
Przewlekłe białaczki
limfatyczne

Przewlekłe białaczki
limfatyczne
• Przewlekła białaczka limfocytowa
B-komórkowa (B-CLL)
• Białaczka prolimfocytowa (PLL)
• Białaczka włochatokomórkowa
(HCL)
• Białaczka z dużych ziarnistych
limfocytów(LGL)

Epidemiologia CLL
• Najczęściej występująca białaczka w
Europie
i Ameryce Płn u dorosłych
• Zapadalność roczna 3,5/100.000 a po 60
r.ż. 20/100.000
• Mediana wieku w trakcie rozpoznania ~
65lat
• M:K=2:1
• >50% przypadkowych rozpoznań
• ~10% występuje rodzinnie (CLL+inne
limfoproliferacje)

Diagnostyka CLL
1. Badanie podmiotowe
2. Badanie przedmiotowe
3. Badania laboratoryjne
4. Badania obrazowe

Badanie podmiotowe
• Objawy B ~10 % chorych
• Objawy wynikające z organomegalii
• Objawy wynikające z cytopenii:
-małopłytkowość
skaza krwotoczna
-niedokrwistość
osłabienie
-hemoliza żółtaczka
• Objawy wynikające z niedoborów
odporności
-zakażenia bakteryjne, grzybicze, wirusowe

Badanie przedmiotowe
• limfadenopatia ~90% chorych
• splenomegalia ~50% chorych
• hepatomegalia ~15% chorych
• powiększenie migdałków
• zajęcie pierścienia Waldeyera
• zmiany skórne
-nacieki
-żółtaczka

Badania laboratoryjne (1)
• Krew obwodowa
-limfocytoza
-cienie Gumprechta
-niedokrwistość ~10% chorych
-małopłytkowość ~3% chorych
• Szpik
-biopsja aspiracyjna >30% limfocytów
-trepanobiopsja
typ nacieku

Badania laboratoryjne(2)
• LDH
• Β2-mikroglobulina
• Immunoglobuliny
• Odczyn Coombsa
• Kinaza tymidynowa
• sCD23

Badania laboratoryjne (3)
• Immunofenotypizacja
CD
CLL
PLL
HCL
MCL
FL
CD5
++
-/+
-
++
-/+
CD10 -
-/+
-
-/+
++
CD19
++
++
+++
++
++
CD20 +
+++
+++
++
++
CD23
++
++
-
-
-/+
CD25 -
-/+
+++
-
-

Badania laboratoryjne(3)
Immunofenotypizacja w CLL
- minimalny panel antygenów CD5, CD19,
CD20,
CD23,
immunoglobuliny
powierzchniowe
- brak innych poza CD5 antygenów pan-T
- niska
ekspresja
powierzchniowych
immunoglobulin,
CD20,
CD79b
w
porównaniu do prawidłowych komórek B
- ekspresja klonalnych łańcuchów kappa,
lambda
- ZAP70 i CD38 nie są badaniami rutynowymi

Badania laboratoryjne(4)
• Badania cytogenetyczne i
molekularne
zaburzenie
cytogenetyka
FISH
klonalne
del 13q14
trisomia 12q
del
11q23-/ATM
del 17p-/p53
del 6q21
30-50%
7-11%
13-19%
6-13%
1-5%
1%
82%
36-45%
11-14%
8-25%
3-7%
2%

Rozpoznanie B-CLL
krew obwodowa
1996 wg NCI - WG
- limfocytoza > 5 G/l
2005
- stały wzrost bezwzględnej liczby limfocytów o
klonalnym charakterze (B-CLL) /Binet/
- CLL = obecność jakichkolwiek limfocytów o
charakterze klonalnym, ze stale utrzymującym
się wzrostem ich liczby /Rawstron/
*średnia liczba komórek o fenotypie B-CLL 0,013x10
9
/L u 3,1% zdrowych
ludzi=13 komórek B-CLL/µl (3-1458/µl)

Kryteria diagnostyczne
CLL
1.
Limfocytoza krwi obwodowej >5 G/l
2.
Limfocyty morfologicznie dojrzałe, a prolimfocyty
i komórki limfoplazmocytoidalne
stanowią<55%
3. Limfocyty w biopsji aspiracyjnej szpiku >30%
komórek, przy prawidłowej komórkowości szpiku
4. Monoklonalna ekspresja łańcuchów lekkich
immunoglobulin
5. Ekspresja markerów linii B (CD19, CD20, CD23)
6. Ekspresja
CD5
Wg NCI
Konieczne spełnienie wszystkich kryteriów

Klasyfikacja stopnia zaawansowania
w/g Rai i Bineta
Rai
średni czas przeżycia (lata)
0 – limfocytoza > 5 G/l > 12,5
1 – limfocytoza > 5 G/l + powiększenie węzłów chłonnych
8
2 – limfocytoza > 5 G/l + spleno- i/lub hepatomegalia 6
3 – limfocytoza > 5 G/l + Hb < 11 g/dl 1,5 - 2
4 – limfocytoza > 5 G/l + Plt < 100 G/l 1,5 – 2
Binet
A – zajęcie do 2 obszarów tkanki chłonnej
*
> 10
B – zajęcie > 2 obszarów tkanki chłonnej
*
5
C – niedokrwistość – Hb < 10 g/dl
2
i/lub małopłytkowość – Plt < 100 G/l
*powiększenie węzłów chłonnych szyjnych, pachowych, pachwinowych, śledziony,
wątroby
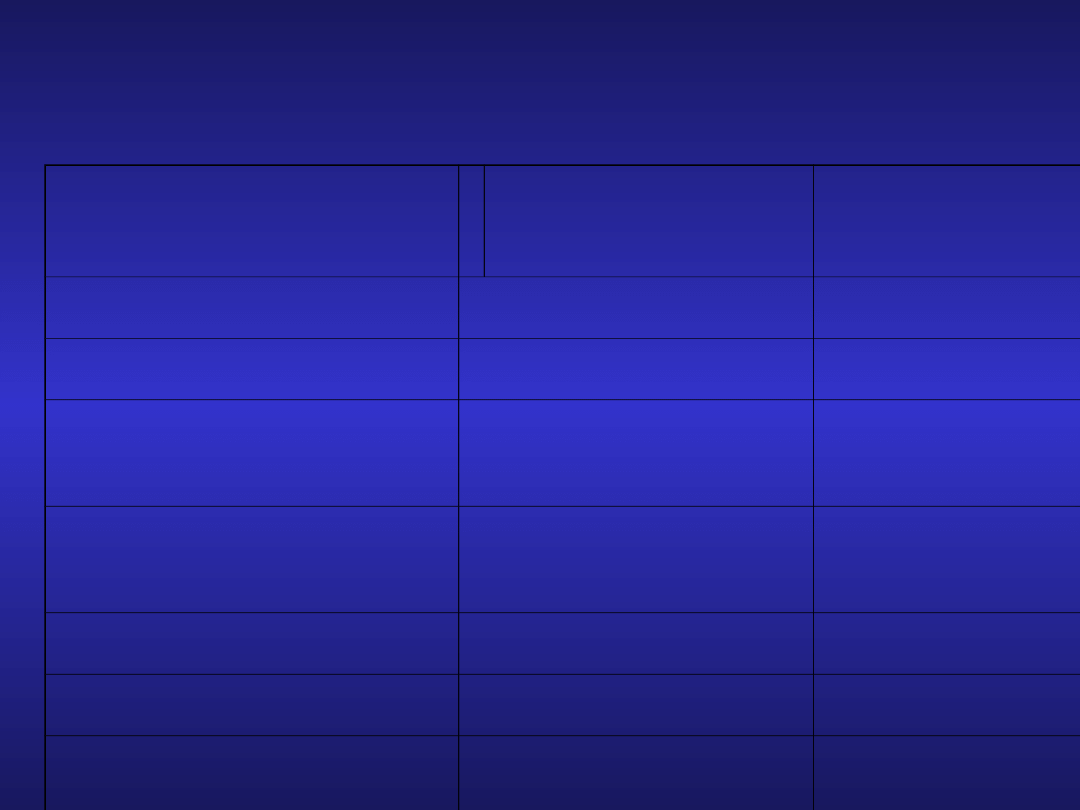
Klasyczne czynniki
prognostyczne
w CLL
czynnik
korzystny
niekorzystn
y
Rai
0
I-IV
Binet
A
B,C
Typ nacieku
w trepanobiopsji
grudkowy
rozlany
Odsetek limfocytów
w szpiku
<80%
>80%
Leukocytoza
<50 G/L
>50 G/L
Prolimfocyty we krwi
<10%
>10%
Czas podwojenia
limfocytozy
<12 miesięcy
>12
miesięcy
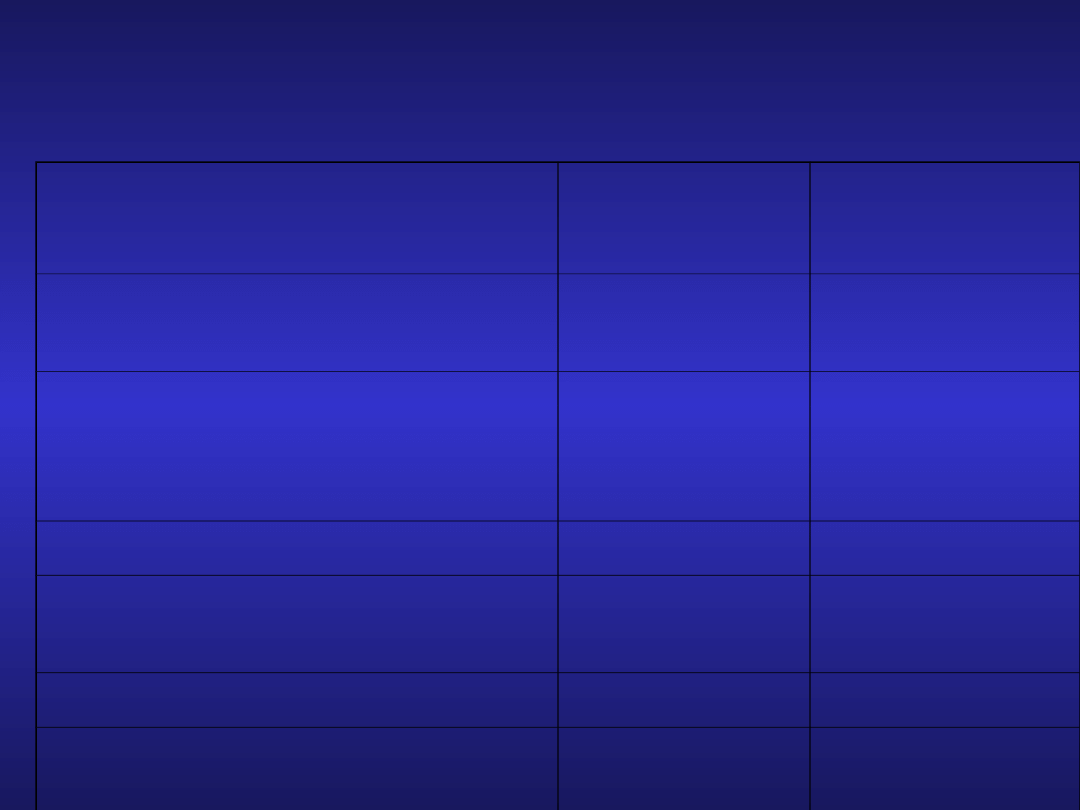
Inne czynniki prognostyczne
w CLL
czynnik
korzystny
niekorzystn
y
LDH, β2-mikroglobulina,
kinaza tymidynowa, sCD23
prawidłowe podwyższone
cytogenetyka
prawidłowa
izol.
del(13q)
del(11q)
del(17p)
ekspresja CD38
<30%
>30%
gen IgV
H
zmutowany
niezmutowan
y
ZAP 70
brak
zwiększona
ekspresja β2 integryny
wskaźnik angiogenezy
VEGF
zwiększone

Czynniki prognostyczne w CLL.
Badania cytogenetyczne i
molekularne
zaburzenie
częstoś
ć
Czas
przeżycia
(miesiące)
uwagi
del
11q23-/ATM
8-25%
79-117
•Brak mutacji IgV
H
•Znaczna limfadenopatia
•Charakter progresywny
•Wczesne nawroty po
autoBMT
del 17p-/p53
3-7%
32-47
•Brak mutacji IgV
H
•Oporność:chlorambucil,
fludarabinę i rituximab
•Atypowa morfologia
limfocytów
trisomia 12q
13-19%
114-122
•Charakter progresywny
•Atypowa morfologia
limfocytów
del 13q14
36-45%
133-292
•Stabilny przebieg
•Obecna mutacja IgV
H
•Typowa morfologia
limfocytów

CLL a mutacja IgV
H
CLL
obecna mutacja IgV
H
(H/L)
brak mutacji
IgV
H
Brak mutacji IgV
H
Obecna mutacja IgV
H
(H/L)
•Ch.progresywna
•Krótszy czas przeżycia
•Korelacja z:
-CD38 w 75%
-ZAP 70 w 92%
-mediana przeżycia
ZAP(+)/ZAP(-)=9,8:24,4 lat
CLL-postępowanie
terapeutyczne
obserwacja
Leczenie wg
standardów
Wcześniejsza terapia
w zależności od
czynników prognostycznych

CLL-postępowanie
terapeutyczne
Watch and wait
-Rai 0 – II, Binet A
-Brak objawów ogólnych
-Brak masywnej limfadenopatii (> 3 cm)
i hepatosplenomegalii (>5 cm)

CLL-postępowanie
terapeutyczne
Rozpoczęcie terapii
1. Rai III-IV, Binet C
2. Aktywna/progresywna postać choroby
- objawy B
-
czas podwojenia leukocytozy < 6(12) miesięcy
-
wzrost limfocytozy > 50%w ciągu 2 miesięcy
lub przewidywany czas podwojenia limfocytozy < 6 miesięcy
- zespół nadlepkości z powodu b.wysokiej limfocytozy
- cytopenie wtórne do infiltracji szpiku
-
znaczne (>10cm) lub narastające powiększenie węzłów chłonnych
bądź znacznego stopnia splenomegalia z objawami uciskowymi
-
zespół Richtera
-
autoimmunologiczna niedokrwistość i małopłytkowość
nie reagujące na glikokortykoterapię
-
hpogammaglobulinemia ze zwiększoną podatnością na zakażenia

CLL-postępowanie
terapeutyczne
Wcześniejsza terapia
w zależności od
czynników prognostycznych
•Mutacja IgV
H
•Obraz cytogenetyczny
•Ekspresja CD38
•ZAP 70
•Markery surowicze
•Rodzaj nacieku
•Stopień infiltracji szpiku

CLL-postępowanie
terapeutyczne
Leczenie w zależności od wieku
Konwencjonalna terapia nie
doprowadza do wyleczenia
Wczesne i agresywne leczenie
może prowadzić do wyleczenia
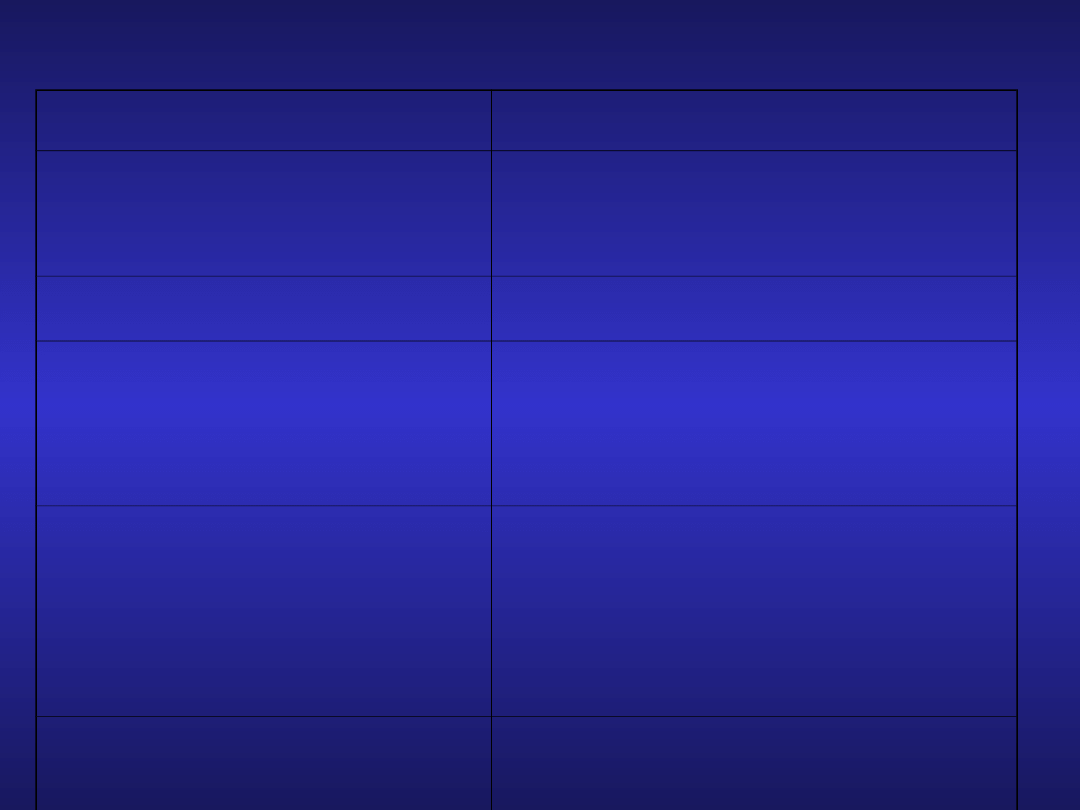
CLL-stosowane leki
Grupa leków
preparaty
Leki alkilujące
Chlorambucil
Cyclophosphamid
polichemioterapia
COP, CHOP
Analogi nukleozydów
purynowych
Fludarabina
Kladrybina (2-CdA)
Pentostatyna
Przeciwciała
monoklonalne
Rituximab
Ofatumumab
Alemtuzumab
Luminiximab
Inne
Bendamustyna,Thalidomid,
Antysense, szczepionki

CLL-leczenie pierwszego
rzutu
Chlorambucil +/- prednison
Analogi puryn
-monoterapia
-z cyclophosphamidem (FC)
-z cyclophosphamidem i rituximabem
(FCR)
Inne
-bendamustyna(2008-FDA)

Chlorambucil w CLL
• W zależności od dawki 60-90% OR
(overall
response)
• Monoterapia+/- prednison
• Dołączenie prednisonu zwiększa OR lecz
nie wydłuża czasu przeżycia
• Niski odsetek CR (<5%)
• Różne schematy dawkowania

Sposoby dawkowania
chlorambucilu
4-8 mg/m
2
przez 4-8 tygodni
15-30 mg/m
2
co 2-4 tygodnie
12 mg/m
2
przez 5-7 dni, 1 raz w miesiącu
0,1 mg/kg/dobę
15 mg/dobę do osiągnięcia CR lub
wystąpienia toksyczności 3 stopnia,
maksymalnie 6 miesięcy+leczenie
podtrzymujące 5-15 mg 2x/tydzień (OR-
90%)

Polichemioterapia w CLL
Nie stwierdzono większej skuteczności
schematów skojarzonych nad
chlorambucilem
• Brak różnic w czasie przeżycia pacjentów leczonych
CHOP vs chlorambucil w średnich dawkach
• Wyższy odsetek OR i wyższa mediana czasu przeżycia
po chlorambucilu w dużych dawkach vs CHOP
(89,5%vs75%; 68 vs 47 miesięcy)
*CLL Trialists Collaborative Group

Analogi nukleozydów
purynowych w CLL
Monoterapia
• Fludarabina 25-30 mg/m
2
x 5 dni
i.v.
• Fludarabina 40 mg/m
2
x 5 dni p.o.
• Kladrybina 0,12 mg/kg x 5 dni i.v.
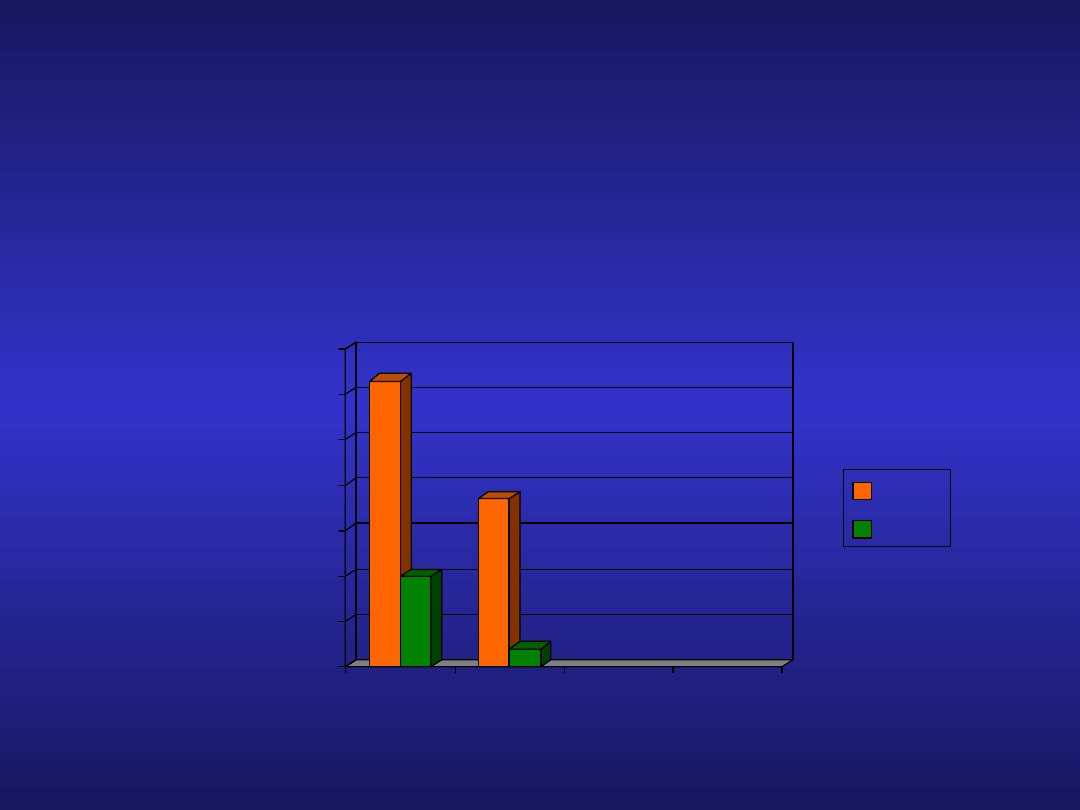
Analogi nukleozydów purynowych w
CLL
Badanie Rai
Fludara vs chlorambucil w terapii
pierwszego rzutu
0,00%
10,00%
20,00%
30,00%
40,00%
50,00%
60,00%
70,00%
Setki
F(n=170)
Clb(n=181)
ORR
CR

Analogi nukleozydów
purynowych w CLL
Leczenie skojarzone
• FC-Fludarabina 25 mg/m
2
i.v. +
cyclophosphamid 250 mg/m
2
i.v. x 3 dni
• FC-Fludarabina 24 mg/m
2
p.o. +
cyclophosphamid 150 mg/m
2
x 5 dni
• CC-kladrybina 0,12 mg/kg +
cyclophosphamid 250 mg/m
2
i.v. x 3
dni

Analogi nukleozydów purynowych w
CLL
Badanie CLL4
• Porównanie leczenia Clb, F, FC w leczeniu pierwszego
rzutu
(Catovsky i wsp.,ICR, UK)
CLL
Chlorambucil
10 mg/m
2
x 7 dni p.o.
Fludara 25 mg/m
2
i.v.
x 5 dni
lub
Fludara 40 mg/m
2
p.o.
x 5dni
Fludara 25 mg/m
2
+
Cyclophosphamid 250 mg/m
2
i.v.
x 3 dni
lub
Fludara 24 mg/m
2
p.o. +
Cyclophosphamid 150 mg/m
2
x 5 dni
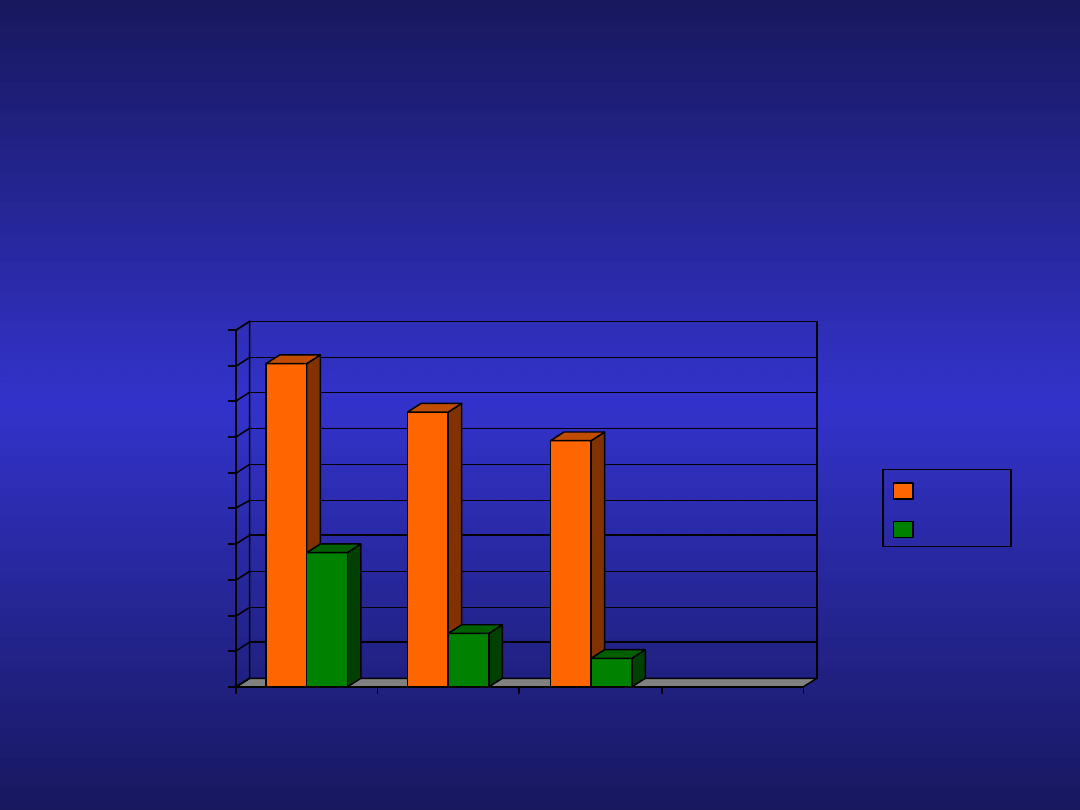
Analogi nukleozydów purynowych w
CLL
Badanie CLL4
• Skuteczność terapii
0,00%
10,00%
20,00%
30,00%
40,00%
50,00%
60,00%
70,00%
80,00%
90,00%
100,00%
Setki
FC(n=176)
F(n=176)
Clb(n=309)
ORR
CR
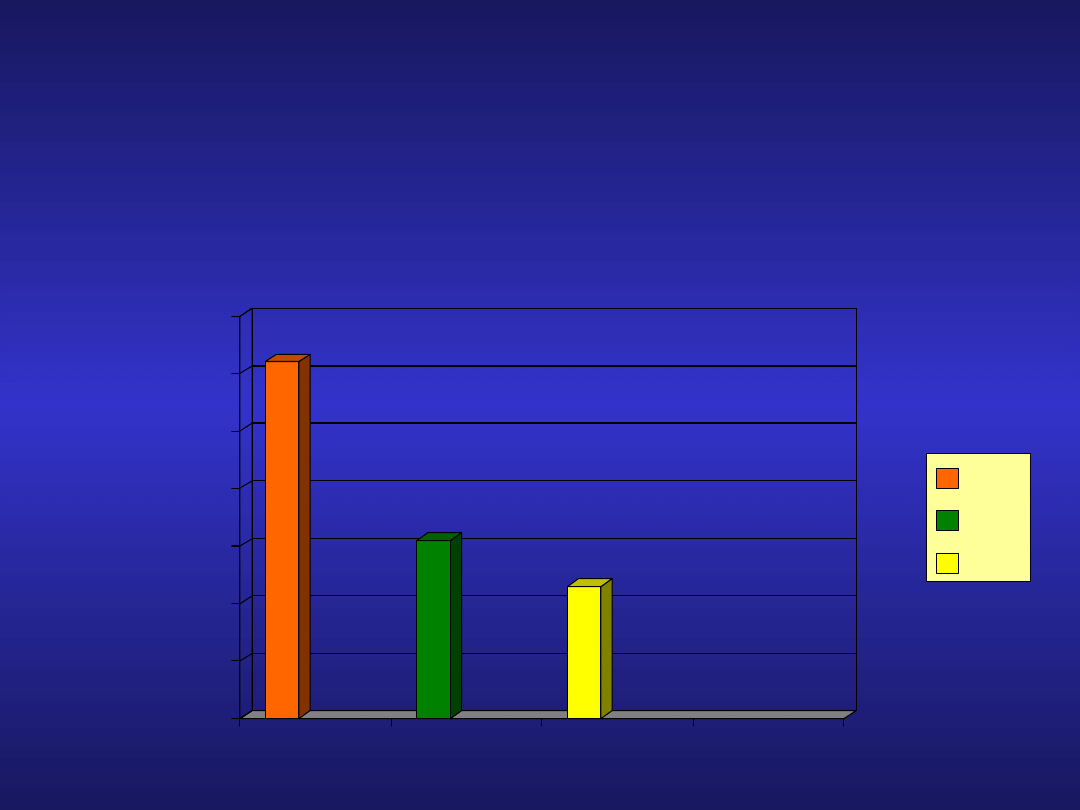
Analogi nukleozydów purynowych w
CLL
Badanie CLL4
• 3
letni czas przeżycia bez progresji choroby
0,00%
10,00%
20,00%
30,00%
40,00%
50,00%
60,00%
70,00%
Setki
FC
F
Clb
FC
F
Clb

Analogi nukleozydów purynowych w
CLL - leczenie pierwszej linii.
Podsumowanie.
Większa skuteczność w porównaniu
z lekami alkilującymi w monoterapii
Korzystne może być kojarzenie ze steroidami
Brak różnicy w medianie czasu przeżycia pacjentów
leczonych fludarabiną vs chlorambucilem (Rai)
Większy odsetek odpowiedzi na leczenie i CR
w terapii skojarzonej z
cyclophoshamidem(>90%;21%)
Podobna skuteczność fludarabiny i kladrybiny

Bendamustyna
• Związek cytotoksyczny łączący cechy leków
alkilujących i analogów puryn
• Hamuje syntezę DNA, RNA, białek
• Stosowana w pierwszej i kolejnych liniach
leczenia
• Lepsze wyniki leczenia w porównaniu z Clb
• Możliwość łączenia z innymi cytostatykami
i rituximabem
• Dawka 100 mg/m
2
iv przez 2 dni co 4 tyg.
Leczenie nawrotu lub oporności na
terapię pierwszego wyboru
Nawrót lub progresja choroby
>12 miesięcy
<12
miesięcy
Powtórzenie
tej samej terapii
Analogi puryn
u leczonych uprzednio chlorambucilem
Dodanie cyclophosphamidu
u leczonych uprzednio
analogiem puryn w monoterapii
+/-rituximab, alemtuzumab

Chronic lymphocytic lekumia with high risk
genetics; disease progression and resistance to
treatment
S. Stilgenbauer
11q - limfadenopatia
- szybka progresja choroby
17q – związek z opornością na leki alkilujące, fludarabinę i
rituximab
- krótki czas przeżycia
Mutacje genów dla zmiennego regionu łańcucha ciężkiego
immunoglobulin (IgV
H
)
(+) dłuższy czas przeżycia
(-) krótszy czas przeżycia
Ograniczona wartość ZAP70 w przypadkach CLL z dodatkowymi
czynnikami ryzyka (11q-, 17p-, V3-21 usage)
np. V3-21 usage, V
H
zmutowane a ZAP70 +
11q-, 17p-, V
H
niezmutowane a ZAP70-
Leuk Lymphoma 46 (suppl 1): S6, 2005

Wnioski
1.
Spodziewana progresja w niskozaawansowanych CLL
(Binet A) w przypadkach V
H
niezm, +12q, 11q-, 17p-,
2.
Oporność na leczenie fludarabiną u chorych z 17p-
3.
Skuteczność terapii alemtuzumabem w przypadku del.
17p-
4.
Rozpatrzenie wskazań do auto/alloBMT u chorych ze
złymi genetycznymi czynnikami ryzyka

Wnioski
Chemioimmunoterapia
- ↑ odsetka remisji
- Nie doprowadza do wyleczenia
- Nowy kierunek terapii w przypadkach
zaburzeń cytogenetycznych 17p-, 11q-

Non-myeloablative stem cell
transplantation for chronic lymphocytic
leukemia
I. Khouri
Korzyści i ograniczenia auto i allo SCT w CLL
Auto SCT
Konw. Allo SCT
NMA allo SCT
Bariera wieku
+
+++ +
Kontaminacja kom. neo +++ -
-
Wtórny MDS +
-
-
GVHD -+++
++
Ryzyko zgonu < 5% 30-40% < 15%
GVL
-+++
+++
Potencjalne wyleczenie -
+++
+++
Leuk Lymphoma 46 (suppl 1): S11, 2005

Chemotherapy with or without oblimersen
sodium (Bcl-2 antisense; Genasense, G3139) in
advanced chronic lymphocytic leukemia: report
of large radomized study
K. Rai i wsp.
•
Bcl-2 - proteina antyapoptotyczna
•
Antisense – zahamowanie produkcji Bcl-2 i wzrost apoptozy
•
Bax
•
•
Terapia: Genasense 3 mg/kg/d w dniach 1- 7
FC w dniach 5 – 7
max. 6 cykli
Wnioski
Dołączenie do terapii FC Genasense znacznie
podwyższa odsetek CR/PR (17% vs 7%) u pacjentów
ze wznową lub oporną postacią CLL
Leuk Lymphoma 46 (suppl 1): S11, 2005
Bcl-2
Kaspazy apoptoza

Przewlekłe białaczki
limfatyczne
• Przewlekła białaczka limfocytowa
B-komórkowa (B-CLL)
• Białaczka włochatokomórkowa
(HCL)
• Białaczka prolimfocytowa (PLL)
• Białaczka z dużych ziarnistych
limfocytów(LGL)

Białaczka
włochatokomórkowa
(hairy cell leukemia)
• Choroba dotycząca dojrzałych
limfocytów B (rzadko T) z
obecnością charakterystycznych
komórek limfoidalnych z
wypustkami cytoplazmatycznymi
• M:K=4:1
• Średni wiek przy rozpoznaniu 52
lata

Białaczka
włochatokomórkowa-
cechy charakterystyczne
• Powiększenie śledziony (>80 % chorych)
• Pancytopenia (60-80%)
• „włochate” komórki (do 20 % leukocytów)
• Często nie możliwa do uzyskania biopsja
aspiracyjna szpiku
• Bardzo skuteczne leczenie kladrybiną
(remisja >75% chorych)
• Inne leki stosowane w terapii: Ifα,
Fludarabina, rituksimab

Przewlekłe białaczki
limfatyczne
• Przewlekła białaczka limfocytowa
B-komórkowa (B-CLL)
• Białaczka włochatokomórkowa
(HCL)
• Białaczka prolimfocytowa (PLL)
• Białaczka z dużych ziarnistych
limfocytów(LGL)

Białaczka prolimfocytowa
(PLL)
B-PLL (80%)
• Średni wiek przy
rozpoznaniu-70 lat
• Znaczna slenomegalia
• Prolimfocyty we krwi
obwodowej>55%
• Często WBC>100G/L
• Lifadenopatia u ok.30%
chorych
• Oporność na Clb i
cyklofosfamid
• Krótki czas
przeżycia<3lat
• Leczenie: analogi puryn,
immunoterapia
(rituksimab,
alemtuzumab)
T-PLL (20%)
• Częściej limfadenopatia
• Nacieki w skórze
• Wysięki w jamach
surowiczych
• Przebieg bardziej
agresywny niż B-PLL
• Leczenie: analogi puryn,
alemtuzumab

Przewlekłe białaczki
limfatyczne
• Przewlekła białaczka limfocytowa
B-komórkowa (B-CLL)
• Białaczka włochatokomórkowa
(HCL)
• Białaczka prolimfocytowa (PLL)
• Białaczka z dużych ziarnistych
limfocytów(LGL)

Białaczka z dużych ziarnistych
limfocytów
large granular lymphocyte leukemia
(LGL)
T-LGL (85%)
• Występuje głównie u ludzi
starszych
• Neutropenia powikłana
infekcjami bakteryjnymi
• Współistniejące zaburzenia
immunologiczne, RZS
• Limfocytoza zwykle<20 G/L
• Leczenie postaci łagodnej:
leki immunosupresyjne
(Mtx, CyA,
prednison,cyclofosfamid)-
wskazane leczenie
podtrzymujące
• Leczenie postaci
agresywnej:
polichemioterapia(CHOP,
Ara-C)
NK-LGL
• Średni wiek przy
rozpoznaniu-40 lat
• Często objawy ogólne
• Znaczna
hepatosplenomegalia
• Przebieg b.agresywny niż
T-LGL
• WBC>niż w T-LGL
• Rzadko neutropenia a
zawsze niedokrwistość+/-
małopłytkowość
• Leczenie jak T-LGL
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
Wyszukiwarka
Podobne podstrony:
przewlekła białaczka limfocytowa
przewlekła białaczka limfocytowa 2
Farmakoterapia przewlekłej białaczki szpikowej new
Białaczki limfocytowe
Przewlekła białaczka limfatyczna
Przewlekła białaczka szpikowa jest chorobą nowotworową szpiku kostnego i krwi
Przewlekła białaczka szpikowa(1), Pierwsza Pomoc Przedmedyczna, Medycyna, Onkologia
Przewlekła białaczka szpikowa
Bialaczki limfocytowePlasmocytoma, Analityka Medyczna, V semestr, Patofizjologia
Współczesne zasady leczenia przewlekłej białaczki szpikowej
13 Przewlekła białaczka szpikowa
Wykład 11 Przewlekłe białaczki
Etiopatogeneza przewleklej bialaczki limfatycznej AB
Ostre białaczki i przewlekłe zespoły mieloproliferacyjne
więcej podobnych podstron