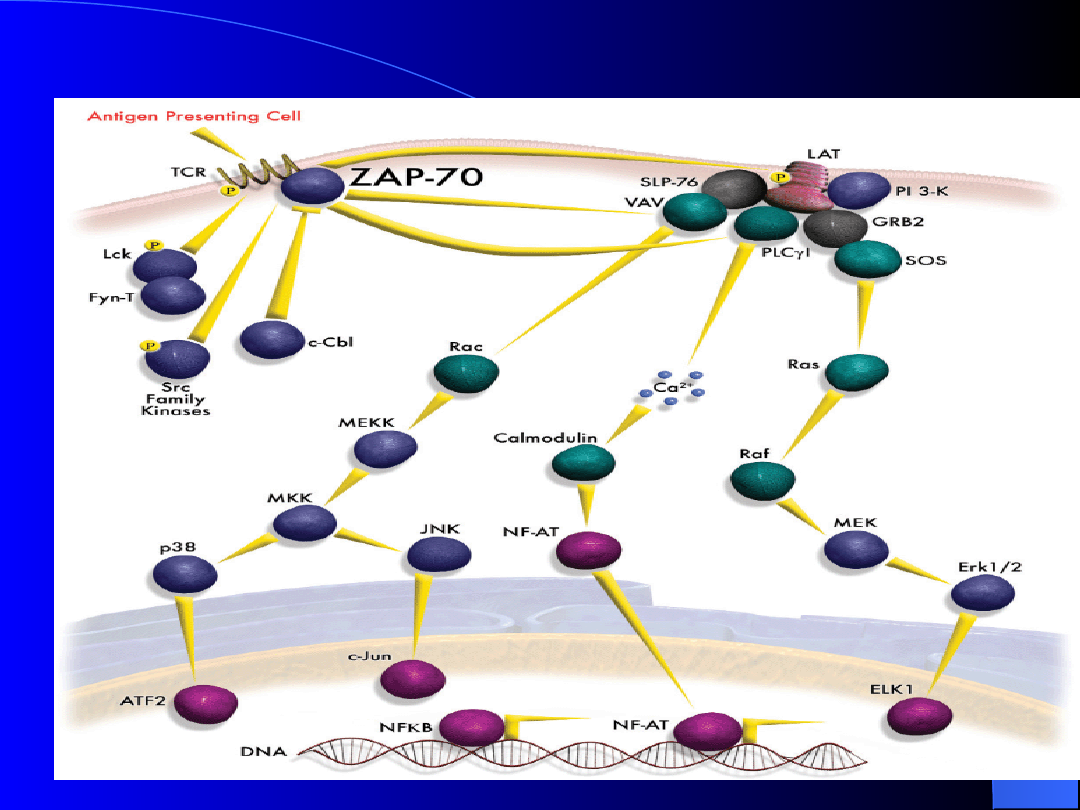
ZAP-70 - fizjologia
ZAP-70 - fizjologia

Czynniki związane z
Czynniki związane z
mutacją IgVH:
mutacją IgVH:
ZAP-70,
LPL,
Dystrofina,
Gravina,
BCL-7A
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
Wyszukiwarka
Podobne podstrony:
przewlekłe białaczki limfocytowe
przewlekła białaczka limfocytowa
Farmakoterapia przewlekłej białaczki szpikowej new
Białaczki limfocytowe
Przewlekła białaczka limfatyczna
Przewlekła białaczka szpikowa jest chorobą nowotworową szpiku kostnego i krwi
Przewlekła białaczka szpikowa(1), Pierwsza Pomoc Przedmedyczna, Medycyna, Onkologia
Przewlekła białaczka szpikowa
Bialaczki limfocytowePlasmocytoma, Analityka Medyczna, V semestr, Patofizjologia
Współczesne zasady leczenia przewlekłej białaczki szpikowej
13 Przewlekła białaczka szpikowa
Wykład 11 Przewlekłe białaczki
Etiopatogeneza przewleklej bialaczki limfatycznej AB
Ostre białaczki i przewlekłe zespoły mieloproliferacyjne
więcej podobnych podstron