
Niedokrwistości
hemolityczne

Niejednolity zespół defektów
zewnątrz- i/lub
wewnątrzerytrocytarnych
prowadzących do znacznego
(poniżej 10 dni),
niekompensowanego przez
hiperplazję erytronu w szpiku,
skrócenia przeżycia krwinki
czerwonej
Definicja niedokrwistości
hemolitycznych

Podział niedokrwistości
hemolitycznych
Wrodzone niedokrwistości hemolityczne
defekt błony komórkowej
(sferocytoza, eliptocytoza)
defekty enzymatyczne
hemoglobinopatie i talasemie
Nabyte niedokrwistości hemolityczne
n. autoimmunohemolityczne
n. h. polekowe
n. h. toksyczne
hipersplenizm
n. mikroangiopatyczne
n. makroangiopatyczne
nocna napadowa hemoglobinuria
Wspólna cecha niedokrwistości
hemolitycznych: skrócenie czasu przeżycia
krwinki czerwonej niekompensowane
hiperplazją układu erytroblastycznego

.
Podział Lanzkowsky’ego uwzgledniający
etiologię
i główne cechy diagnostyczne
hemolitycznych
A. Spowodowana defektem wewnątrz-krwinkowym
1. Defekty błony (sferocytoza, eliptocytoza).
Budowa,
oporność
osmotyczna;
2. Defekty enzymatyczne (kinaza pirogronianowa,
dehydrogenaza glukozo-6-fosforanowa - G-6-PD)
autohemoliza
, badania
enzymatyczn
e;
3. Defekty hemoglobiny
a. Hemu
b. Globiny
c. Jakościowe (np. niedokrwistość sierpowato-
komórkowa)
elektroforeza
hemoglobiny;
d. Ilościowe (np. talasemie)
HbF,
obecność
hemoglobiny
A
2

.
Podział Lanzkowsky’ego uwzgledniający
etiologię
i główne cechy diagnostyczne
hemolitycznych
B. Spowodowana defektem pozakrwinkowym
1. Immunohemolityczna
odczyn
Coombsa;
a. Izoimmunohemolityczna
b. Autoimmunohemolityczna
c. Idiopatyczna
odczyn
Coombsa,
identyfikacja
przeciwciał;
d. Makroangiopatyczna
sztuczne
zastawki,
protezy
naczyniowe
e. Mikroangiopatyczne
poszukiwanie
nowotworu!

Podział Lanzkowsky’ego uwzgledniający
etiologię
i główne cechy diagnostyczne
hemolitycznych
C) Wtórna
Zaburzenia immunologiczne (np.
toczen)
zmniejszona zawartość
składowych dopełniacza:
C
3
, C
4
, CH
50
, obecność
przeciwciał
przeciwjądrowych (ANA);
Obejmująca jeden szereg komórkowy
(np. czerwonokrwinkowy)
Niedokrwistość z
dodatnim odczynem
Coombsa;
Obejmująca wiele szeregów
komórkowych (np. białokrwinkowy,
płytkowy)
pancytopenia
neutropenia
trombocytopenia - ITP;
Na tle nieimmunologicznym (idiopatyczna, wtórna)
infekcje (cytomegalia, toksoplazmoza, różyczka, kiła)
niedokrwistość hemolityczna na tle uszkodzeń drobnych naczyń z
zespołem DIC albo bez: rozsiane zakażenie herpes simplex,
infekcje wywołane wirusami Coxacckie B, posocznica w zakażeniu
bakteriami Gram - ujemnymi, zakrzepica żył nerkowych,
nieprawidłowo przebiegający poród, niektóre przypadki zespołów
błon szklistych

Objawy kliniczne
Objawy ogólne: gorączka, dreszcze, bóle
mięśni i stawów
Objawy niedokrwistości
Objawy żółtaczki
Splenomegalia i/lub hepatomegalia
Cechy laboratoryjne
stężenie bilirubiny
stężenia urobilinogenu w
moczu
stężenia sterkobilinogenu w
kale
retikulocytozy
aktywności LDH
stężenia Fe
haptoglobiny
czasu przeżycia erytrocytów

Przebieg niedokrwistości
hemolitycznej:
Ostry
Podostry
Przewlekły
Przełom
hemolityczny
ostre bóle w okolicy
lędźwiowej
i kończyn dolnych
bóle głowy, brzucha
dreszcze
gorączka
pogłębienie
niedokrwistości ze
wzrostem retikulocytozy
nasilenie bilirubinemii
wzrost leukocytozy z
przesunięciem w lewo w
rozmazie
wystąpienie lub
nasilenie splenomegalii
Przełom
aplastyczny
ciężki stan ogólnych
chorego
gorączka
wymioty
hipo- lub aplazja układu
czerwonokrwinkowego
pancytopenia lub
duocytopenia
zmniejszenie odsetka
retikulocytów
narasta stężenie żelaza
obniżenie stężenia
bilirubiny
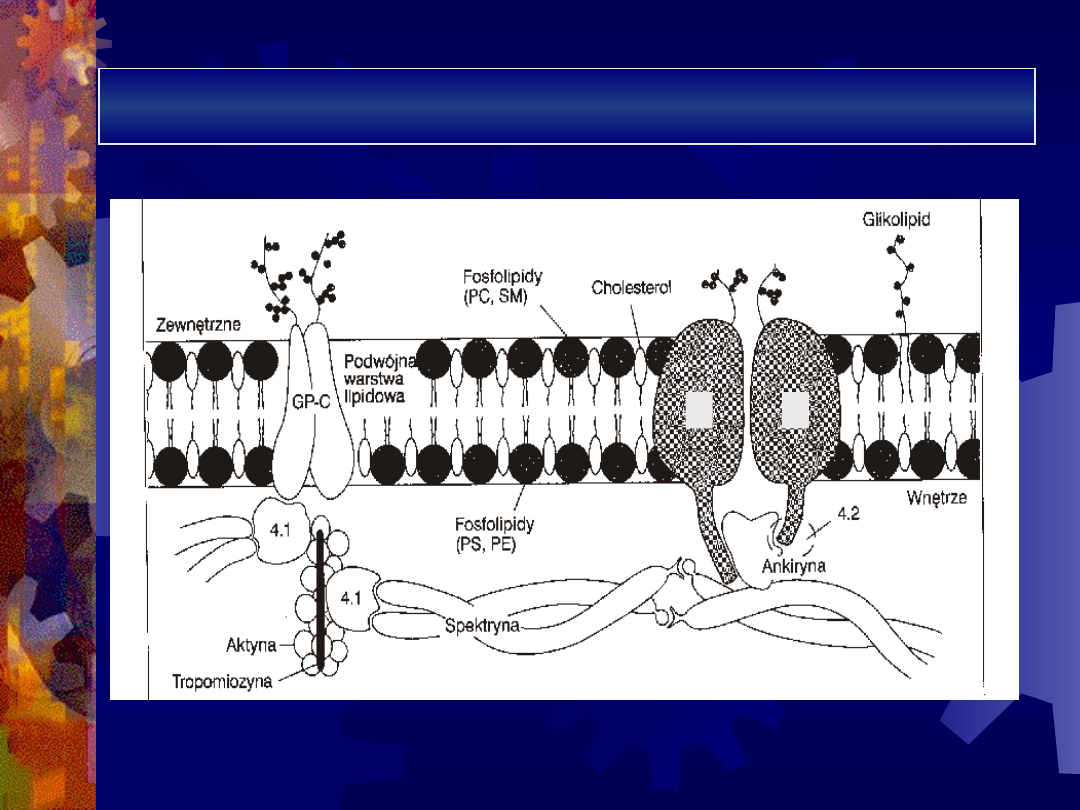
Budowa błony erytrocyta i białka
szkieletu
3
3

Sferocytoza wrodzona
Występowanie: Europa północna i USA
(1/5000)
Druga co do czestości wystepowania
wrodzoną niedokrwistością hemolityczną po
niedoborze dehydrogenazy
glukozo-6-fosforanowej.
Defekt: niedobór/mutacja spektryny -
autosom. recesywne niedobór/mutacja
spektryny - autosom. dominujące
nieprawidłowości ankiryny
nieprawidłowości białka 3
zwiększenie przepuszczalności błony k.
dla Na+ i K+
Anomalie białek błony komórkowej prowadzą
do wytwarzania sferocytów, które ulegają
sekwestracji
w śledzionie.

PRZEBIEG: Choroba ujawnia sie niekiedy już w
okresie noworodkowym pod postacią cieżkiej
żółtaczki i niedokrwistości.
Umiarkowaną niedokrwistość, dyskretną
żółtaczkę i powiekszoną śledzionę obserwuje
sie na ogół już od 2 roku życia.
Kamica żółciowa wystepuje u 50% chorych.
Najcięższym powikłaniem choroby w
dziecinstwie są zagrażające życiu epizody
aplastyczne.
W tym czasie we krwi obwodowej stwierdza
się brak retikulocytów, a w szpiku wystepuje
zanik układu czerwonokrwinkowego. Epizody
aplastyczne rozwijają się w związku z
zakażeniami wirusowymi, szczególnie
parwowirusem i trwają zwykle około 14 dni.
Sferocytoza wrodzona

Oporność osmotyczna krwinek czerwonych
jest obniżona.
Test autohemolizy wykazuje destrukcje 15-
45% erytrocytów po 48 godzinach.
W celu odróżnienia wrodzonej sferocytozy
od autoimmunohemolitycznych
niedokrwistości należy wykonać odczyn
Coombsa.
Przy hiperbilirubinemii okresu
noworodkowego należy różnicować z
konfliktem serologicznym w grupach
głównych.
W przypadku negatywnego wywiadu
rodzinnego właściwą diagnoza możliwa jest
po dłuższym okresie obserwacji.
Sferocytoza wrodzona

Badania dodatkowe: sferocyty (+ akantocyty)
zmniejszenie oporności osmotycznej,
powiększenie retikulocytozy, hiperplazja
erytronu
Leczenie: splenektomia po 5 roku życia
Po usunieciu śledziony ustepują objawy
choroby, ale nadal utrzymuje sie sferocytoza i
obniżona odporność osmotyczna.
Usuniecie śledziony zapobiega wystepowaniu
pprzełomów aplastycznych, kamicy żółciowej,
hemosyderozy i uszkodzeń wątroby.
Po wykonaniu splenektomii istnieje
niebezpieczenstwo wystąpienia piorunujących
zakażen pneumokokowych. Uodpornienie
szczepionką przeciwpneumokokową przed
zabiegiem i profilaktyczne podawanie
penicyliny o przedłużonym działaniu w
pierwszych latach po splenektomii chroni
dzieci przed niebezpieczenstwem wystąpienia
piorunujących posocznic.
Sferocytoza wrodzona
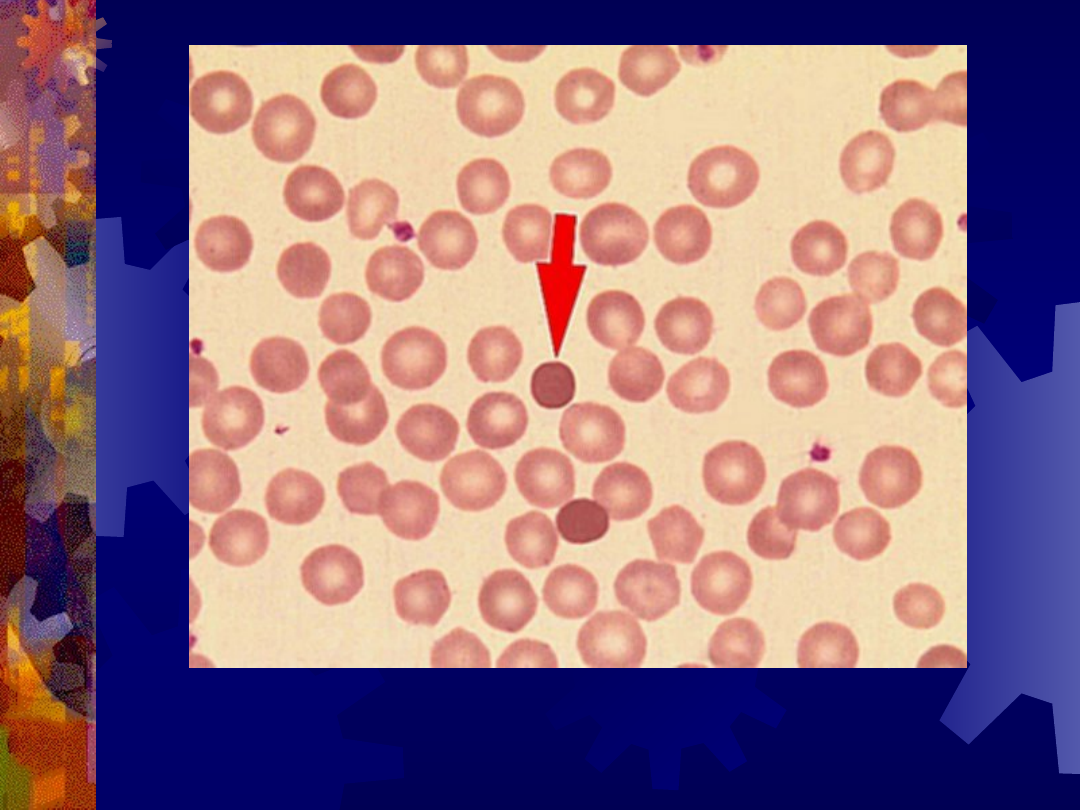
Sferocyty (strzałka)
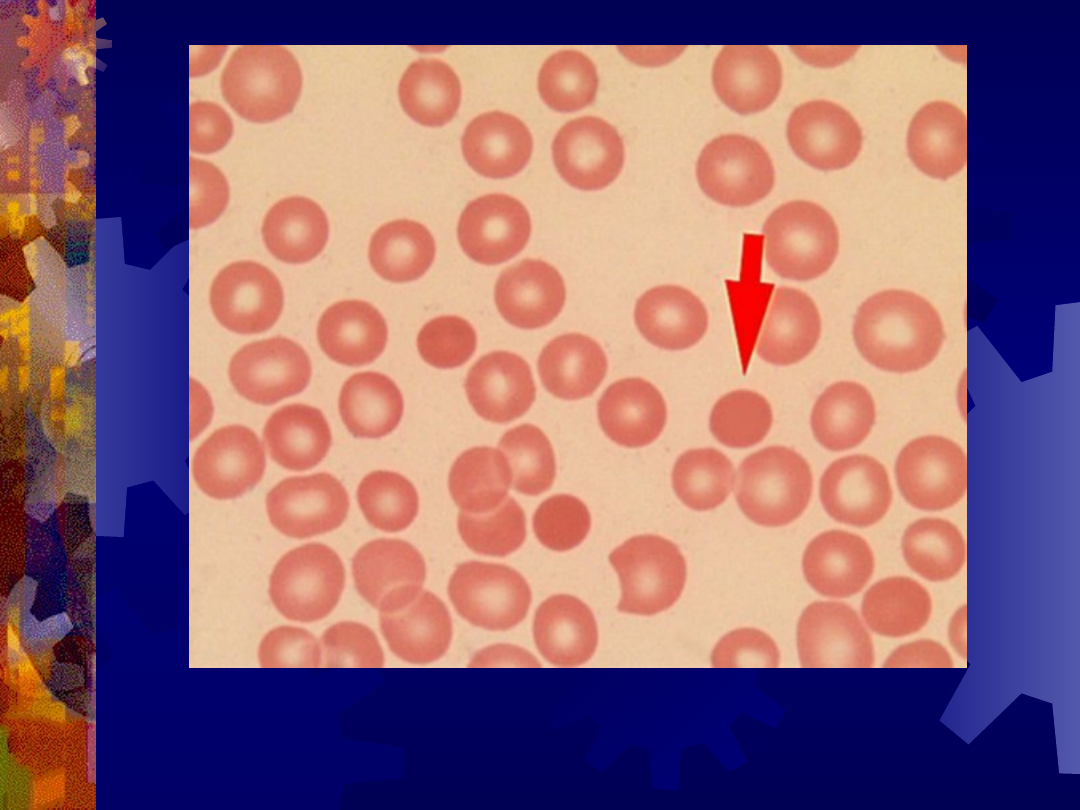
Sferocyty (strzałka)
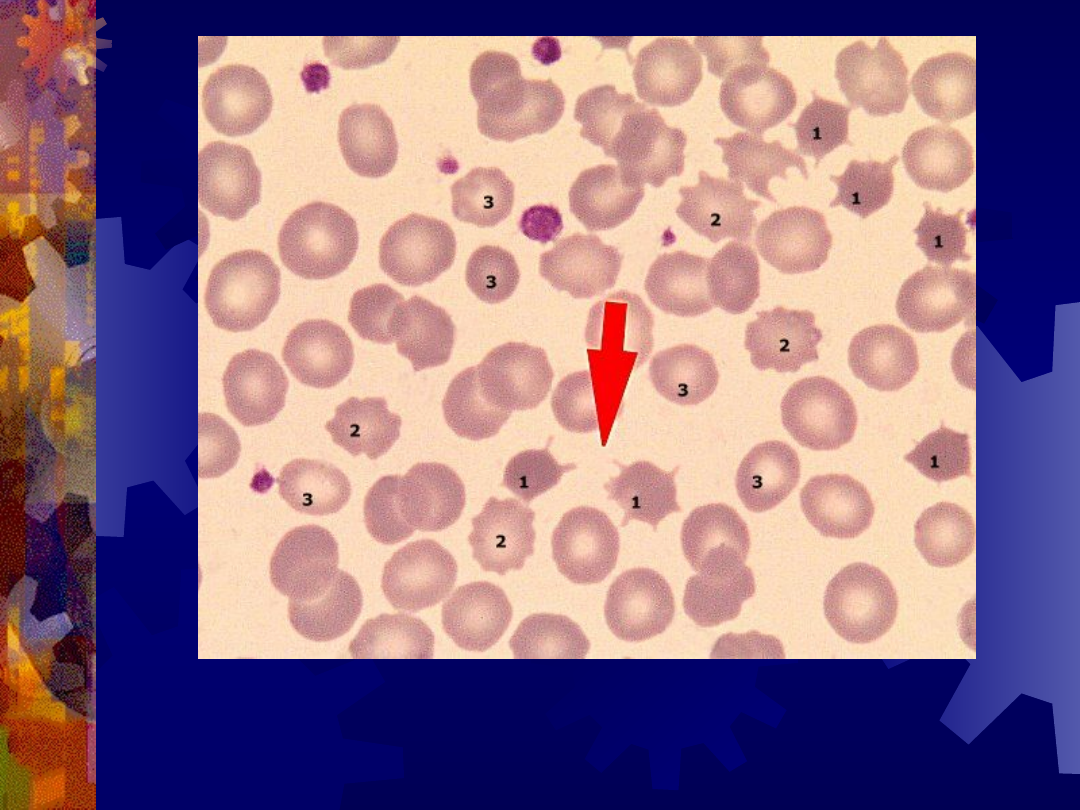
1 – akantocyty
2 – echinocyty
3 – mikrocyty

Eliptocytoza wrodzona
Występowanie: powszechne (1/5000)
Defekt: nieprawidłowe warianty
spektryny (a. dominujące) niedobór białka
4,1, glikoforyny C, białka 4,9
nieprawidłowości białka 3
zmniejszenie wiązania ankiryny do
białka 3
Odmiany: eliptocytoza pospolita,
sferocytowa
i stomatocytowa
Objawy: przebieg najczęściej łagodny,
bezobjawowy,
możliwe
zaostrzenia w postaci przełomów
aplstycznego, hemolitycznego
(przebieg ciężki w postaci
homozygotycznej)
Badania dodatkowe: eliptocyty
(owalocyty) sferocyty, fragmentocyty,
prawidłowa lub zwiększona
oporność osmotyczna,
powiększenie retikulocytozy,
hiperplazja erytronu
Leczenie: splenektomia ?
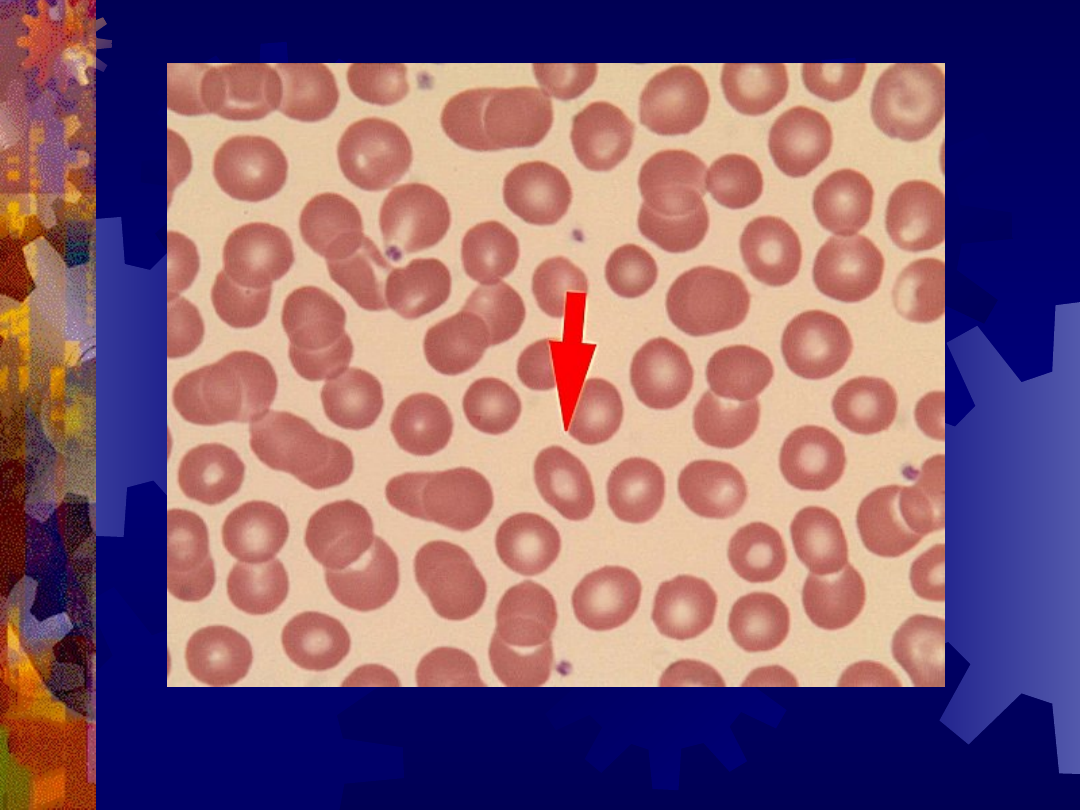
Eliptocyt (strzałka)
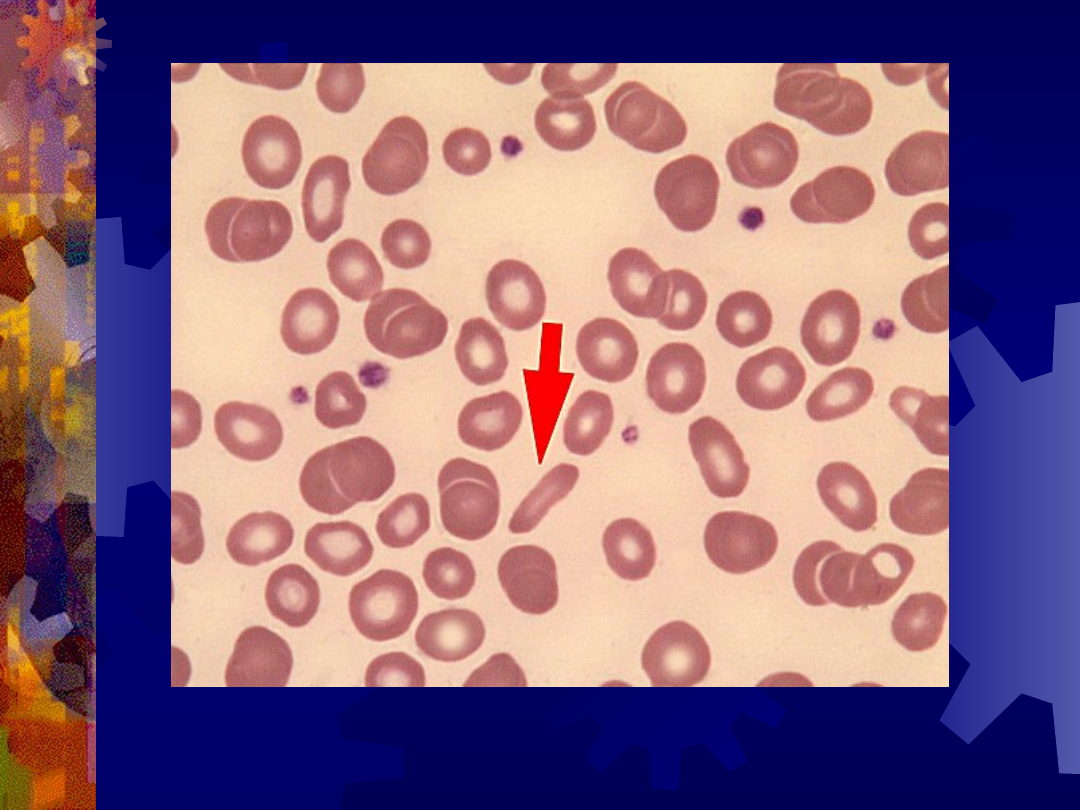
Eliptocyt (strzałka)
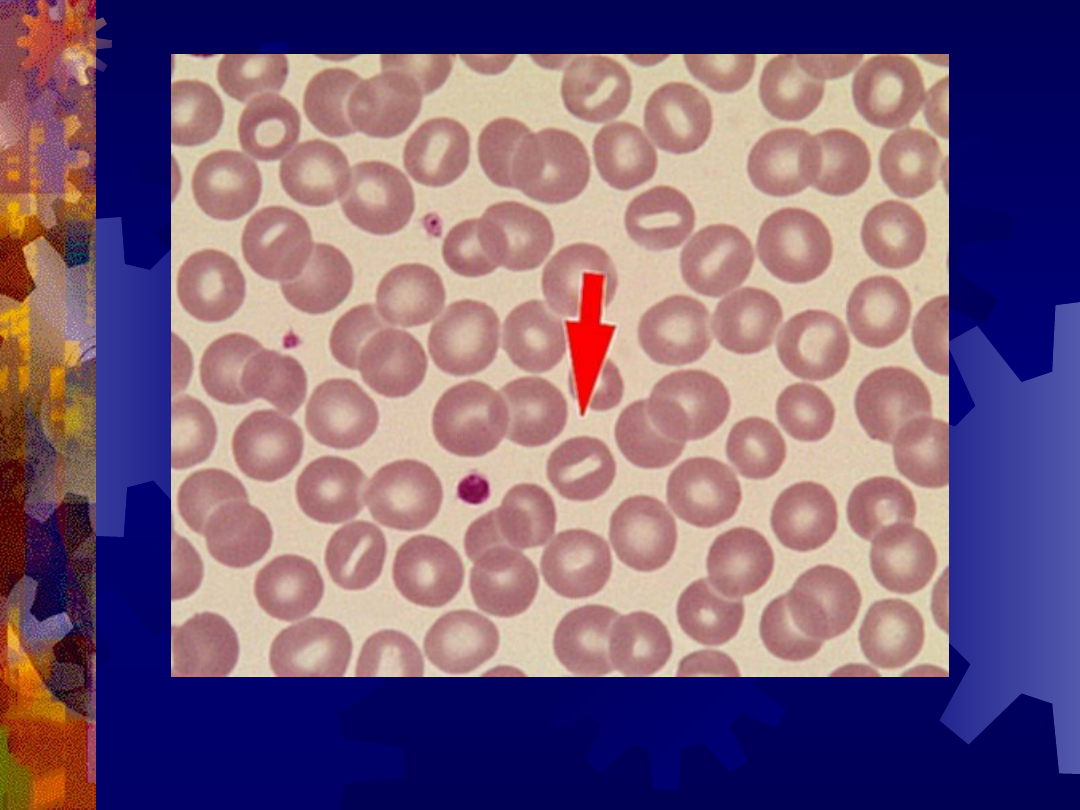
Stomatocyt (strzałka)

Enzymopatie
Anomalie enzymów cyklu glikolizy
beztlenowej
(Embdena-Meyerhofa
Niedobór kinazy pirogronianowej (autos.
recesywne)
Niedobór fosfoheksoizomerazy
Anomalie enzymów cyklu pentozowego
Niedobór dehydrogenazy glukozo-6-
fosforanowej
Anomalie enzymów syntezy glutationu
Niedobór reduktazy glutationu
Anomalie przemiany nukleotydów
Niedobór pirymidyno-5-nukleotydazy
Nadmiar dezaminazy adenozyny
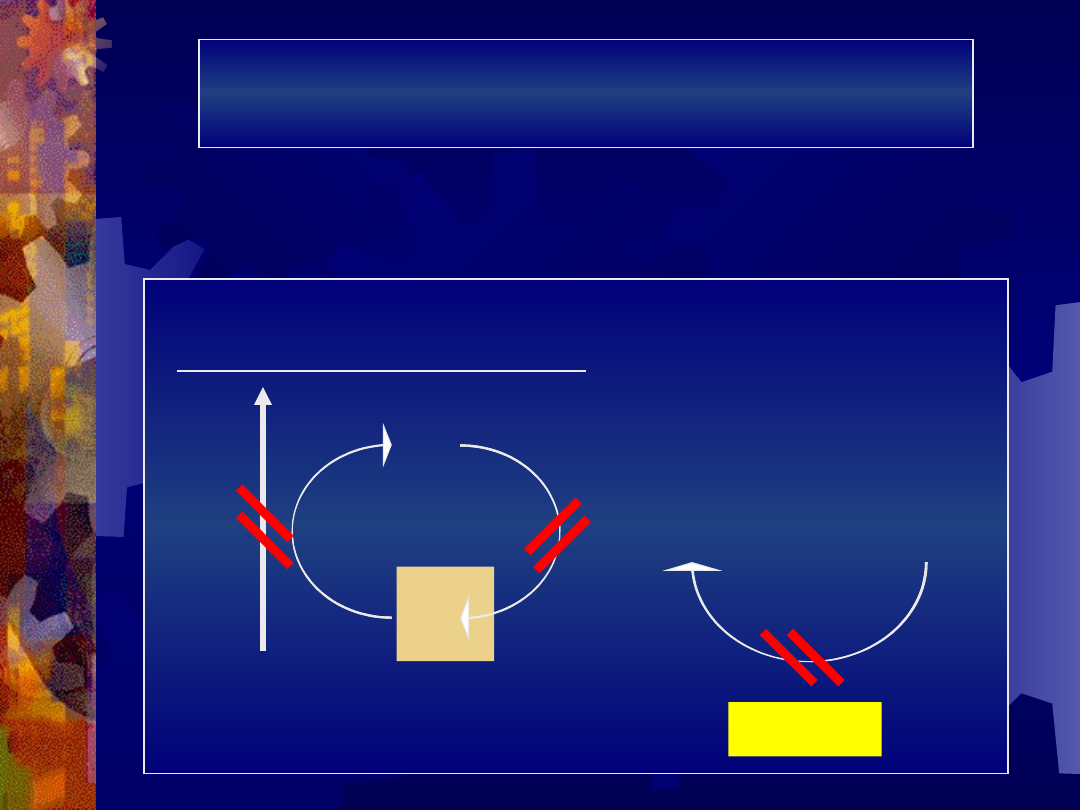
Anomalie enzymów cyklu
pentozowego
Niedobór dehydrogenazy glukozo-
6-fosforanowej
Częstość – 2%
Dziedziczenie związane z chromosomem X – pełna
ekspresja wady tylko u mężczyzn
U kobiet aktywność bardzo różna – mozaikowe populacje
erytrocytów
H
2
O
2
H
2
O
G
S
H
G
S
S
G
P
e
ro
k
s
y
d
a
za
g
lu
ta
ti
o
n
u
R
e
d
u
k
ta
za
g
lu
ta
ti
o
n
u
NADPH
NADP
G-6-PD
Zmniejszenie stężenia zredukowanego glutationu (GSH)
DENATURACJA GLOBINY: ciałka HEINZA
methemoglobinemia

Opisano około 150 wariantów niedoboru G-6-PD.
Najcześciej wystepującymi wariantami defektywnego
enzymu są wariant A oraz wariant B, zwany
śródziemnomorskim.
• Wariant A wystepuje głównie u Murzynów
afrykanskich i amerykanskich. Enzym ulega szybko
inaktywacji (jego półokres biologiczny wynosi 13
dni).
• Wariant śródziemnomorski B jest spotykany u
Greków oraz u Włochów,
a polega on na niemal zupełnym braku aktywności
enzymu, nawet w młodych erytrocytach. Przyczyną
jest skrajna niestabilnośa G-6-PD (jej okres
biologiczny wynosi zaledwie kilka godzin).
Anomalie enzymów cyklu
pentozowego
Niedobór dehydrogenazy glukozo-
6-fosforanowej

Anomalie enzymów cyklu
pentozowego
Niedobór dehydrogenazy glukozo-
6-fosforanowej
Objawy kliniczne – hemoliza indukowana przez leki
(
wewnątrznaczyniowa)
fawizm
hemoliza indukowana zakażeniami
(Salmonella, E.coli, EBV)
przewlekła
niedokrwistość hemolityczna
hiperbilirubinemia noworodków
niedokrwistość towarzysząca kwasicy
Badania dodatkowe – zmniejszenie aktywności G-6-PD
(badanie w okresie zacisza)
niedokrwistość
zwiększenie retikulocytozy
hiperbilirubinemia
hemoglobinuria
c. Heinza
sferocytoza, fragmentocytoza
Leczenie – unikanie czynników wywołujacych
transfuzja wymienna u noworodków
regularne transfuzje ME
antyoksydanty (Vit E)
splenektomia ?
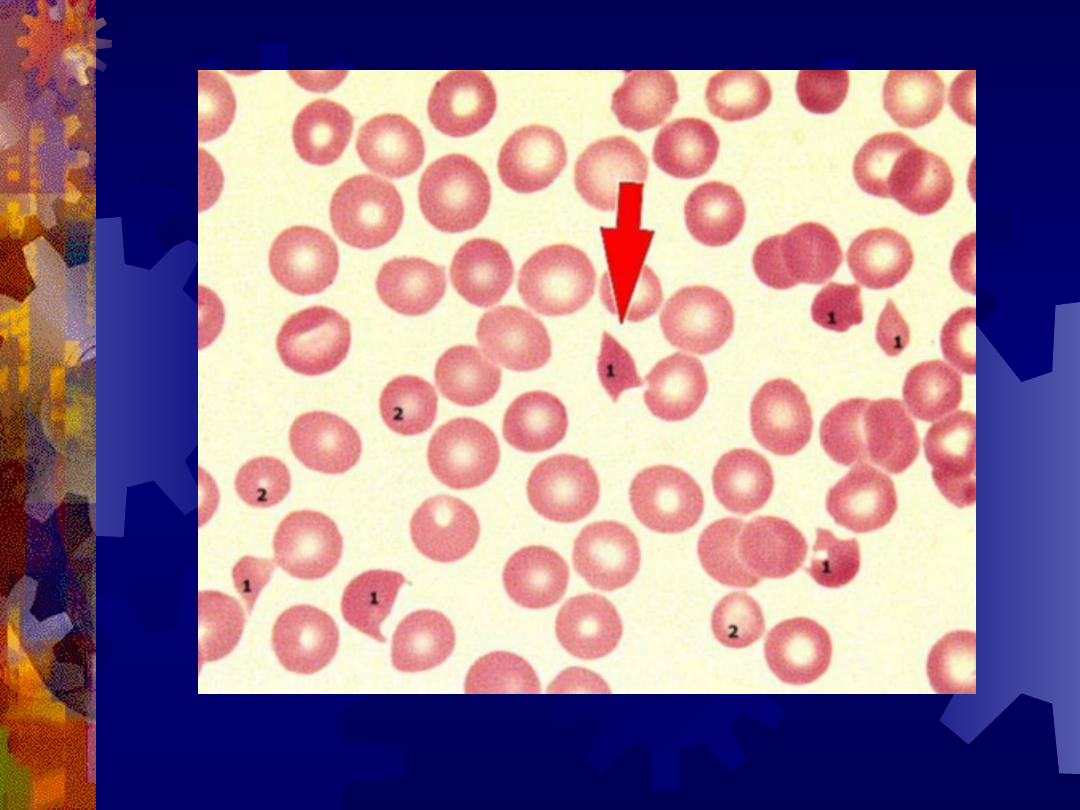
Fragmentocyty (strzałka)

Anomalie enzymów cyklu
pentozowego
Niedobór dehydrogenazy glukozo-
6-fosforanowej
Leki wywołujące hemolizę – acetaminofen
chinina
chloramfenikol
chlorochina
fenylobutazon
fenytoina
izoniazyd
kolchicyna
kwas
acetylosalicylowy
lewodopa
nitrofurantoina
sulfonamidy
streptomycyna
trimetoprym
Vit C
Vit K

• Zbyt mała aktywność G-6-PD w erytrocytach prowadzi
do zmniejszenia liczby cząsteczek zredukowanego
glutationu w ilościach wystarczających do ochrony
przed działaniem czynników utleniających.
• „Wolne rodniki” powodują utlenianie grup
sulfhydrylowych hemoglobiny,
a w nastepstwie tego jej denaturacje.
• Cząsteczki hemu i globiny dysocjują. Globina
precypituje w postaci ciałek Heinza, tworzących mostki
dwusiarczkowe z błoną krwinkową.
• Uszkodzone erytrocyty są usuwane z krwi przez
komórki układu siateczkowo - śródbłonkowego.
Masywnie uszkodzone krwinki
czerwone mogą hemolizować śródnaczyniowo.
• Klasycznym objawem niedoboru G-6-PD jest
wystepowanie ostrych przełomów hemolitycznych,
zwykle wywołanych przez leki. Może także objawiać sie
hemolizą w przebiegu zakażeń i żółtaczki noworodków,
w przewlekłej niesferocytowej niedokrwistości
hemolitycznej lub jako fawizm.
Anomalie enzymów cyklu pentozowego
Niedobór dehydrogenazy glukozo-6-
fosforanowej – podsumowanie

•U chorych z wariantem A lub B, po okresie latencji
trwającym 1-3 dni od chwili podania leków utleniających
(np. sulfonamidów, salicylanów, fenacetyny), wystepuje
nagle hemoliza krwinek czerwonych. Jej przebieg zależy
od wariantu defektu G-6-PD.
•U chorych z wariantem A hemolizie ulega tyko populacja
starych erytrocytów. Zdrowienie nastepuje wraz z
pojawieniem sie w krwi obwodowej krwinek młodych z G-
6-PD o aktywności wystarczającej do ochrony przed
działaniem czynników utleniających .
•U chorych z wariantem śródziemnomorskim hemolizie
ulegają wszystkie populacje erytrocytów w jednakowym
stopniu. Chorzy ci wymagają przetoczeń do czasu
usunięcia leków z organizmu.
•Chorym z niedoborem G-6-PD, prowadzącym do ostrych
przełomów hemolitycznych, nie należy podawać leków
indukujących hemolize. Splenektomia nie poprawia stanu
klinicznego tych chorych.
Anomalie enzymów cyklu pentozowego
Niedobór dehydrogenazy glukozo-6-
fosforanowej – podsumowanie

Hemoglobinopatie
Błędy w budowie hemoglobiny
prowadzące do
zmiany powinowactwa do tlenu
powstania nietrwałej
hemoglobiny
Zaburzenia sierpowatokrwinkowe: wywołane
odziedziczonym genem drepanocytozy
Niedokrwistość sierpowatokrwinkowa:
homozygoty
z hemoglobiną S
Defekt: -globina (walina kwas glutaminowy)
taktoidy
Klinika: owrzodzenia podudzi, zatorowość
płucna, udary mózgu,
nadciśnienie płucne, niewydolność krążenia,
choroby siatkówki, niewydolność nerek,
skłonność do infekcji pałeczkami Gram(-),
Przełom bólowy, przełom sekwestracyjny
Początkowa splenomegalia cofa sie ponieważ
wielokrotne zatory prowadzą do marskości
śledziony.

Chorobę spotyka się przeważnie u
Murzynów. Homozygoty mają 80-100% HbS,
2-3% HbA
2
i 20% HbF. Chorzy umierają
przeważnie w dzieciństwie.
Heterozygoty mają 30-45% HbS, 70-55%HbF
i małą ilość HbA
2
. Te osoby, z powodu
skróconego czasu przeżycia krwinek
czerwonych, są chronione przed
zachorowaniem na zimnice
i są lepiej dostosowane do rejonów
malarycznych, dlatego cześciej wystepują w
danej społeczności. Czesto te osoby
wykazują zapalenie kości wywołane przez
Salmonella.
Test na „sierpowacenie" ujawnia sie
wyraźnie dopiero wtedy, gdy krew umieści
sie na 6-24 godziny w środowisku bez
dostepu tlenu (pod uszczelnionym
szkiełkiem nakrywkowym).
Sierpowatokrwinkowość
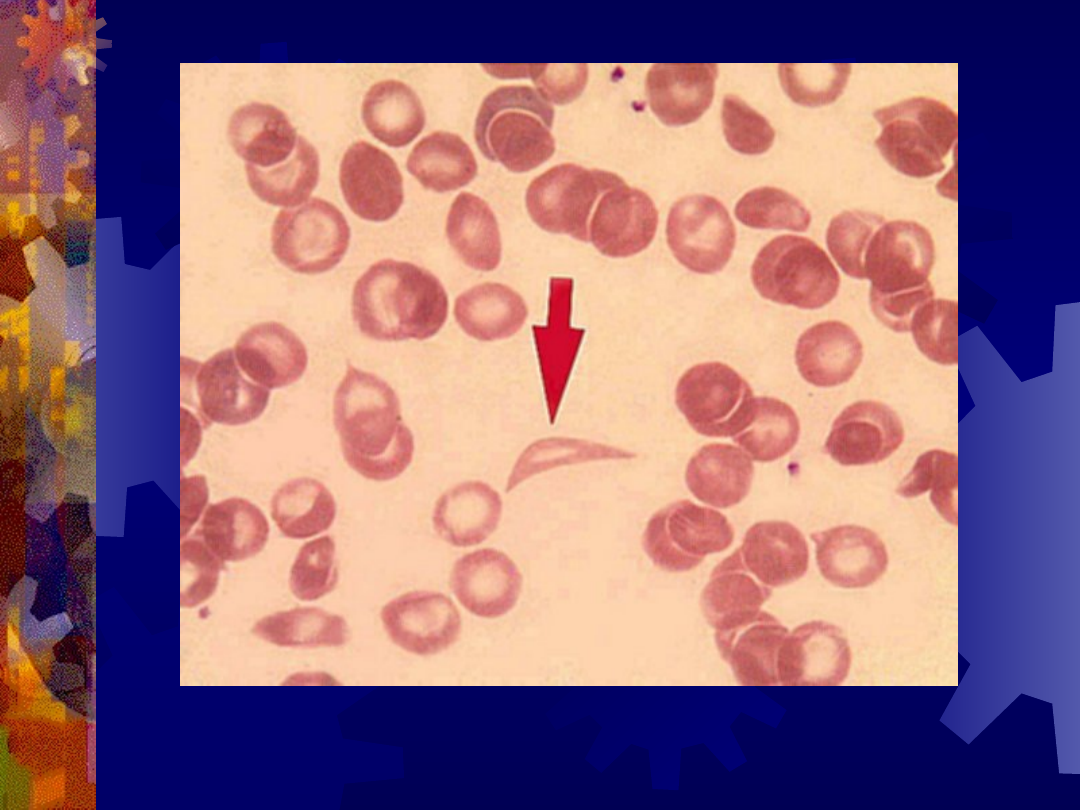
Drepanocyt (strzałka)

Methemoglobinemia
1. Dziedziczna aut. dominująco wada
hemoglobiny utrudniająca jej
redukcję methemoglobiny do
hemoglobiny
2. Dziedziczna aut. Recesywnie
niedobór reduktazy
methemoglobinowej
3. Nabyta: ekspozycja na utleniacze
(lidokaina, nitraty, sulfasalazyna,
sulfonamidy, pochodne benzenu)
Klinika: przewlekła hemoliza
wewnątrznaczyniowa
30-40% methemoglobinemii –
śmiertelne
Leczenie: infuzja błękitu
metylenowego 1-2mg/kg

Karboksyhemoglobina
Wadliwa instalacja wentylacyjna i
kominowa
Palacze cygar i papierosów
50% karboksyhemoglobinemii –
śmiertelne
Leczenie – tlenoterapia,
transfuzje ME

Talasemia
Talasemia - zaburzenia syntezy łańcucha
hemoglobiny, nadprodukcja łańcucha
Delecja 4 loci łańcucha hemoglobiny -
powstają tetramery łańcucha -
hemoglobina Barta – postać śmiertelna w
okresie płodowym lub noworodkowym
Delecja 3 loci łańcucha hemoglobiny –
powstaje hemoglobina H –nietrwała –
krwinki tarczowate, ciałka Heinza;
objawy w okresie dzieciństwa lub
dojrzewania
Delecja 2 loci łańcucha hemoglobiny –
nosicielstwo talasemii – umiarkowana
niedokrwistość niedobarwliwa,
mikrocytarna – często mylona z n.
sideropeniczną
Delecja 1 loci łańcucha hemoglobiny –
bezobjawowe nosicielstwo talasemii

Talasemia
Talasemia - zaburzenia syntezy łańcucha
hemoglobiny, nadprodukcja łańcucha
Powstają:
hemoglobina A
2
–
2
2
hemoglobina F –
2
2
Klinika: niedokrwistość mikrocytarna,
obecność krwinek tarczowatych,
hepatosplenomegalia, erytropoeza
pozaszpikowa (kości czaszki – mongołowaty
wyraz twarzy), niewydolność krążenia,
nerek, kamica żółciowa, zahamowanie
wzrostu, wtórna hemosyderoza
Postać major (homozygoty) – ciężki
przebieg ze zgonem w ciągu kilku
miesięcy/lat
Postać minor (heterozygoty) – postać
łagodna
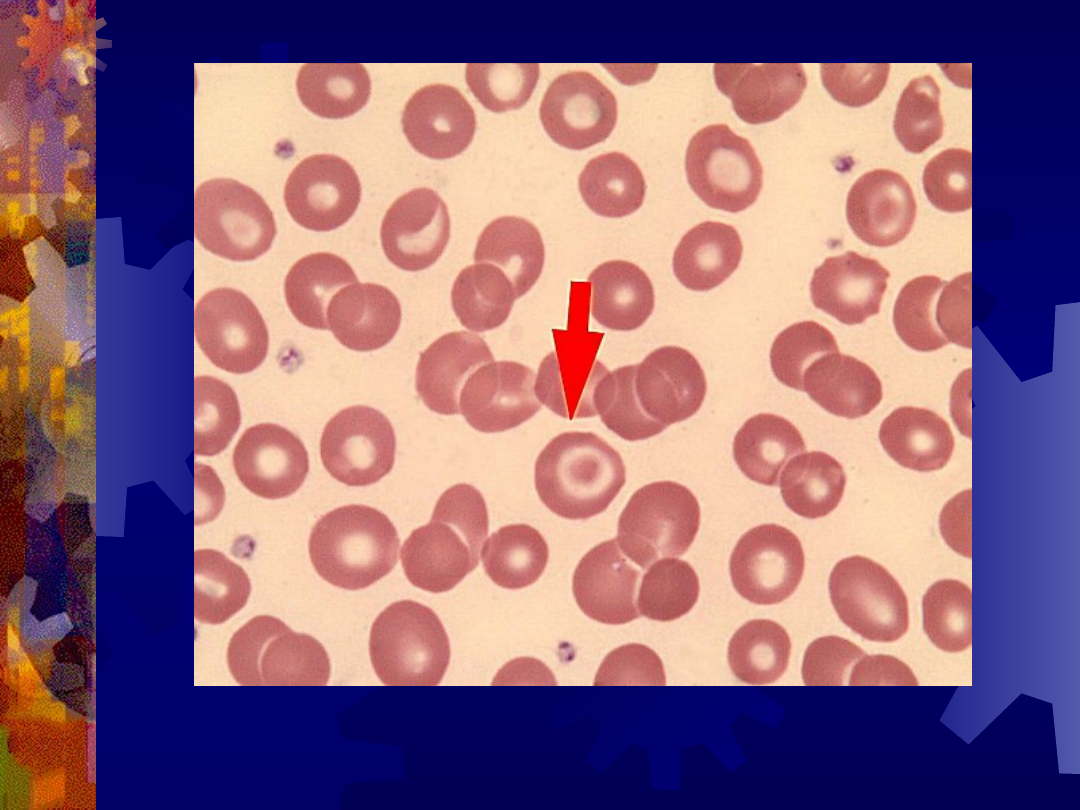
Erytrocyt tarczowaty (strzałka)
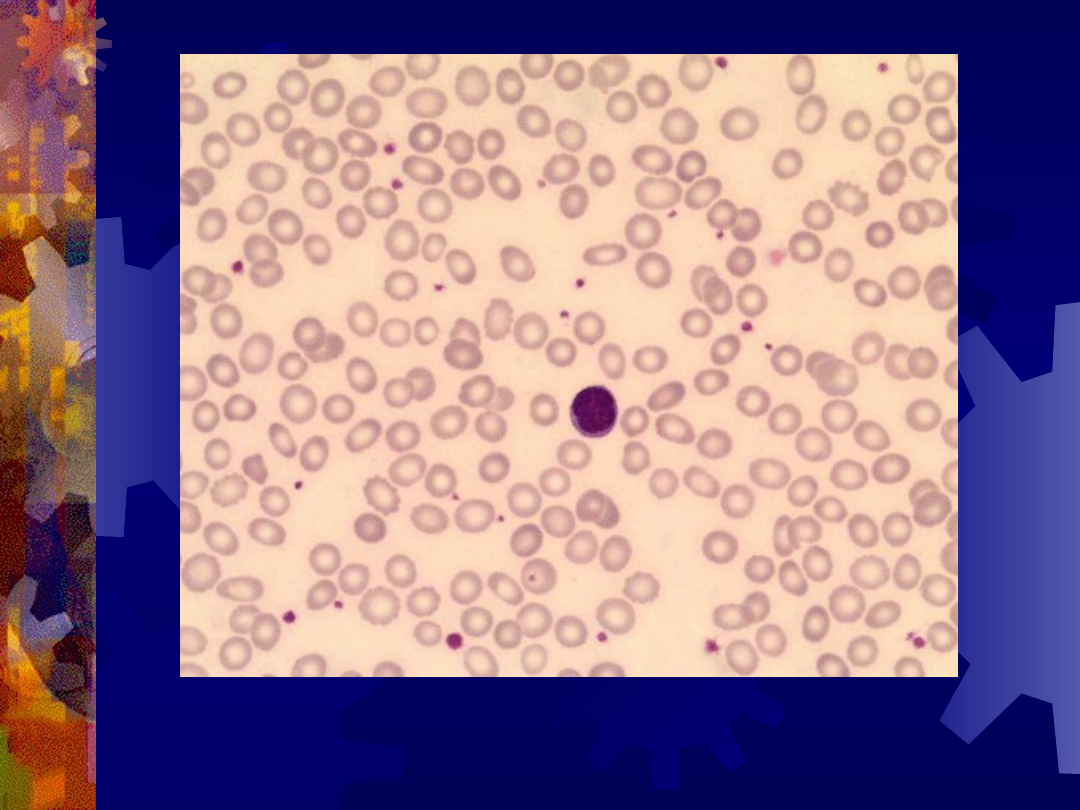
Mikrocyty
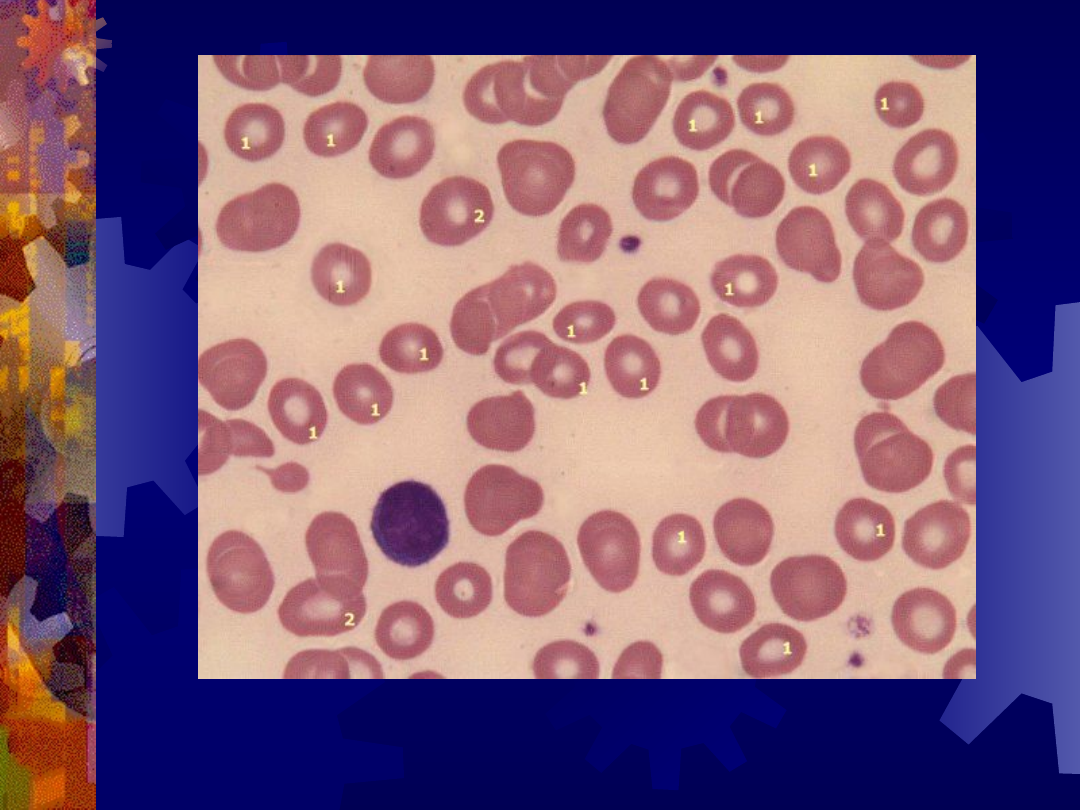
1 – mikrocyty
2 – normocyty

Podział nabytych niedokrwistości
hemolitycznych
zewnątrzkrwinkowe niedokrwistości
hemolityczne
n. autoimmunohemolityczne AIHA
n. h. polekowe
n. h. toksyczne
hipersplenizm
n. mikroangiopatyczne
n. makroangiopatyczne
nocna napadowa hemoglobinuria
wewnątrzkrwinkowe niedokrwistości
hemolityczne
nocna napadowa
hemoglobinuria
zewnątrzkrwinkowe i
wewnątrzkrwinkowe niedokrwistości
hemolityczne
niedokrwistość megaloblastyczna,
sideropeniczna

Przyczyny niedokrwistości
autoimmunohemolitycznych
Samoistne (70%)
Objawowe: NHL
(41,5%)
SLE
(24,5%)
przewlekłe zapalenie wątroby
(10,7%)
przewlekłe zakażenia (8,9%)
grypa
RZS
(3,6%)
gruźlica
ziarnica złośliwa
sarkoidoza
rakowiak (1,8%)
Choroba zimnych aglutynin: samoistna
Mycoplasma pneumoniae
mononukleoza zakaźna
NHL
Zimna napadowa hemoglobinuria: samoistna
kiła
gruźlica
P
rz
e
ci
w
ci
a
ła
t
y
p
u
ci
e
p
łe
g
o
P
rz
e
ci
w
ci
a
ła
t
y
p
u
zi
m
n
e
g
o

Charakterystyka autoprzeciwciał
Typ
Ciepłe
Zimne
częstość
84%
16%
zakres temp.
37
o
C
0 - 32
o
C
typ reakcji
aglutynacja lub
hemoliza
aglutynacja lub
hemoliza
kompletność
niekompletne
Kompletne
klasa Ig
IgG
IgM
typ łańcucha
lekkiego
kappa/lambda
kappa
wiązanie
dopełniacza
+/-
+
swoistość
anty-Rh, Kell
I, i
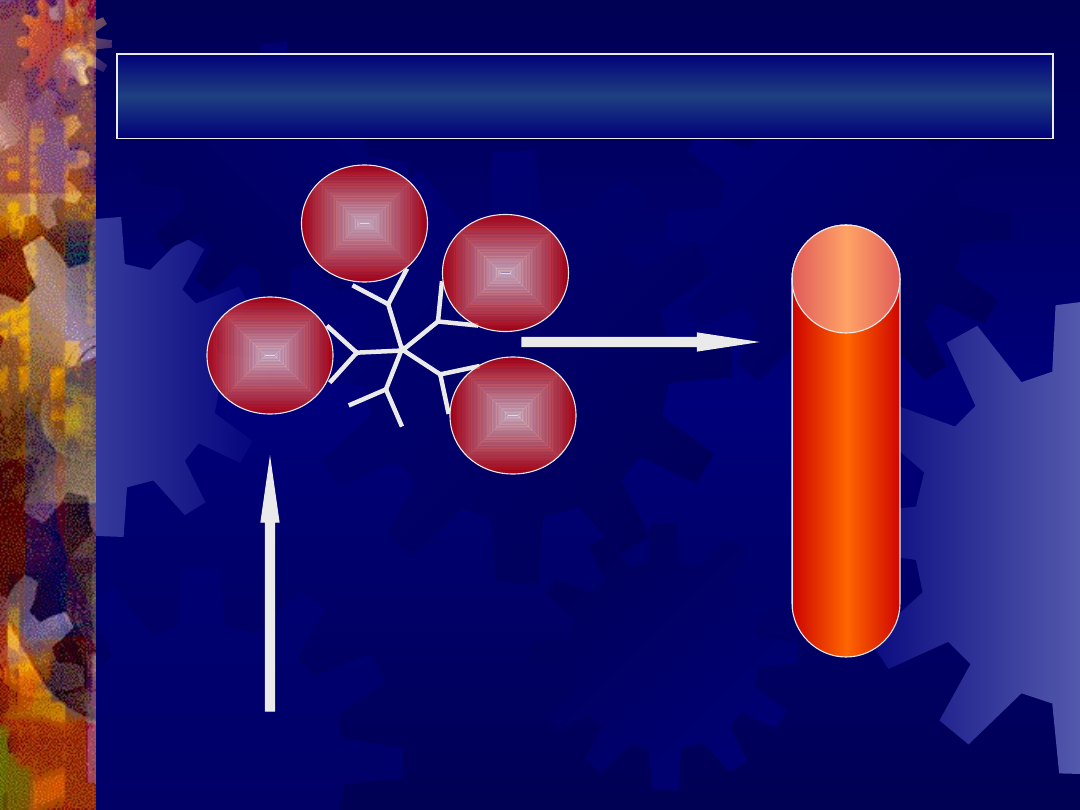
Mechanizm niszczenia erytrocytów przez
autoprzeciwciała IgM
IgM -
kompletne
DOPEŁNIACZ
aktywacja
cytolitycznych
składowych
dopełniacza
RBC
hemoliza
wewnątrznaczyni
owa
RBC
RBC
RBC
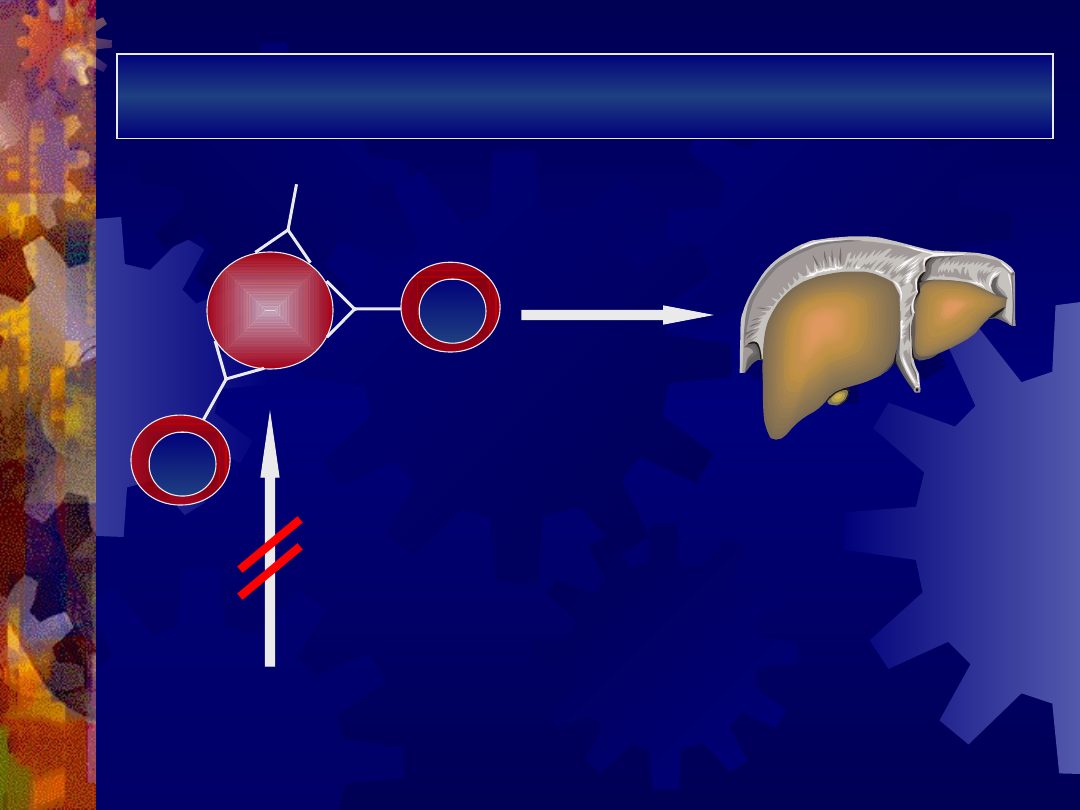
Mechanizm niszczenia erytrocytów przez
autoprzeciwciała IgG
IgG -
niekompletne
DOPEŁNIACZ
brak możliwości
aktywacji
dopełniacza
RBC
hemoliza w
układzie
siateczkowo-
śródbłonkowym
makrofa
g
makrofa
g

Polekowe niedokrwistości
hemolityczne - mechanizmy
Zainicjowanie reakcji immunologicznej
Działanie utleniające z powstaniem
methemoglobiny
Pogłębianie defektów metabolicznych
Denaturacja hemoglobiny

Polekowe niedokrwistości
autoimmunohemolityczne
1. Typ metyldopy – indukcja „odpowiedzi
immunologicznej” skierowanej
przeciwko autologicznym erytrocytom
poprzez:
modyfikację białka błony erytrocyta
modyfikację białka surowicy
modyfikację odczynowości
komórkowej
(ibuprofen, mefacit, cymetydyna,
fenytoina, metotrexat)
2. Bezpośredni udział leku w reakcji
antygen-przeciwciało
lek + białko błony erytrocyta =
antygen (penicylina, ampicylina,
cefalosporyny, cisplatyna)
lek + białko surowicy = kompleks
immunologiczny = antygen
(chinina, pyralgin, fenacetyna,
ryfampicyna, triamteren)

Niedokrwistość hemolityczna mikro-
(makro)angiopatyczna
Przyczyny:
nowotwory złośliwe – gruczolakorak
żoładka,
j. grubego, jajnika , sutka, trzustki,
płuca, prostaty, jądra,
chłoniaki nieziarnicze
z. hemolityczno-mocznicowy
nadciśnienie tętnicze faza złośliwa
naczyniaki mnogie lub olbrzymie
kolagenozy
toksyny bakteryjne
reakcja odrzucania przeszczepu
angiopatia cukrzycowa
patologia ciąży
protezy zastawek i naczyń
postać samoistna

Niedokrwistość hemolityczna mikro-
(makro)angiopatyczna
Patogeneza:
wewnątrznaczyniowa destrukcja
erytrocytów
w mechanizmie:
mechanicznym – zwiększone opory
krążenia erytrocytów przez
mikrokrążenie
chemicznym – wydzielanie substancji
tromboplastycznych, agregacji płytek,
aktywacji krzepnięcia, hamowanie
fibrynolizy, wytrącanie fibryny,
aktywacja dopełniacza
Klinika:
przebieg ostry (z. h.m.) lub przewlekły
objawy choroby podstawowej,
niedokrwistość, żółtaczka, skaza
krwotoczna patologiczne formy
erytrocytów we krwi, małopłytowość,
leukocytoza, BTA(-), PTA (-)

Nocna napadowa hemoglobinuria:
Zespół mieloproliferacyjny
defekt komórki
macierzystej
somatyczna mutacja genu PIG-A na chr.
X
niedobór glikozylo-fosfatydyloinozytolu (GPI)
nadmierna wrażliwość krwinek czerwonych,
płytkowych, granulocytów na cytolityczne działanie
dopełniacza ujawniające się w kwaśnym ph krwi w
nocy
Początek w 30-40rż, epizody głębokiej anemizacji z
oddawaniem ciemnego moczu, bólami brzucha, głowy
ok. lędźwiowej trwające dni lub tygodnie, skłonność
do zakrzepicy w naczyniach m.in. nerek, wątroby
pancytopenia, hipo- lub aplastyczny szpik,
hemoglobinuria,
stężenia żelaza,
retikulocytozy,
obecność fragmentocytów, akantocytów, sferocytów w
rozmazie krwi obwodowej
Rozpoznanie:
wywiad
test Hama
ocena ekspresji CD59 (MIRL, błonowy inhibitor
lizy)na erytrocytach, granulocytach, płytkach krwi
ocena ekspresji CD55 (DAF, czynnik przyspieszonego
rozpadu) na erytrocytach, granulocytach, płytkach
krwi
Leczenie: objawowe, sterydy, transplantacja szpiku

Niedokrwistość hemolityczną należy
podejrzewać:
W każdym przypadku współistnienia
niedokrwistości i żółtaczki
W każdej niedokrwistości o innych podłożu,
możliwy jest komponent hemolityczny
W przypadku następującej konstelacji badań
dodatkowych bez jednoczesnych objawów
krwotoku:
bilirubiny (pośredniej)
retikulocytozy
obecność fragmentocytów, akantocytów,
sferocytów
w rozmazie krwi obwodowej
Mimo braku objawów klinicznych niedokrwistości
występuje:
nadmierna wrażliwość obwodowych części ciała
na zimno
ostre bóle brzucha bez uchwytnej przyczyny
okresowe oddawanie ciemnego moczu
kamica żółciowa w młodym wieku
splenomegalia
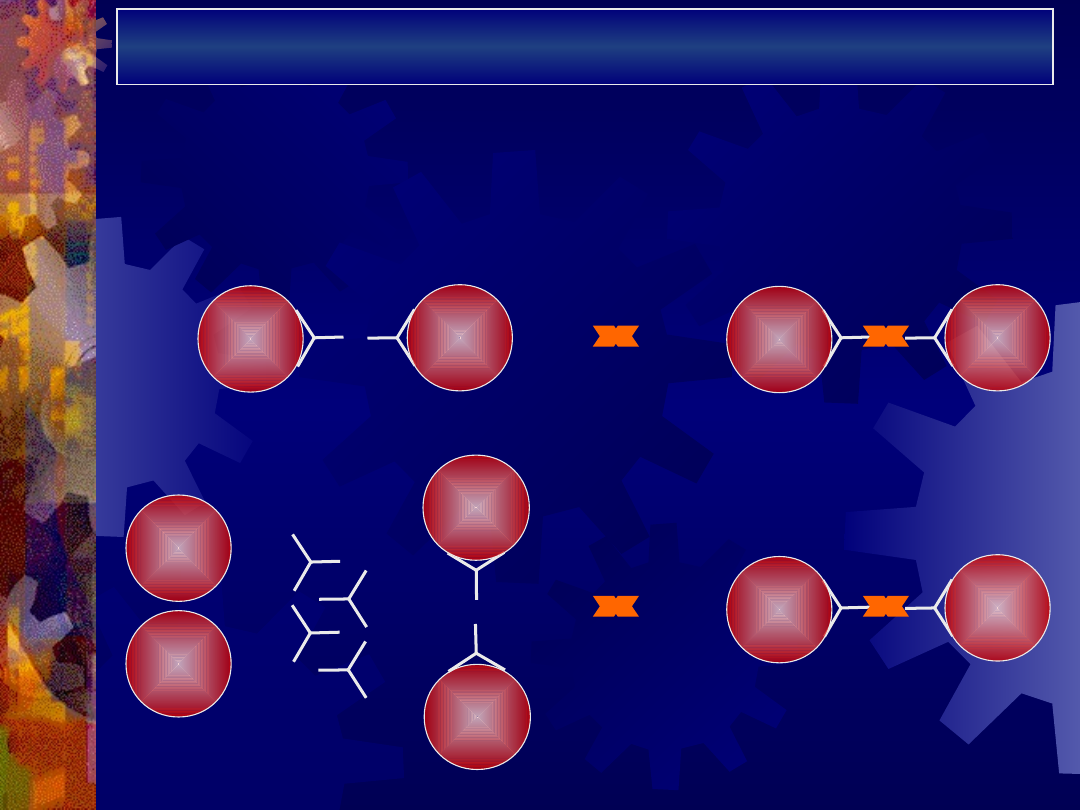
Diagnostyka niedokrwistości
hemolitycznej
Wywiad
Badanie fizykalne
Badanie morfologiczne krwi
Badanie biochemiczne
Badania serologiczne: BTA, PTA, test Hama, test
Donatha-Landsteinera
RBC
anty-
gamma-
globuliny
+
=
BTA
RBC
RBC
RBC
aglutynacja
RBC
anty-
gamma-
globuliny
+
=
PTA
RBC
RBC
RBC
aglutynacja
erytrocyty testowe
+
surowica
badana
R
B
C
R
B
C
=

Diagnostyka niedokrwistości
hemolitycznej
Oporność osmotyczna erytrocytów
(n = 4,5
– 3,0g/l NaCl)
Zmniejszenie oporności osmotycznej:
nabyte niedokrwistości hemolityczne,
sferocytoza wrodzona
Zwiększenie oporności osmotycznej:
niedokrwistość sideropeniczna
po splenektomii
talasemie
niedokrwistość sierpowatokrwinkowa
Czas przeżycia krwinek czerwonych
(n
51
Cr
28dni)
Wskazania:
potwierdzenie rozpoznania w
przypadkach niejasnych
różnicowanie przyczyn wewnątrz- i
zewnątrzkrwinkowych (krzyżowe
przetaczanie
51
Cr erytrocytów)
ocena wskaźnika śledzionowo-
wątrobowego – kwalifikacja do
splenektomii (n=1,3:1)

W przypadku podejrzenia niedokrwistości
hemolitycznej
Dowody rozpadu krwinek czerwonych:
Badanie rozmazu krwi
Oznaczanie steżenia bilirubiny w surowicy
Oznaczanie wydalania urobilinogenu z moczem
Oznaczanie haptoglobiny w osoczu
Dowody odnowy w układzie krwinek czerwonych:
Oznaczanie liczby retikulocytów
Badanie rozmazu krwi
Badanie rentgenowskie układu kostnego
Diagnostyka niedokrwistości
hemolitycznej

Stwierdzenie niedokrwistości hemolitycznej typu
krwinkowego
Rozmaz krwi
Test oporności osmotycznej
Test autohemolizy
Hemoglobina
Test na sierpowatość
Elektroforeza hemoglobiny
Oznaczanie hemoglobiny F
Test termostabilności
Test Hama
CD59, CD55
Zaburzenia enzymatyczne
Udokumentowanie obecności ciałek Heinza
Testy enzymatyczne
Diagnostyka niedokrwistości
hemolitycznej

Stwierdzenie pozakrwinkowego typu
niedokrwistości hemolitycznej
Odczyn Coombsa
Test na obecność kwaśnych hemolizyn
Test sacharozowy
Test na obecność przeciwciał typu Donatha-
Landsteinera
Test na obecność przeciwciał przeciwjądrowych (ANA)
Diagnostyka niedokrwistości
hemolitycznej

Leczenie niedokrwistości
hemolitycznej
1. Przyczynowe: leczenie choroby
podstawowej,
eliminacja leku lub innej
substancji
unikanie oziębienia ciała
2. Objawowe:
glikokortokosteriody
(prednizon 1mg/kgmc.)
puls sterydowy: 0,5-1,0g
metylprednisolonu/dz.
cyklofosfamid 1-1,5 mg/kg.mc.
azatiopryna 2mg/kgmc.
plazmafereza
transfuzja (wymienna) ME
unikanie transfuzji w AIHA
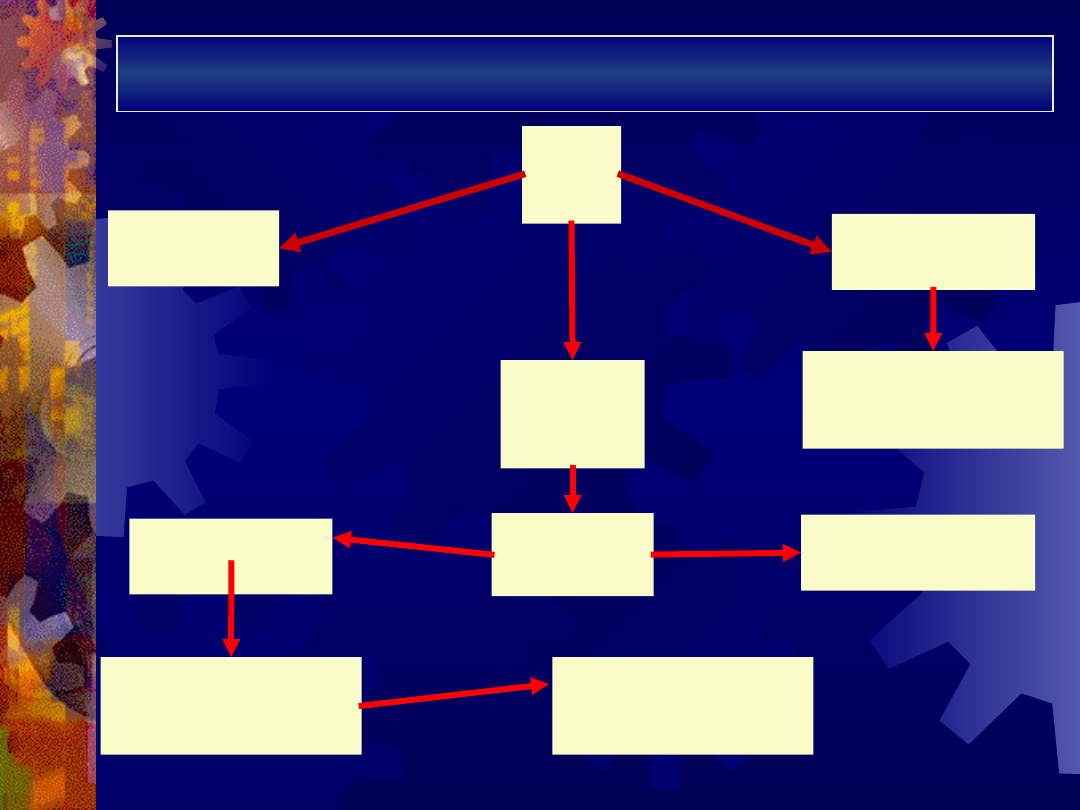
Algorytm leczenia AIHA
AIH
A
splenektomi
a
Prednis
on
1mg/kg
odpowi
edź
redukcja
dawki
Puls
sterydowy
obserwac
ja
ciężka
łagodna
2-3
tyg.
TAK
NIE
Puls
sterydowy
splenektomi
a
chemioterapi
a
brak
odpowiedz
i
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
Wyszukiwarka
Podobne podstrony:
Choroba hemolityczna p odu na kurs
Anemie hemolityczne nieimmunologiczne
Niedokrwistości hemolityczne, Analityka medyczna, Hematologia
34. Żółtaczka hemolityczna, MEDYCYNA VI rok, Pediatria, PEDIATRIA CAŁOŚĆ, Ustny PEDIATRIA Balwierz
NIEDOKRWISTO¦Ć HEMOLITYCZNA
choroba hemolityczna(1) ppt
Zespół hemolityczno mocznicowy
Hemoliza, studia pielęgniarstwo
KONFLIKT SEROLOGICZNY choroba hemolityczna p odu
Niedokrwistości hemolityczne, MEDYCYNA i RATOWNICTWO, Pediatria
Niedokrwistości hemolityczne 3
(32) Leki stosowane w niedokrwistościach niedobrowolnych i hemolitycznych
choroba hemolityczna
Choroba hemolityczna noworodków, MEDYCYNA i RATOWNICTWO, Pediatria
Niedokrwistości hemolityczne
więcej podobnych podstron