
ZABURZENIA UKŁADU
HEMOSTAZY

ELEMENTY HEMOSTAZY
• Ściana naczyń krwionośnych
– Śródbłonek: m.in. uwalnianie
prostacykliny, NO, t-PA, vWf,
ekspresja trombomoduliny
• Płytki krwi
– Utworzenie czopu płytkowego
– Udział w krzepnięciu krwi
• Układ krzepnięcia

Szlaki aktywacji
Szlaki aktywacji
krzepnięcia
krzepnięcia
XI
I
XIIa
XI
XIa
X
Xa
X
Protrombin
a
Trombina
Fibrynog
en
Fibryna
XIII
Ca
XIIIa
IX
IXa
VII
VII
a TF
V
Va
VII
I
VIIIa
Ca
PL
Ca
PL
Mech.
wewnątrzpochodny
Mech.
zewnątrzpochodny
TF
PK
WK

UKŁAD INHIBITORÓW
KRZEPNIĘCIA
• Antytrombina (AT)
– unieczynnia większość czynników krzepnięcia, gł.
trombinę, Xa, IXa, XIIa, XIa
• Białko C i białko S
– białko C unieczynnia czynniki Va i VIIIa, białko S jest
jego kofaktorem
• Inhibitor zewnątrzpochodnego toru krzepnięcia
– – TFPI w obecności Xa wiąże i inaktywuje kompleks
TF-VIIa
• Neksyny proteazowe, np. aneksyna V obecna w ścianie
naczyń krwionośnych, wpółzawodniczy z czynnikami
krzepnięcia o fosfolipidy

WRODZONA TROMBOFILIA
• Najczęstsza przyczyna wrodzonej trombofilii -
oporność na aktywowane białko C (activated
protein C resistance - APC-r)
– najczęściej związana z mutacją Leiden cz. V
• Inne:
• mutacja genu protrombiny
• niedobór antytrombiny III
• niedobór białka C
• mutacja genu protrombiny
UKŁAD
FIBRYNOLITYCZNY
Prekalikreina
Kalikreina
XIIa
Plazminogen
Plazmina
t-PA
u-PA
PAI-1
Fibrynogen,
Fibryna Fibryna
FDP = fragmenty X, Y,
D, E
D-dimer
2-
antyplazmina

INHIBITORY
FIBRYNOLIZY
• Inhibitor aktywatora
plazminogenu (PAI)
2
-antyplazmina
• Inhibitor fibrynolizy aktywowany
przez trombinę (TAFI) –
odszczepia od fibryny C-końcowe
reszty lizyny i argininy

PRZYCZYNY SKAZY
KRWOTOCZNEJ
• Najczęstszą nabytą przyczyną
krwawień jest małopłytkowość
• Najczęstszą wrodzoną przyczyną
krwawień jest choroba von
Willebranda

PODEJŚCIE DO CHOREGO
ZE SKAZĄ
• Wywiad
– Czas i okoliczności wystąpienia
– Krwawienia po zabiegach i ekstrakcjach
zębów
– Charakter miesiączek
– Zwyczaje żywieniowe, alkohol
– Leki: antybiotyki i inne, zioła
– Czynniki toksyczne
– Współistniejące choroby

Ciężkie
Możliwe do
opanowania
uciskiem
Krwawienia po
operacjach
Z
opóźnieniem
Bezpośrednio po
urazie
Czas
wystąpienia
krwawienia
Siniaki,
krwiaki,
krwawienia
do tkanek
miękkich,
dostawowe
Śluzówkowe: z
nosa, śluzowek j.
ustnej, dróg
moczowych, p.
pok.,
przedłużone
miesiączki,
wybroczyny
Charakter
krwawień
Skazy
osoczowe
Skazy
płytkowo-
włośniczkowe
Objawy

PODSTAWOWE BADANIA
UKŁADU HEMOSTAZY
• Liczba płytek krwi
• Czas krwawienia
• Czas protrombinowy
• Czas aktywowanej częściowo
tromboplastyny
• Czas trombinowy

CZAS KRWAWIENIA
• Czas który upływa od momentu zranienia skóry do
momentu ustania upływu krwi
• Metoda Duke`a – do 5 minut, metoda Ivy– do 8 minut
• Zależy od sprawności hemostatycznej płytek krwi i
naczyń, nie zależy od osoczowych czynników krzepnięcia
• Przyczyny wydłużenia:
– małopłytkowość
– zaburzenia funkcji płytek (wrodzone, np. trombastenia
Glanzmanna, nabyte, np. mocznica, leki p-płytkowe)
– choroba von Willebranda
– skazy naczyniowe

CZAS PROTROMBINOWY
VII
+ TF
X
V
Pl
Ca
II
Trombina
I
Skrzep

CZAS PROTROMBINOWY
Przyczyny wydłużenia:
– Izolowany niedobór (najczęściej wrodzony)
czynników: VII, X, II, V lub fibrynogenu
– Złożony niedobór czynników zależnych (II,
VII, IX, X) lub niezależnych od witaminy K:
• niedobór witaminy K: antybiotykoterapia,
cholestaza, złe odżywianie
• leczenie doustnymi antykoagulantami
• choroby wątroby
– Antykoagulant tocznia
– DIC

CZAS PROTROMBINOWY
Sposoby wyrażania wyniku PT
• Wskaźnik Quicka:
– PT kontrolny (sek) : PT badany (sek) x 100%
– Norma 80-110%
• INR: międzynarodowy znormalizowany
współczynnik
– Uniezależnia wartość PT od rodzaju preparatu
tromboplastyny użytego w teście
– Umożliwia ujednolicenie pomiarów PT i
porównywanie wyników z różnych laboratoriów

CZAS CZĘŚCIOWEJ
CZAS CZĘŚCIOWEJ
TROMBOPLASTYNY PO
TROMBOPLASTYNY PO
AKTYWACJI (APTT)
AKTYWACJI (APTT)
XII
IX + VIII
XI
X
V
Pl
Ca
II
Trombina
I
Skrzep

CZAS CZĘŚCIOWEJ
TROMBOPLASTYNY PO AKTYWACJI
(APTT)
Przyczyny wydłużenia:
• Niedobór któregokolwiek z czynników
krzepnięcia za wyjątkiem czynnika VII
– np. hemofilia A – niedobór czynnika VIII lub
hemofilia B – niedobór czynnika IX, ciężka
postać choroby von Willebranda
• Obecność inhibitora: heparyny lub
krążącego antykoagulantu, np. toczniowego
• Choroby wątroby

CZAS TROMBINOWY
CZAS TROMBINOWY
Protrombina
Trombina
Monomer fibryny
+ FBP A + FBPB
Skrzep
Fibrynog
en

CZAS TROMBINOWY
CZAS TROMBINOWY
Przyczyny wydłużenia:
• Niedobór fibrynogenu: wrodzony lub
nabyty
choroby wątroby
leczenie fibrynolityczne
pierwotne skazy fibrynolityczne
DIC
• Obecność heparyny

PRZYCZYNY WYDŁUŻENIA
PT
I APTT
Wrodzone:
niedobór cz. VIII,
IX, XI, XII, prekalikreiny,
kininogenu
wielkocząsteczkowego, ch. von
Willebranda
Nabyte:
heparyna, inhibitory
cz. VIII, IX, XI lub XII,
antykoagulant tocznia
norm
a
Wrodzone:
niedobór czynnika
VII
Nabyte:
niedobór witaminy K,
choroby wątroby, poch.
dikumarolu, inhibitor cz. VII
norma
PRZYCZYNY
APTT
PT

PRZYCZYNY WYDŁUŻENIA
PT
I APTT
PRZYCZYNY
APTT
PT
Wrodzone:
niedobór
protrombiny, fibrynogenu,
cz. V lub X
Nabyte:
choroby wątroby,
DIC, przedawkowanie
heparyny lub doustnych
antykoagulantów,
inhibitory protrombiny,
fibrynogenu, cz. V lub X

INTERPRETACJA BADAŃ
UKŁADU KRZEPNIĘCIA
n
n
n
n
n
hemofilia
n
n
n
n
n
trombopa
t
n
n
n
n
małopłyt
k
n lub
n
n
n
n
naczynio
wa
Fibr
TT
APTT
PT
Plt
cz.
krwaw
schorze
nie

INTERPRETACJA BADAŃ
UKŁADU KRZEPNIĘCIA
n
n
n
n
n
v Willebr
DIC
Fibr
TT
APTT
PT
Plt
cz.
krwaw
schorzen
ie

SKAZY KRWOTOCZNE
OSOCZOWE
Wrodzone
• Choroba von
Willebranda
• Hemofilia A
• Hemofilia B
Nabyte
• Skazy na tle niedoborów
czynników krzepnięcia
zależnych od wit. K
• W chorobach wątroby
• W wyniku pojawienia
się patologicznych
antykoagulantów
• DIC
• Skazy fibrynolityczne

NIEDOBÓR CZYNNIKÓW
KRZEPNIĘCIA ZALEŻNYCH OD WIT.
K
• choroby wątroby
• niedobór wit. K:
– upośledzone wytwarzanie wit. K:
• brak flory bakteryjnej jelit (choroba krwotoczna
noworodków)
• wyjałowienie jelita przez antybiotyki
– upośledzone wchłanianie wit. K
• zespół złego wchłaniania
• zahamowanie wydzielania żółci do światła jelita (kamica,
nowotwór)
• wpływ leków (cholestyramina)
– upośledzone wykorzystanie wit. K:
• Leczenie doustnymi antykoagulantami

NIEDOBÓR CZYNNIKÓW
KRZEPNIĘCIA ZALEŻNYCH OD WIT.
K
• Objawy: objawy choroby podstawowej +
skaza: krwawienia z nosa, dziąseł, p.
pokarmowego, dróg rodnych, moczowych
• Badania laboratoryjne:
– PT, ewent. APTT
• Leczenie:
– leczenie choroby podstawowej
– wit. K iv
– FFP
– rVIIa

SKAZY W WYNIKU POJAWIENIA SIĘ
PATOLOGICZNYCH
ANTYKOAGULANTÓW
Alloprzeciwciała:
p-czynnikowi VIII:
• hemofilia A
• u kobiet po porodzie
• w przebiegu chorób
autoimmunologicz.
– objawy jak w
ciężkiej hemofilii
– leczenie:
immunosupresyjne,
immunoglobuliny
Autoprzeciwciała:
zsp.
antyfosfolipidowy
:
• p-ciała
antyfosfolipidowe
+ objawy
kliniczne

DIC
Zespół wykrzepiania
wewnątrznaczyniowego jest to
nabyty zespół zaburzeń hemostazy
wtórny do różnych schorzeń i
charakteryzujący się
jednoczesnym występowaniem
zakrzepicy i skazy krwotocznej

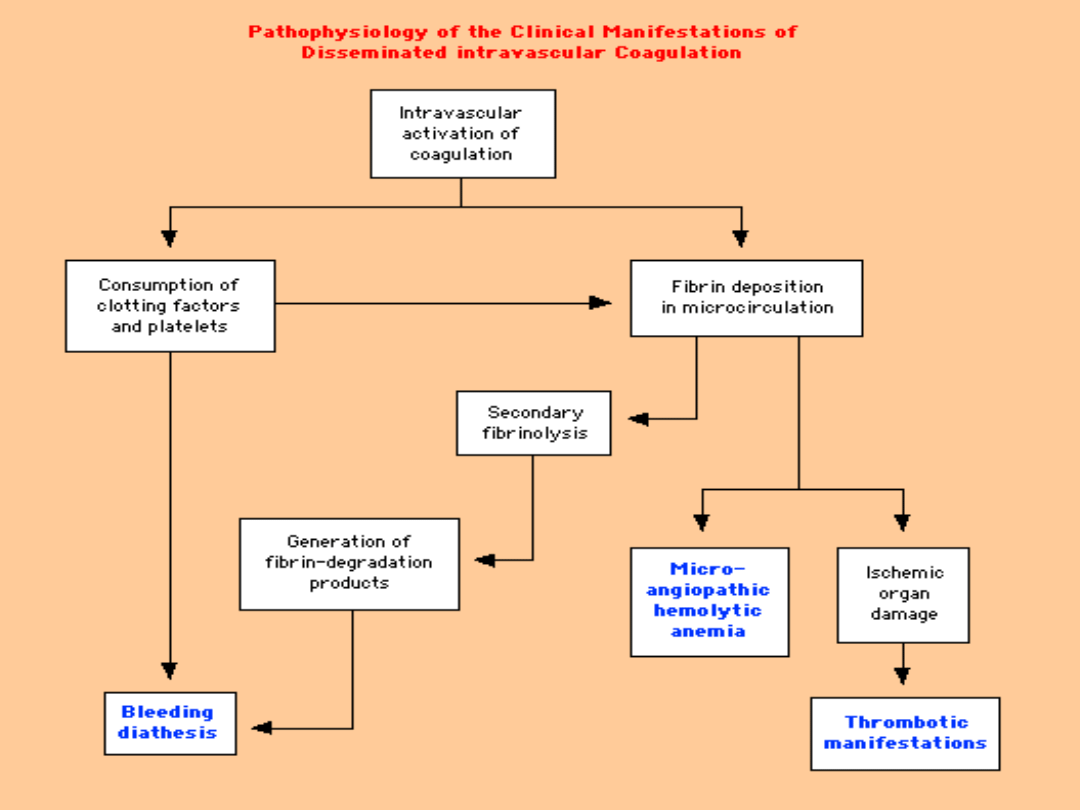
DIC - PATOGENEZA
• Fizjologicznie – produkcja trombiny i
odkładanie się fibryny ograniczone do
miejsca uszkodzenia. Proces ten jest
regulowany przez różne mechanizmy, jak np.
AT III lub PAI
• Gdy produkcja trombiny i mechanizmy
obronne wyczerpane trombina pojawia się
w krążeniu
DIC:
masywna
zakrzepica
gł. w
mikrokrążeniu niewydolność narządowa
zużywanie płytek i czynników krzepnięcia
wtórna aktywacja fibrynolizy
krwawienie
Pathop6.gif

DIC - PATOGENEZA
DIC – proces dynamiczny. Przebieg zależy od:
– Przyczyny
– Szybkości szerzenia się
•
Gdy powoli pojawia się nadmiar
prokoagulantów tendencja do zakrzepicy.
Dopóki wątroba kompensuje utratę
czynników krzepnięcia a szpik kostny utratę
płytek krwawienie nie występuje
obraz
przewlekłego skompensowanego DIC.
Klinicznie bez objawów albo powikłania
zakrzepowe żylne lub tętnicze

DIC - PATOGENEZA
• Gdy przebieg gwałtowny
masywna zakrzepica,
niewydolność narządowa, skaza
krwotoczna
obraz ostrego
zdekompensowanego DIC
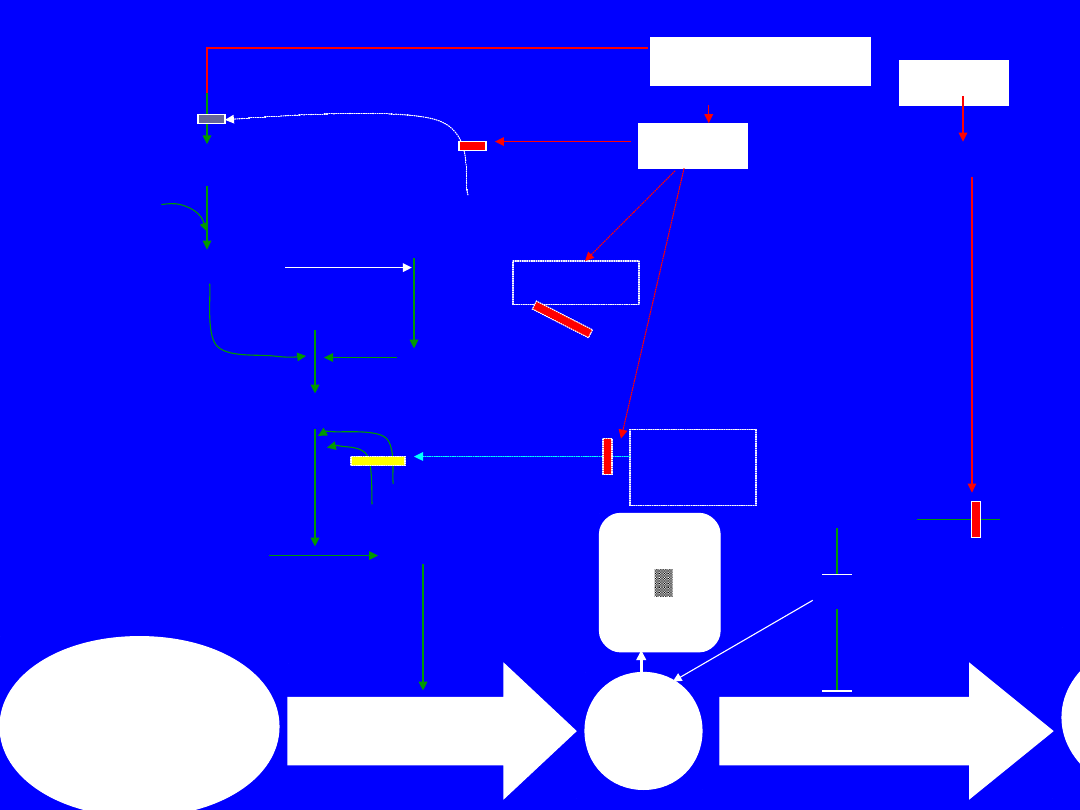
TF
IL-1 IL-6,
IL-8
TNF
TNF
Aktywatory
plazminoge
nu
Plazmino
gen
Plazmina
VIII
Va
TF-VIIa
IX
IXa
X
Xa
Protrombi
na
Trombina
VII
Fibrynog
en
Zwiększone powstawanie
włóknika
Włóknik
Niedostateczne usuwanie
włóknika
TFPI
Białko
C
PAI-
1
AT III
a
D-
dimer
D
D

DIC - ETIOLOGIA
• Posocznica
– Gł. G(-): jawny DIC u 30-50%, ale też G(+)
• Uraz lub rozległe zabiegi operacyjne
– Urazy głowy: DIC u 41% gdy w CT uszkodzenie
mózgu, u 25% gdy nie ma uszkodzenia mózgu. DIC w
1-4 h po urazie, duża śmiertelność
• Nowotwory
– DIC jest najczęstszą koagulopatią w przebiegu
nowotworów
– Występuje u 15% pacjentów z rozsianą ch.n. i u
większości z ostrą białaczką promielocyt. - M3
– Ostra postać – najczęściej M3, w guzach litych
najczęściej przewlekła

DIC - ETIOLOGIA
Powikłania położnicze
– Zator płynem owodniowym-u 50% DIC
– Łożysko przodujące – u 50%
– Zespół HELLP – u 20%
– Ciąża obumarła
– Septyczne poronienie
• Choroby układu krążenia
– olbrzymie naczyniaki, tętniak rozwarstwiający
aorty,
protezy naczyniowe
• Hemoliza wewnątrznaczyniowa

DIC - ETIOLOGIA
• Nocna napadowa hemoglobinuria
• Ukąszenie węża
• Marskość wątroby
• Udar cieplny

OSTRY DIC – OBRAZ
KLINICZNY
• Krwawienie
– Wybroczyny, krwiaki, krwawienie z ran,
wkłuć dożylnych, z p. pokarmowego, do OUN,
po operacjach – krwawienie wokół drenów,
cewników, tracheostomii, do jam ciała
• Ostra niewydolność nerek u 25-40%
• Uszkodzenie wątroby – u 19%
– Często żółtaczka z powodu uszkodzenia
wątroby i hemolizy

OSTRY DIC – OBRAZ
KLINICZNY
• Powikłania pulmonologiczne – u 16%
– Krwotok płucny, krwioplucie, duszność,
ARDS
• Zajęcie OUN – u 2%
– Drgawki, śpiączka objawy ogniskowe
– Krwawienie do OUN szczególnie częste w
M3 (u 40%)

PRZEWLEKŁY DIC – OBRAZ
KLINICZNY
• Pacjenci często bezobjawowi
• Powikłania zakrzepowe:
– Żylne: zakrzepica żył kończyn dolnych
– Wędrujące zapalenie żył
powierzchownych (zsp. Trousseau)
– Tętnicze: w obrębie kończyn, naczyń
nerkowych, OUN
• Rzadko krwawienia, gł. śluzówkowe

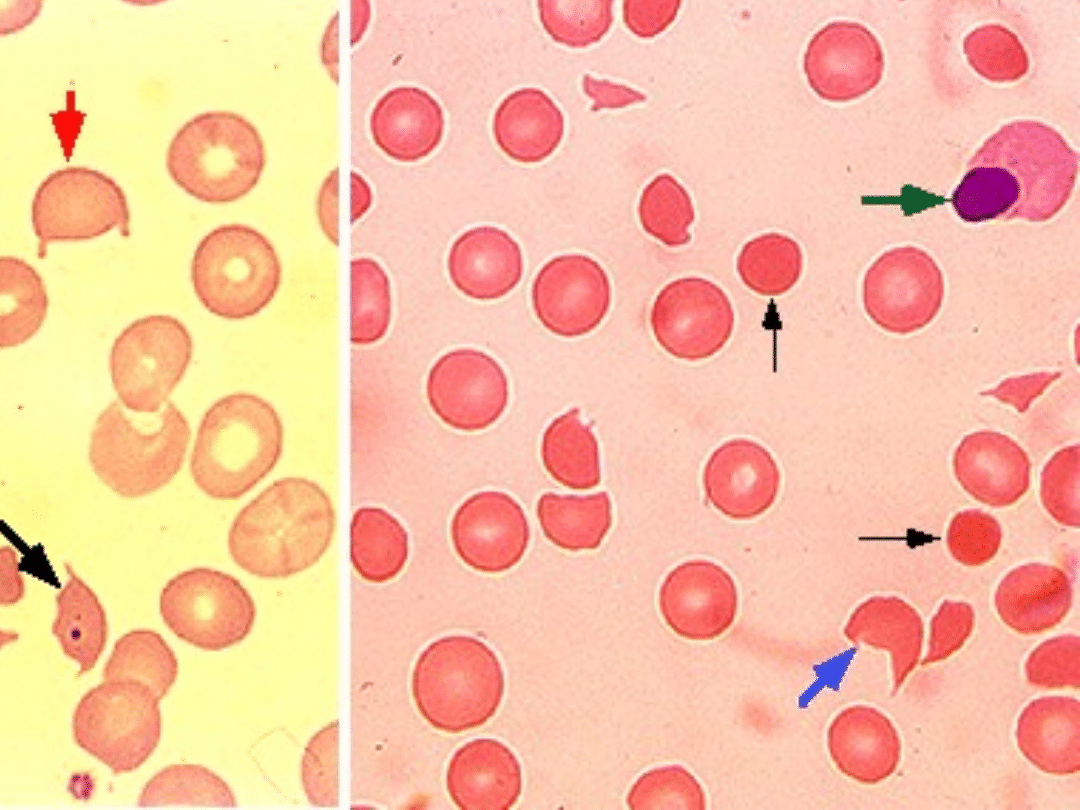
OSTRY DIC -
ROZPOZNANIE
• Obraz kliniczny
• Małopłytkowość
• Anemia hemolityczna
mikroangiopatyczna (rozmazy krwi
obwodowej)
• Badania układu krzepnięcia
wykazujące zarówno powstawanie
trombiny jak i pobudzenie fibrynolizy
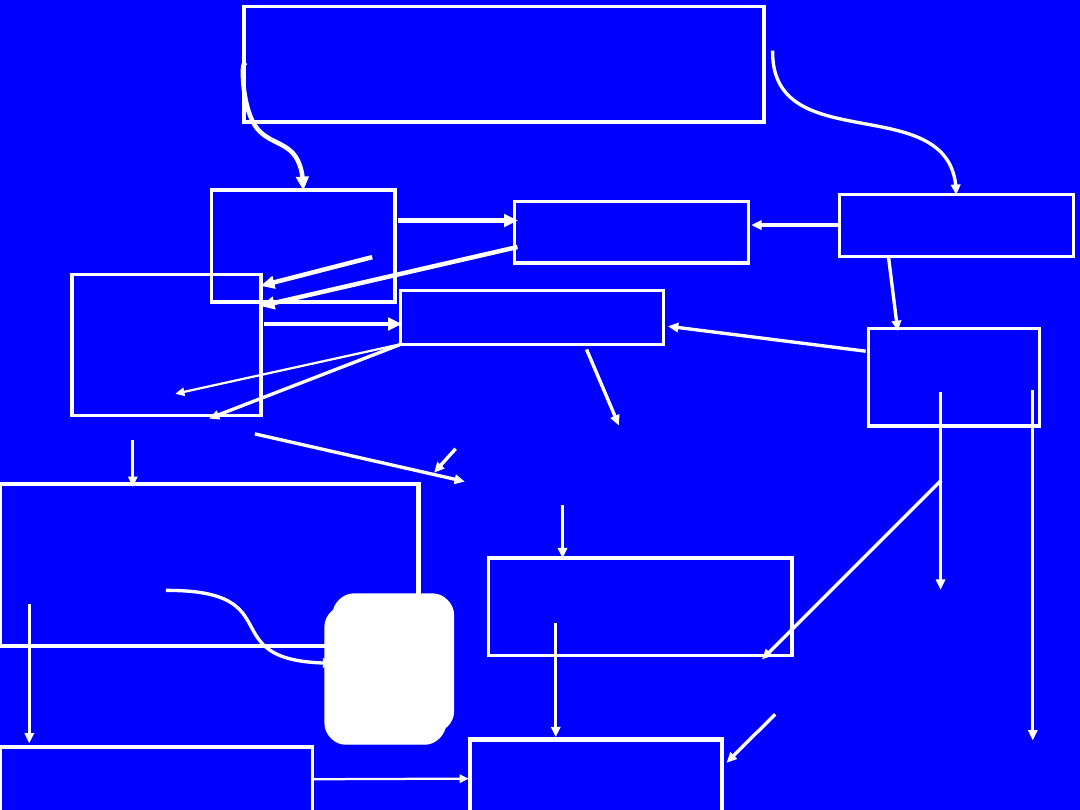
Trombi
na
+ F
1+2
Plazmi
na
Fibrynogen
Monomery fibryny
Fragmenty X, Y, D, E (=FDP)
Fibryna
stabilizowana
(zakrzepica)
Rozpuszczalne monomery
fibryny
Trombocytopen
ia
Krwawie
nia
TF-
VIIa
Choroba
podstawowa
XIIXIIa
FPA, FPB
Trombocytopat
ia
Biodegrada
cja V, VIII,
IX, XI
Aktywacja
dopełniacz
a
Generacja
kinin
XXIa
D-
dimer
D
D
Plazmin
a

OSTRY DIC -
ROZPOZNANIE
D-dimer (ELISA)– najczęstszy
nieprawidłowy parametr u pacjentów
z DIC
FDP
PT i APTT
fibrynogen (w niektórych
sytuacjach bo białko ostrej fazy)
TT

OSTRY DIC -
ROZPOZNANIE
poziom fizjologicznych
inhibitorów krzepnięcia:
– AT III ( aktywność na początku
wstrząsu septycznego zły czynnik
prognostyczny), białko C i S
• Testy parakoagulacji – pomiar
rozpuszczalnych monomerów
fibryny

INNE STANY PRZEBIEGAJĄCE
ZE WZROSTEM FDP LUB D-
DIMERÓW
• Zatorowość płucna
• Zawał serca
• Niektóre choroby nerek
• Zakrzepica żył kończyn dolnych
• Uszkodzenie wątroby
• Doustne leki antykoncepcyjne

DIC - ROZPOZNANIE
D-dimer
FDP
N lub
Fibrynogen
N
TT
N
APTT
N
PT
N lub
Liczba płytek
Przewlekły
DIC
Ostry DIC
Parametr

DIC - LECZENIE
• Część pacjentów nie wymaga
szczególnego leczenia
koagulopatii ze względu na krótki
czas jej trwania lub małe ryzyko
krwawienia lub zakrzepicy
• Zasadnicze znaczenie ma leczenie
przyczynowe

DIC - LECZENIE
• Koncentraty krwinek płytkowych i
czynników krzepnięcia
– Nie jest celowe ich podawanie u pacjentów,
którzy nie krwawią
– Wskazane u chorych krwawiących lub
poddawanych procedurom inwazyjnym
– Świeżo mrożone osocze lub krioprecypitat
gdy znaczne PT lub fibrynogen < 50 mg/dl
– Wskazane utrzymywanie stężenia
fibrynogenu na poziomie > 100 mg/dl

DIC - LECZENIE
Heparyna
• Teoretycznie podawanie w celu przerwania
wykrzepiania logiczne
• Ograniczenia: nasilenie krwawienia,
niedostateczny efekt p-zakrzepowy ze
względu na brak AT III
• Praktycznie brak kontrolowanych badań
wykazujących jej skuteczność w hamowaniu
wykrzepiania i mało dowodów, że poprawia
funkcję narządów wewnętrznych

DIC - LECZENIE
Heparyna – dawkowanie:
• iv
– Aktywność AT III powinna być blisko
normy (tj. 80-100%)
– Bez bolusu
– Dawka początkowa 500j / h APTT ok.
45s. Gdy widoczny efekt uzupełnienie
czynników krzepnięcia
• Heparyny niskocząsteczkowe: skuteczność
podobna, mniejsze ryzyko krwawień

DIC - LECZENIE
Koncentrat aktywowanego białka C
• Aktywność przeciwzapalna i
przeciwzakrzepowa
• Przeciwzapalna: poprzez bezpośredni
wpływ na komórki śródbłonka i
hamowanie ekspresji cząstek adhezyjnych
• Wskazane gł. we wstrząsie septycznym
• Drotrekogin alfa (Xigris), dawka 24 g/kg
mc./h

DIC - LECZENIE
Antytrombina III
• Oprócz działania p-zakrzepowego może mieć też
działanie p-zapalne poprzez poziomu
interleukiny 6 i TNF
• Przeprowadzone wieloośrodkowe
randomizowane badania wykazały, że AT III nie
zmniejsza śmiertelności u pacjentów z sepsą,
rośnie natomiast ryzyko powikłań krwotocznych
przy jednoczesnych stosowaniu heparyny
*
*Warren BL i wsp. Caring for the critically ill patient.
High-dose antithrombin III in severe sepsis: a
randomized controlled trial. JAMA 2001; 286, 1869

DIC - ROKOWANIE
• Ostry DIC – śmiertelność 40 – 80%
w zależności od choroby
podstawowej
• Czynniki ryzyka zgonu: wiek,
stopień niewydolności narządowej

CHOROBA VON
WILLEBRANDA
• Najczęstsza wrodzona skaza
krwotoczna
• Autosomalny sposób dziedziczenia
• Przyczyna: niedobór lub nieprawidłowa
struktura czynnika von Willebranda
(vWf)
• Przebieg w większości łagodny, tylko u
części krwawienia, głównie śluzówkowe

CHOROBA VON
WILLEBRANDA
• Vwf jest syntezowany w komórkach śródbłonka
i megakariocytach. Występuje w krążeniu w
postaci multimerów różnej wielkości
• 2 funkcje Vwf:
– Wiąże się z GP Ib-IX-V i ułatwia adhezję
płytek do podśródbłonkowej tkanki łącznej
– nośnik dla czynnika VIII w krążeniu - chroni
cz. VIII przed proteolizą
• Przy niedoborze: zaburzenia hemostazy
pierwotnej i czas krwawienia oraz zaburzenia
krzepnięcia krwi i APTT

CHOROBA VON
WILLEBRANDA
• Klasyfikacja:
• 3 typy choroby
– w typie 1 i 3 rzeczywisty niedobór
vWf (w typie 1 niewielki, w typie 3
ciężki)
– w typie 2: anomalie struktury i
funkcji vWf
•typ 2A, typ 2B, typ 2M, typ 2N

CHOROBA VON
WILLEBRANDA -
DIAGNOSTYKA
• Badania laboratoryjne:
– Czas krwawienia
– APTT
– Antygen cz. vWf (vWf:Ag)
– Aktywność kofaktora rystocetyny (R:Cof )
– Aktywność cz. VIII
• Jeśli te badania nieprawidłowe:
– Badanie multimerów cz. vWf
– Aglutynacja płytek pod wpływem rystocetyny
(RIPA
)

CHOROBA VON
WILLEBRANDA
• Typ 1
• Dziedziczenie
autosomalne
dominujące
• 75% wszystkich
przypadków ch.
vWf
• łagodny ilościowy
niedobór vWf
• Skaza łagodna lub
umiarkowana
• vWf:Ag, R:Cof,
VIII:C
proporcjonalnie
• prawidłowy rozkład
multimerów
• Leczenie: DDAVP,
koncentraty VIII-
vWf,

CHOROBA VON
WILLEBRANDA
• Typ 3
• Znaczny ilościowy
niedobór vWf
• Ciężki przebieg
kliniczny
• vWf:Ag, VIII:C,
R:Cof (<5%),
brak lub śladowa
ilość wszystkich
multimerów
• Leczenie: VIII-
vWf,

CHOROBA VON
WILLEBRANDA - LECZENIE
Desmopresyna (DDAVP)
• syntetyczna pochodna wazopresyny, która
uwalnianie vWf i cz. VIII z śródbłonków,
pobudza też fibrynolizę poprzez uwalnianie t-
PA
• Podawana iv, sc, donosowo.
Dawka iv i sc: 0,3 g/kg, 2-5 x vWf i cz. VIII
w 30-60 min. po podaniu, utrzymuje się 6-12h.
Stosuje się 2x dz przez kilka dni.
• po zastosowaniu DDAVP zaleca się podanie
EACA 5-7,5 g i.v. lub 10 g p.o.

CHOROBA VON
WILLEBRANDA - LECZENIE
Leczenie substytucyjne vWf:
• Koncentraty cz. VIII średnio
oczyszczone, np. Humate P
– Cel: utrzymanie aktywności vWf na
poziomie 50-100% przez 3-10 dni w razie
ciężkiego krwawienia lub operacji
– Dawki: najczęściej 20-30 IU/kg 2x dz
• Krioprecypitat: zawiera kompleks cz.
VIII z vWf, fibrynogen i cz. XIII

CHOROBA VON
WILLEBRANDA - LECZENIE
Leki hamujące fibrynolizę:
• EACA, kwas traneksamowy: głównie w
krwawieniach śluzówkowych, w celu
zmniejszenia krwawienia po ekstrakcji zęba.
Gdy łagodne krwawienie – monoterapia
• Doustnie: 4 x dz 50 mg/kg EACA lub 25
mg/kg kwasu traneksamowego
Leczenie miejscowe:
• Gł. krwawienie z nosa: spongostan z
trombiną, środki z kolagenem

CHOROBA VON
WILLEBRANDA – LECZENIE
• Estrogeny:
syntezę czynnika vWf
• Rekombinowany czynnik VIIa – w
typie 3 z obecnością przeciwciał p-
vWf.
– rVIIa inicjuje zewnątrzpochodny tor
krzepnięcia z pominięciem cz. VIII

HEMOFILIE
• Hemofilia A - wrodzony brak lub
niedobór VIII czynnika krzepnięcia
• Hemofilia B - wrodzony brak lub
niedobór IX czynnika krzepnięcia
• Hemofilia A 4-8 x częściej niż B
• Dziedziczenie recesywne sprzężone z
płcią, w około 30% przypadków
hemofilia A występuje sporadycznie,
czyli po raz pierwszy w danej rodzinie

HEMOFILIE
Krwawienia
pourazowe
> 5%
Łagodna
Wylewy dostawowe
rzadziej
1 – 5%
Umiarkow
ana
Wylewy śródstawowe,
do mięśni, wylewy
podskórne, krwiomocz,
wylewy śródczaszkowe
< 1%
Ciężka
Objawy
Aktywnoś
ć czynnika
Postać

HEMOFILIE
• Ciężka hemofilia: pierwsze objawy
koniec pierwszego, drugi rok życia,
u 5 % krwawienie śródczaszkowe
w okresie okołoporodowym, często
powikłane drgawkami
• Łagodna i umiarkowana hemofilia:
objawy później, mogą być nie
wykryte nawet do starości

HEMOFILIE
• Miejsca krwawień:
–
Do stawów, gł. kolanowych, skokowych, łokciowych
–
Do mięśni, gł. mięsień czworogłowy, biodrowo-
lędźwiowy, przedramion
–
OUN
–
W obrębie głowy i szyi, np. krwiak tylnej ściany
gardła
–
Z przewodu pokarmowego, do ściany brzucha, do
przestrzeni zaotrzewnowej, krwiaki w obrębie
ściany jelita
–
Z dróg moczowych
–
Po urazach, zabiegach

HEMOFILIE
APTT
– liczba płytek krwi, czas krwawienia,
czas protrombinowy - prawidłowe
• Hemofilia A:
VIII:C
– vWF:Ag, R:Cof - prawidłowe
• Hemofilia B:
IX

HEMOFILIE
Hemofilia A
•
koncentraty czynnika
VIII
•
DDAVP
•
krioprecypitat
•
1j. cz. VIII na 1 kg m.c.
aktywność w osoczu o
2%
•
dawka wstępna: 0,5 x
masa ciała x pożądany
wzrost cz. VIII
Hemofilia B
• koncentraty czynnika
IX
• koncentraty zespołu
protrombiny
• 1j. cz.IX na 1 kg m.c.
aktywność w osoczu o
1%
• dawka wstępna: masa
ciała x pożądany
wzrost cz. IX

•
HEMOFILIE - LECZENIE
• Koncentraty czynnika VIII
– Średnio oczyszczone: zawierają 6-10 U/mg białka
– Wysoko oczyszczone: zawierają przynajmniej 50 U/mg
białka
– Bardzo wysoko oczyszczone: otrzymywane przy użyciu
p-ciał monoklonalnych (3000 U cz. VIII/ mg białka)
lub rekombinowane
• Koncentraty czynnika IX
– Koncentraty zespołu protrombiny
– Oczyszczone koncentraty cz. IX
– Rekombinowany czynnik IX

HEMOFILIE
• Powikłania późne hemofilii:
– Artropatia hemofilowa
• Profilaktyczne wstrzyknięcia koncentratów od
najmłodszych lat życia istotnie ryzyko
artropatii
– Zakażenia wirusowe
• Częstość poprzez restrykcyjny dobór
dawców, lepsze metody eliminacji wirusów i
rozwój produktów rekombinowanych
– Powstanie inhibitorów przeciwko
czynnikom krzepnięcia

HEMOFILIA A I KRĄŻĄCY
ANTYKOAGULANT
• U 15-30% chorych na ciężką hemofilię A
i u < 5% chorych na hemofilię B
pojawiają się p-ciała przeciw cz. VIII lub
IX
• związek z leczeniem substytucyjnym,
niektóre mutacje genu cz. VIII
predysponują do ich wystąpienia
• inhibitor nie częstości krwawień, ale
krwawienia w jego obecności są trudne
do opanowania

Rekombinowany czynnik
VIIa
• Wiąże się z płytkami krwi i bezpośrednio aktywuje cz. X
• Zastosowanie:
– krwawienia, zabiegi operacyjne u chorych na hemofilię
A i B powikłaną inhibitorem (w zagrażających życiu
krwawieniach + FEIBA)
– nabyty inhibitor cz. VIII
– niedobór cz. XI, VII
– choroby wątroby
– zaburzenia czynności płytek, małopłytkowości oporne
na leczenie
• Koszt!, ryzyko powikłań zakrzepowych, zwł. gdy czynniki
ryzyka

HEMOFILIA A I KRĄŻĄCY
ANTYKOAGULANT
• Indukowanie tolerancji
immunologicznej wobec niedoborowego
czynnika krzepnięcia
– częste wstrzykiwanie koncentratu
niedoborowego cz. krzepnięcia w
odpowiednio dużych dawkach, czasem z
cyklofosfamidem
– czynniki sprzyjające uzyskaniu tolerancji:
niskie miano inhibitora, młody wiek
– u 90% można uzyskać eliminację inhibitora
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
Wyszukiwarka
Podobne podstrony:
1 Fizjologia gospodarki wapniowo fosforanowej seminarium dla studentówid 9243 ppt
1 wyklad dla studentowid 10099 ppt
13 CUN 09 06 2007r c I komentarz dla studentówid 14624 ppt
12Ana m brzucha 09 06 07 komentarz dla studentówid 14232 ppt
1 Kości, klp 14 10 06 wersja dla studentówid 9361 ppt
Wykład 5 2010 studenci ppt
RKZ studenci(1) ppt
diagnostyka kily studenci ppt
2008 STUDENCI ZARZADZANIE PROCESAMIid 26553 ppt
11 ZABURZENIA UKŁADU HEMOSTAZYid 12267 ppt
Diagnostyka chorob tkanki lacznej studenci ppt
Leki biologiczne w dermatologii studenci ppt
ST1 2010Dla studentów ppt
ZIF2013 2014 WSFiZ Ramowy program wykładów sem letni r ak 2013 2014 v 1 6 4 dla wszystkich studentów
Diagnostyka chorób pęcherzowych studenci ppt
więcej podobnych podstron