
Zespół hemolityczno -
mocznicowy
Magdalena Silska
Klinika Kardiologii i Nefrologii Dziecięcej
Uniwersytet im. K. Marcinkowskiego w Poznaniu

Zespół hemolityczno-mocznicowy
(ang. haemolytic-uraemic syndrome,
HUS)
wielonarządowy zespół chorobowy –
mikroangiopatia zakrzepowa
występowanie - głównie
niemowlęta i małe dzieci
klasyczna triada objawów:
niedokrwistość hemolityczna
mikroangiopatyczna
trombocytopenia
ostra niewydolność nerek
–
najczęstsza
przyczyna ONN u dzieci <3r.ż.
HUS – mikroangiopatia zakrzepowa
mikrozakrzepy z fibryny, trombocytów i
czynnika von Willebranda – uszkodzenie
śródbłonków drobnych naczyń
tętniczych
Różnicowanie z TTP (thrombotic
thrombocytopenic purpura, zakrzepowa
plamica małopłytkowa):
zwykle osoby dorosłe
zajęcie ośrodkowego
układu nerwowego
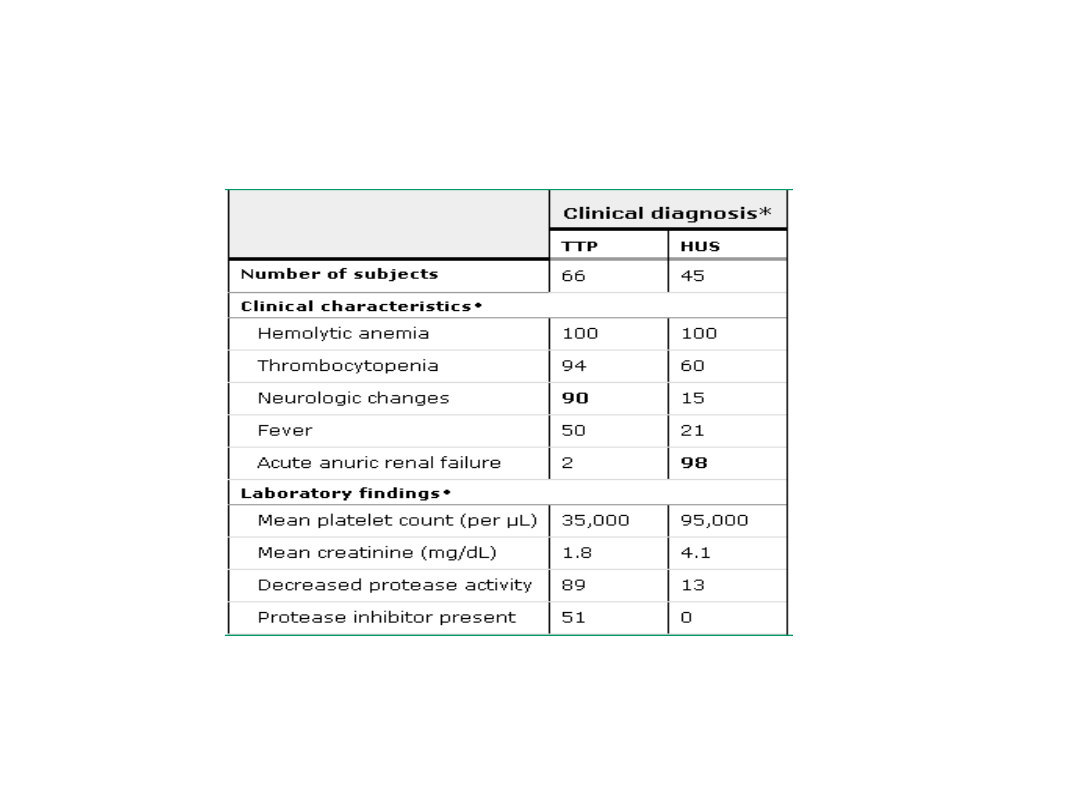
HUS/TTP - Różnicowanie
Veyradier, A, 2001, Blood 98:1765-1772.

Podział
Postać typowa D „+” (ang. diarrhoea
plus )
poprzedzona biegunką infekcyjną
ponad 90% przypadków HUS u dzieci
głównie < 3 r.ż.
Postać atypowa D „-” (ang. diarrhoea
minus)
5-10% przypadków HUS
w każdym wieku, dominuje u dzieci > 3
r.ż. i młodzieży
Postać wtórna

Etiologia
Postać typowa indukowana infekcją bakteryjną:
Escherichia coli serotyp O157:H7, rzadziej inne
serotypy (O111:H8, O103:H2, O121, O145, O26,
0113)
Inne bakterie Gram-ujemne (Schigella, Salmonella,
Campylobacter, Yersinia, Bartonella, Clostridium)
Mechanizm oparty na produkcji toksyn Shiga-
podobnych (Stx, werotoksyna) – 90% przypadków
HUS u dzieci
Egzotoksyna cytotoksyczna wobec
hodowli komórek linii Vero,
zależna od lizogennego zakażenia
bakteriofagiem

Patogenna E.coli
naturalny rezerwuar – bydło domowe (ok. 1%)
transmisja zakażenia:
droga pokarmowa (surowe, niedogotowane
mięso, niepasteryzowane mleko, skażone
owoce i warzywa)
rzadko bezpośredni kontakt ze zwierzętami,
skażone akweny wodne
tzw. choroba brudnych rąk
ok. 15% pacjentów z biegunką krwotoczną
wywołaną
patogenną E. coli rozwija objawy HUS
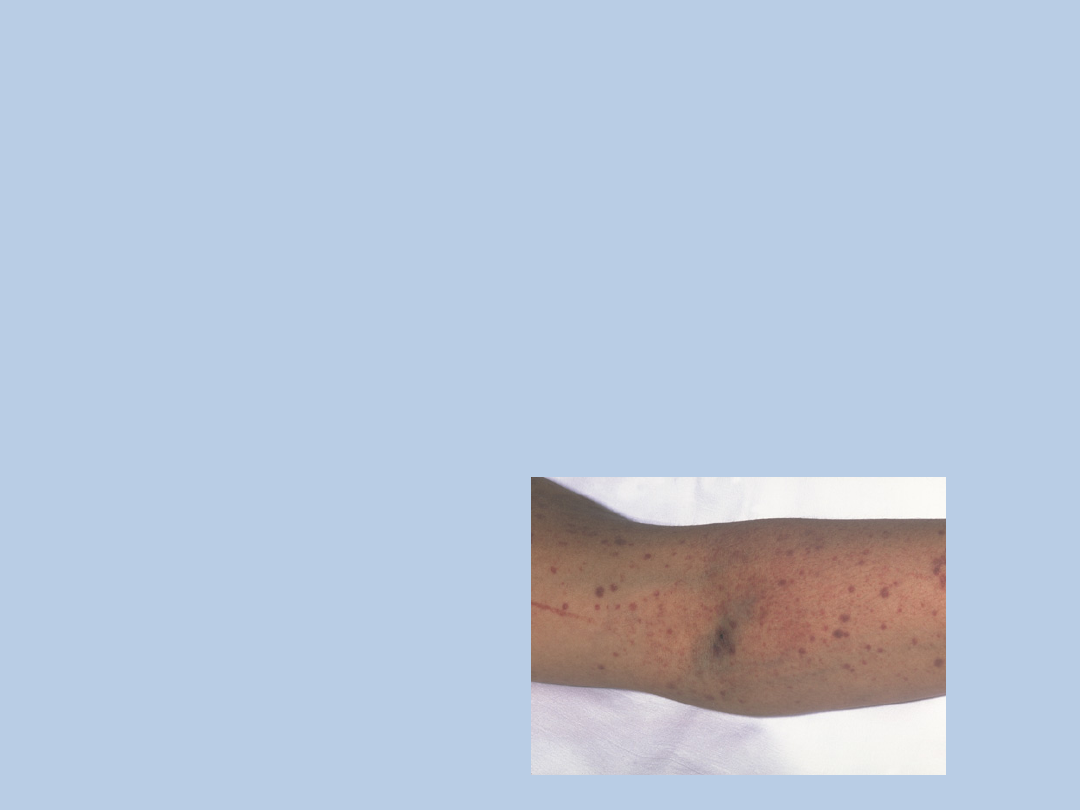
Objawy alarmowe: bladość, żółtaczka,
wybroczyny, krwawienia, pogorszenie stanu
ogólnego pacjenta, spadek diurezy,
nadciśnienie tętnicze, obrzęki
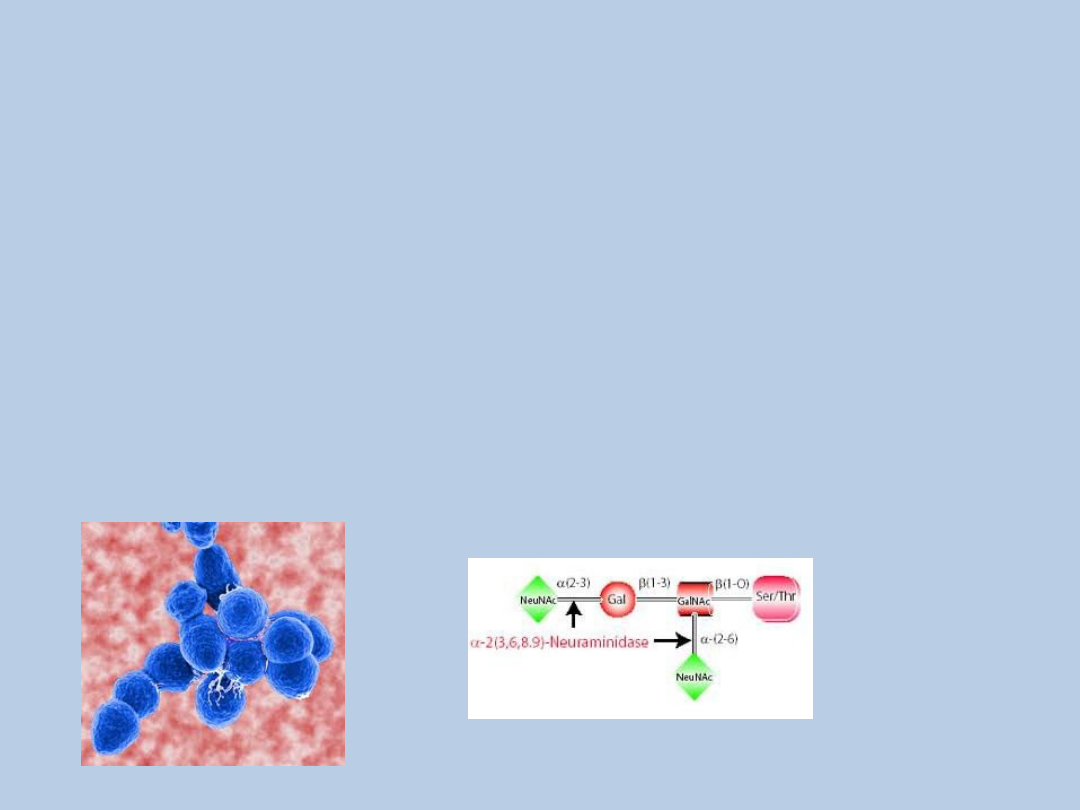
Inne czynniki infekcyjne –
atypowy HUS
Streptococcus pneumoniae –
neuraminidaza ( odsłania antygen
Thomsen-Friedenreich(T)
VZV, ECHO, Coxackie A i B, CMV, HIV
Cochran, JB, 2004, Pediatr Nephrol 19:317-321.

Patomechanizm
połączenie podjednostki B Shiga-toksyny z
receptorem Gb3 błony komórki docelowej
(naczynia kłębuszków i cewek nerkowych)
wniknięcie podjednostki A Shiga-toksyny do
wnętrza komórki
uszkodzenie rybosomalnego RNA –
zahamowanie translacji – śmierć komórki
śródbłonka
agregacja płytek krwi – zwężenie światła
małych naczyń – fragmentacja erytrocytów
Stx uwrażliwia komórki śródbłonka na
toksyczne działanie hemu
HUS - D „+”
Charakterystyka laboratoryjna:
rozmaz krwi obwodowej – schizocyty („skorupki”)
↑LDH
↓haptoglobina
↑ kreatynina
małopłytkowość(wyraźniej zaznaczona w zakrzepowej plamicy
małopłytkowej niż w zespole hemolityczno-mocznicowym)
Powikłania:
30-50% - rozwój przewlekłej choroby nerek
3-5% - śmiertelność
uszkodzenie mózgu
(udar i/lub obrzęk mózgu)
uszkodzenie serca
uszkodzenie płuc
martwica lub perforacja jelit
niewydolność wielonarządowa
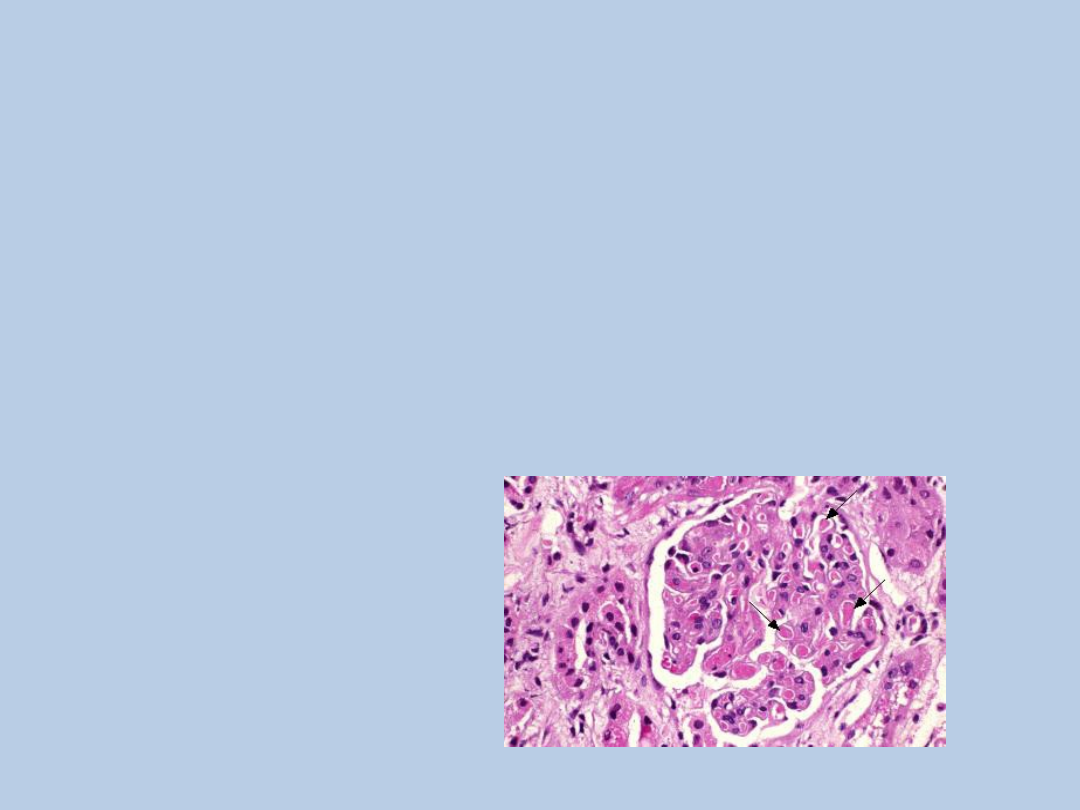
Zakrzep w kapilarach nerkowych
Etiologia
Postać atypowa:
Zaburzenia w obrębie układu dopełniacza:
• Genetycznie uwarunkowane
• Nabyte, przeciwciało przeciwko czynnikowi H
Niedobór proteazy czynnika von Willebranda (vWF-CP):
• Mutacja genu ADAMTS13
• Nabyty niedobór vWF-CP:
zaburzenia autoimmunologiczne,
polekowe
Defekt metabolizmu kobalaminy
Indukowany kininami
Walport M. N Engl J Med
2001;344:1058-1066
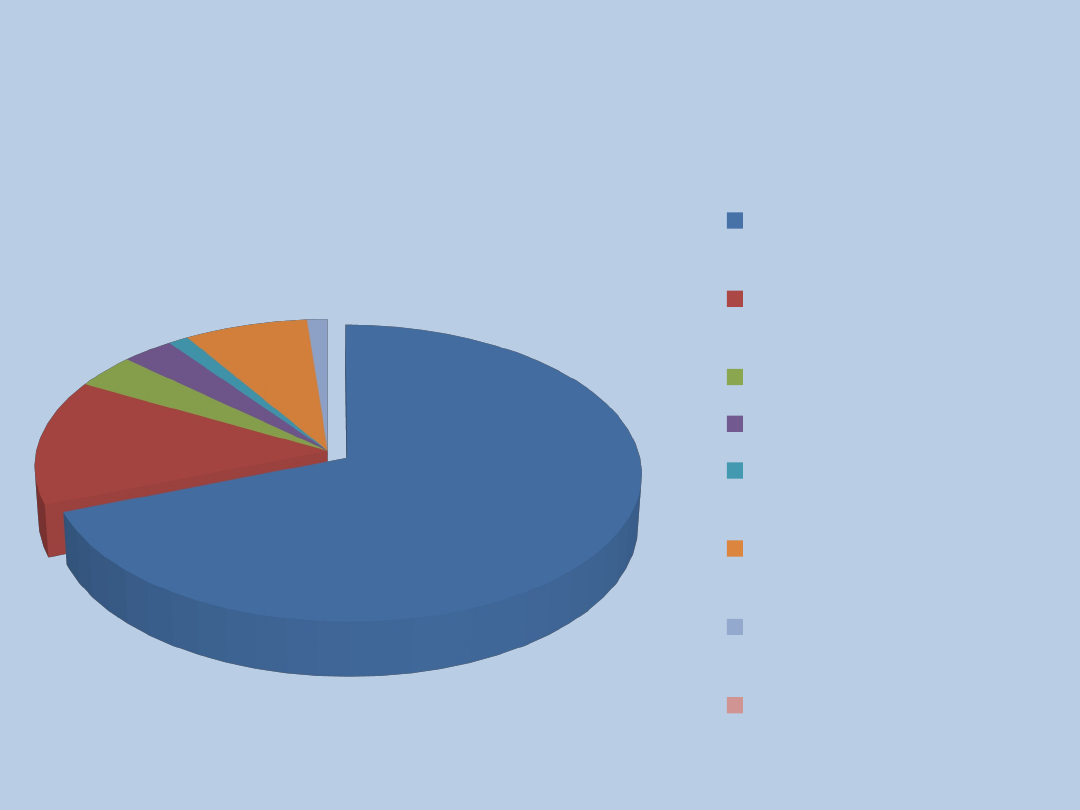
The European Paediatric
Research Group For D(-) HUS
Etiologia nieznana
(110)
Mutacja/niedobór
czynnika H (22)
Mutacja czynnika I (6)
Mutacja MCP (5)
Przeciwciało przeciwko
czynnikowi H (2)
Niedobór protezy vWF
(12)
Posocznica
pneumokokowa (12)
Mutacja FH i nidobór
proteazy (2)
167 pacjentów
Badanie w latach 2000-2004r.
Besbas e.al. , KI
2006
.
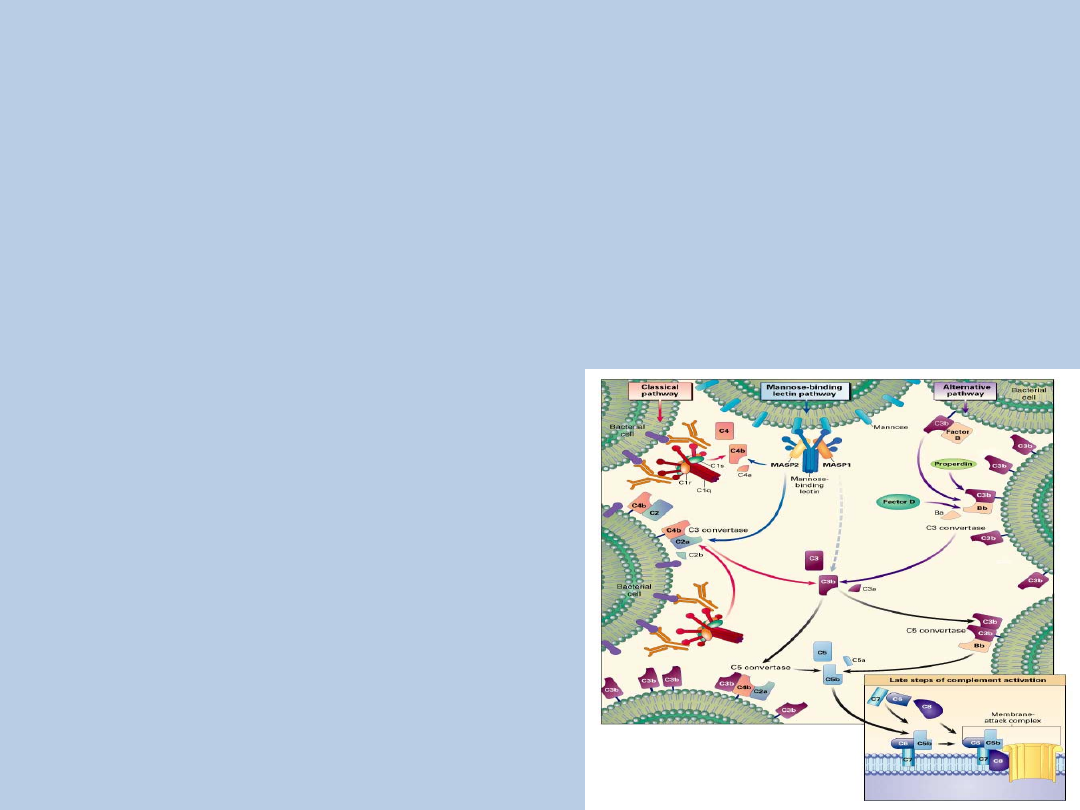
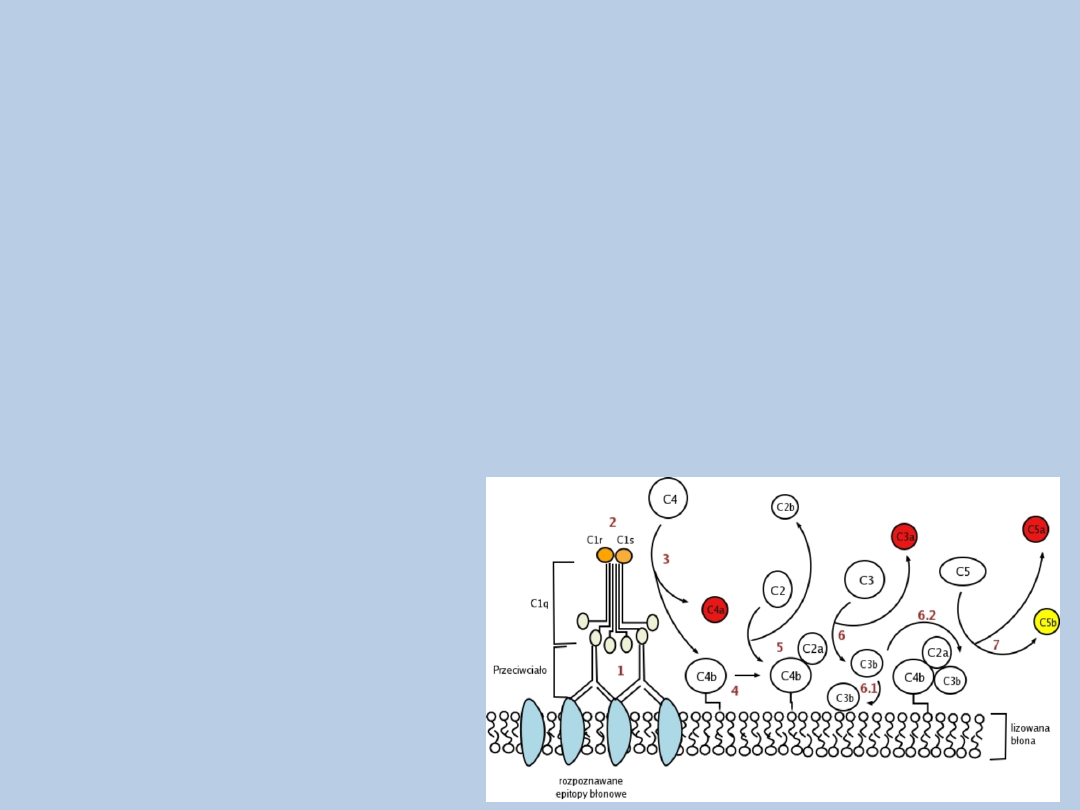
Aktywacja układu dopełniacza
Droga klasyczna
1. Przyłączenie się cząsteczki C1q do przeciwciał związanych z antygenem –
zmiana konformacji
2. Wzbudzenie proteazy serynowej C1r – aktywacja proteazy C1s
3. Aktywowana C1s - rozkład białek C4 i C2
4. Fragment C4a uwalniany do osocza, C4b związany z błoną komórkową
5. Kompleks C4b2a – konwertaza C3 drogi klasycznej
6. Konwertaza C3 – rozkład białka C3
7. C3b – możliwość wiązania do błony komórkowej patogenu (opsonina)
lub konwertazy C3 (konwertaza C5)
8. Konwertaza C5- rozkład białka C5 –
powstawanie MAC
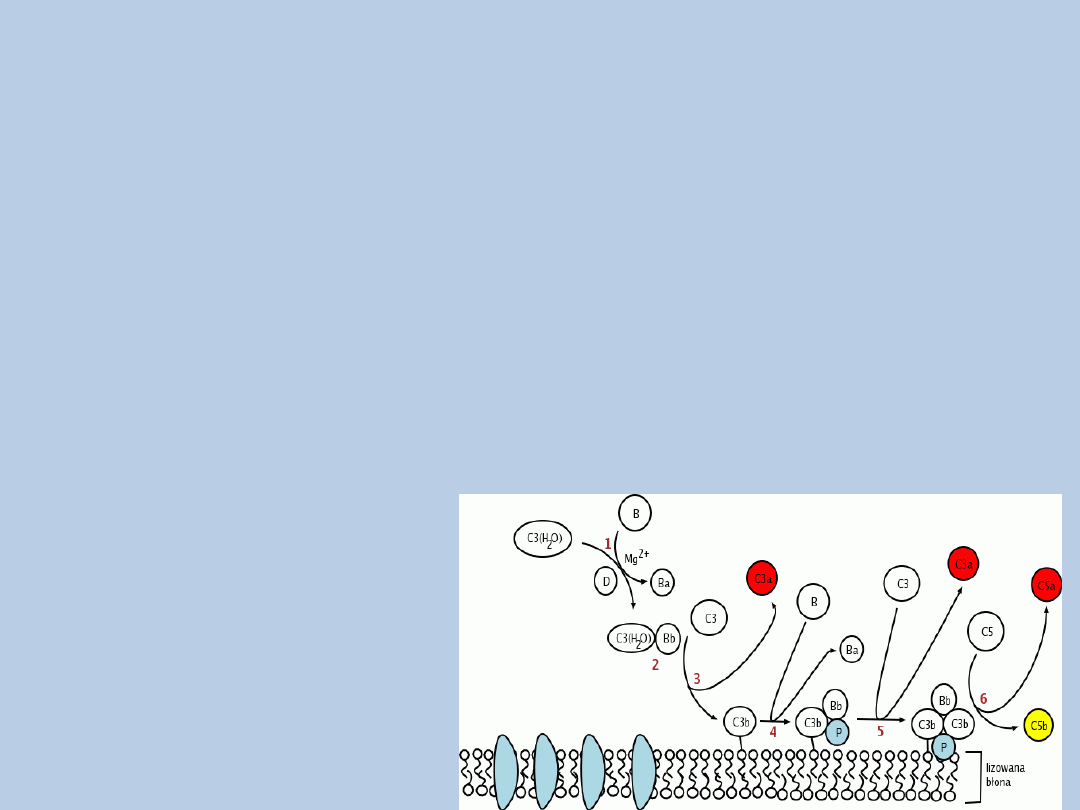
Aktywacja układu dopełniacza
Droga alternatywna
1. Pobudzona forma C3(H2O) - związanie czynnika B
2. Rozkład czynnika B w obecności jonów magnezu i czynnika D
3. Kompleks C3(H2O) i fragment Bb – rozpuszczalna konwertaza C3 drogi alternatywnej
4. Konwertaza C3 – rozkład białka C3
5. Kompleks C3b i czynnik B – rozkład czynnika B
6. Powstanie błonowej konwertazy C3 drogi alternatywnej, a następnie konwertazy C5
7. Czynnik P (properdyna) – stabilizacja błonowej konwertazy C3
8. C5b – tworzenie MAC
C3b - czynnik łączący drogę
klasyczną i alternatywną

Aktywacja układu dopełniacza
Droga lektynowa
1. Zbliżona do drogi klasycznej – odmienne pierwsze etapy aktywacji
2. Swoista reakcja antygen – przeciwciało zastąpiona nieswoistą
reakcją z udziałem kolektyn
3. Kolektyny – białka surfaktantu płucnego A i D oraz
osoczowa lektyna wiążąca mannozę (MBL)
4. MBL - główny czynnik zapoczątkowujący drogę lektynową
5. Kompleks MBL – antygen –zmiana konformacji – aktywacja
proteazy serynowej MASP-1
6. aMAPS-1 – aktywacja MAPS-2
7. aMAPS-2 – rozkład białek C2 i C4
8. Droga lektynowa może zapoczątkować reakcję obronną
bezpośrednio po wniknięciu patogenu do organizmu

Regulacja układu dopełniacza
Czynniki osoczowe
Czynnik H:
• wiąże składową dopełniacza C3b
• wspomaga czynnik I w hamowaniu konwertazy C3 drogi
alternatywnej
Czynnik I:
• rozkłada formy wolne i związane w konwertazach C3b i C4b
• powoduje rozpad konwertaz
Inhibitor C1:
• hamuje aktywne proteazy C1r i C1s
Białko wiążące C4:
• wiąże C4b
• wspomaga czynnik I w hamowaniu konwertazy C3 drogi klasycznej
Białko S
• blokuje C5b67
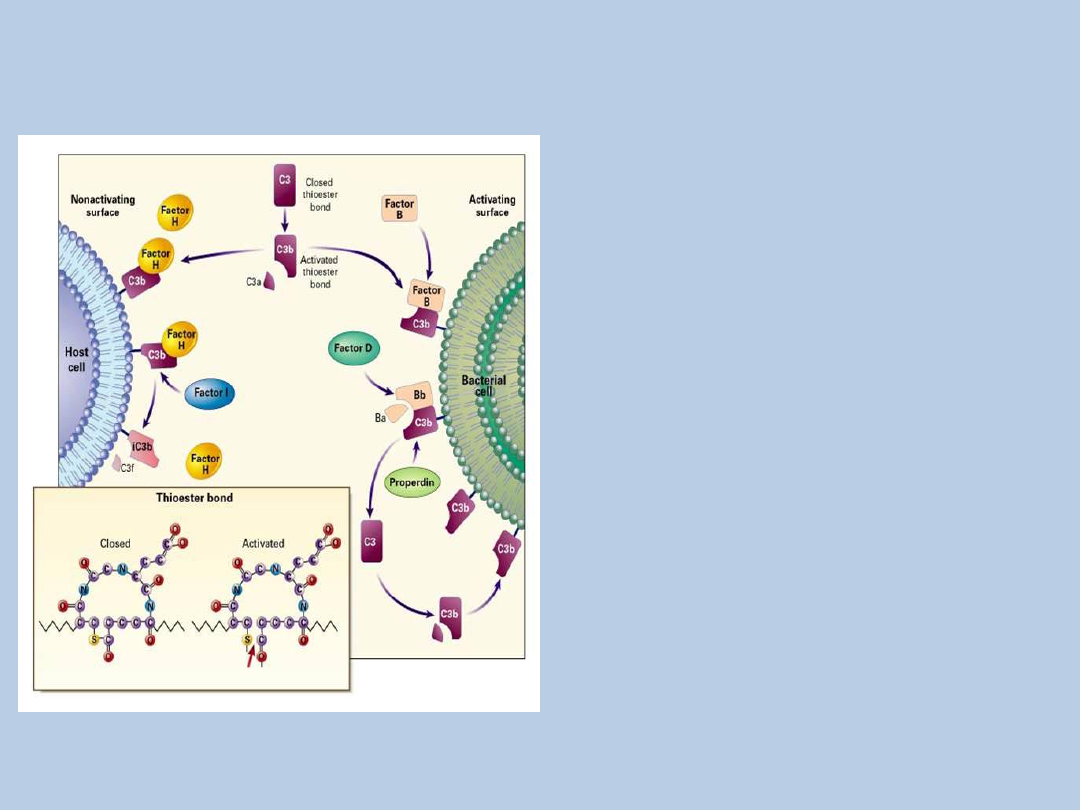
Regulacja rozkładu
Układ dopełniacza
Walport M. N Engl J Med
2001;344:1058-1066
C3 przez czynnik H
oraz czynnik I
Niedobór czynnika H
przyczyna 10-22% przypadków atypowego HUS
postaci sporadyczne i rodzinne
okres niemowlęcy lub wczesne dzieciństwo
sporadycznie osoby dorosłe
poziomy FH, C3, FB i CH
50
:
homozygoty – niskie
heterozygoty – wartości od niskich
do normalnych
nabyty niedobór C3 lub końca C’
infekcja meningokokowa
niedobór FH i C2
SLE
Dragon-Durey, M-A, et al, 2004, J Am Soc Nephrol 15:787-795.

Niedobór czynnika I
sporadyczne przypadki atypowego HUS
najczęściej mutacje heterozygotyczne
bez zwiększonej podatności na infekcje
homozygotyczność niedoboru FI
zwiększona podatność na infekcje
bakterie bezotoczkowe
(meningococcus, pneumococcus, hemophilus)
różnorodność penetracji i ekspresji
wartość C3 może być niska lub normalna
nabyty niedobór C3 spowodowany nadmiernym zużyciem
Dragon-Durey, M-A, et al, 2005, Springer Semin Immun
27:359-374.
Kavanagh, D, et al, 2005, J Am Soc Nephrol 16:2150-2155.


Regulacja układu dopełniacza
Czynniki komórkowe
receptor dla dopełniacza (CR1):
• rozkład C3b i C4b
• inaktywacja konwertazy
błonowy kofaktor białkowy (ang. Membrane Cofactor Protein, MCP, CD46)
• wiązanie C3b i C4b
• obecny na wszystkich komórkach jądrzastych organizmu
czynnik przyspieszający rozkład (ang. Decay Accelerating Factor, DAF)
• skraca okres półtrwania konwertaz
czynniki restrykcji homologicznej(ang. Homologous Restriction Factor, HRF,
CD59)
• wiązanie C8 i C9
• hamowanie tworzenia MAC
MIRL (ang. Membrane Inhibitor of Reactive Lysis)
• blokuje przyłączenie C9 i tworzenie MAC.
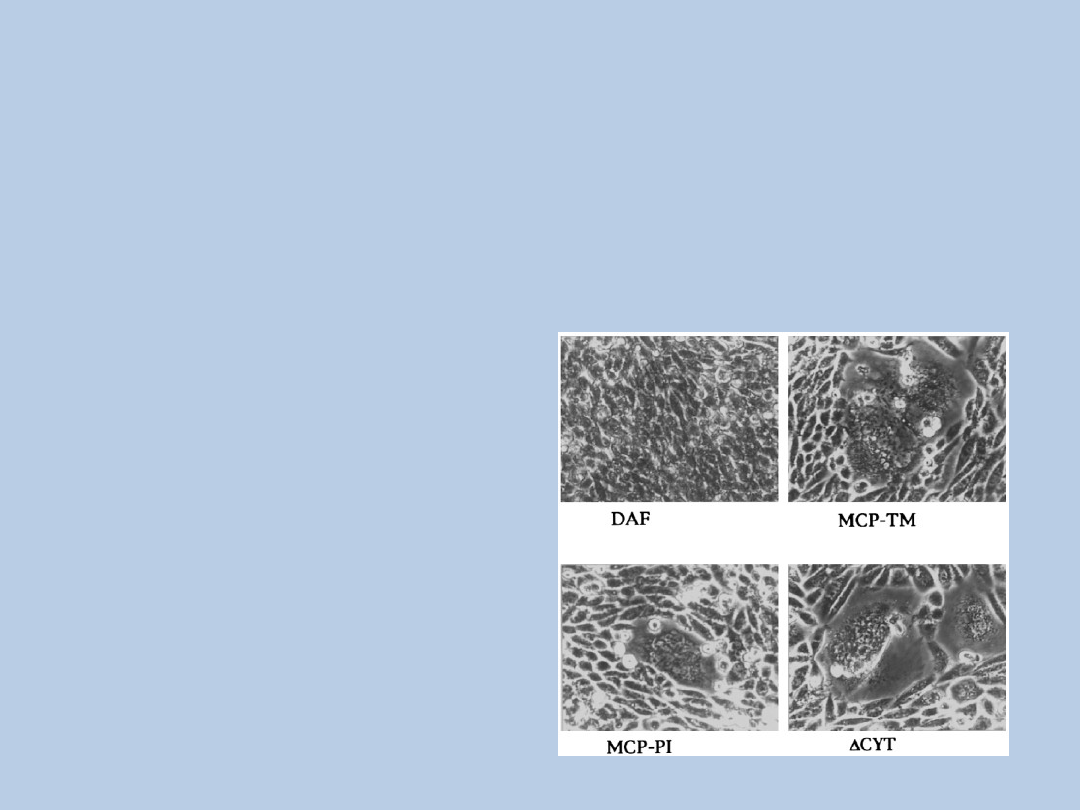
BŁONOWY KOFAKTOR BIAŁKOWY
(MCP = CD46)
~65 kD glikoproteina przezbłonowa
obecny na leukocytach, płytkach krwi, komórkach endotelialnych i
epitelialnych, fibroblastach, w nerkach
kofaktor czynnika osoczowego czynnika I
receptor dla patogenów:
wirus odry
Adenowirusy
HHV-6
dwoinki i streptokoki grupy A
http://www.biochem.ucl.ac.uk/~becky/FH//proteinInfo.php?protein=MCP

Patogeneza-atypowy HUS
infekcja/stan zapalny – wzrost liczby C3b
aktywacja kaskady dopełniacza oraz C3a/C5a
C3a/C5a:
migracja leukocytów
produkcja TNF i IL-8
cytokiny powodują uszkodzenie śródbłonka co prowadzi do ekspozycji macierzy
pozakomórkowej
dalszy wzrost składowej C3b i aktywacja dopełniacza
Niedobór czynnika H, czynnika I lub MCP jest
przyczyną 50% przypadków atypowego HUS
http://www.biochem.ucl.ac.uk/~becky/FH/proteinInfo.php?protein=FH
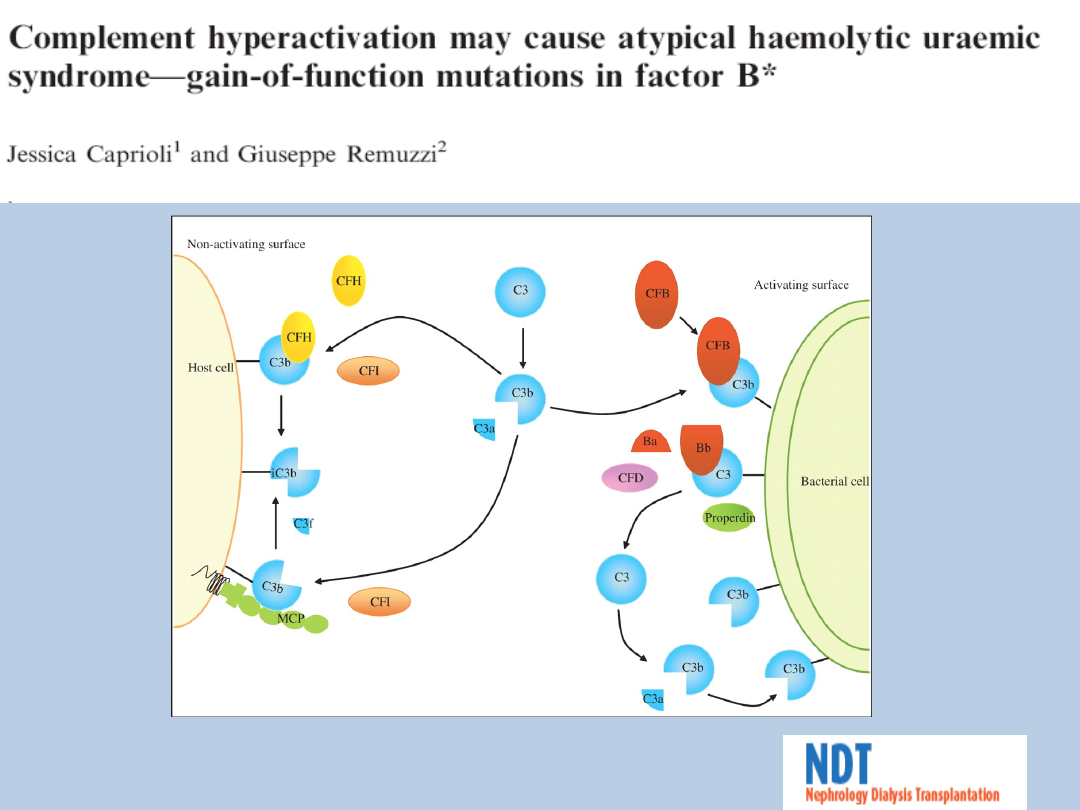
Patogeneza-atypowy HUS
Caprioli, J. et al. Nephrol. Dial. Transplant. 2007 22:2452-2454; doi:
10.1093/ndt/gfm193

ZABURZENIA FUNKCJI DOPEŁNIACZA
niedobory czynnika H, czynnika I oraz MCP mają
niepełną penetrację
Modyfikacja przebiegu choroby lub rola innych czynników:
czynniki środowiskowe
Infekcje, poprzedzające
• 70% przypadków z mutacją FH
• 60% z mutacją FI
• 100% przypadków HUS z mutacją MCP
Ciąża
czynnik wyzwalający w 4% przypadków FH-HUS
40% z FI-HUS
Richards, A, 2007, Mol
Immunol 44:111-122
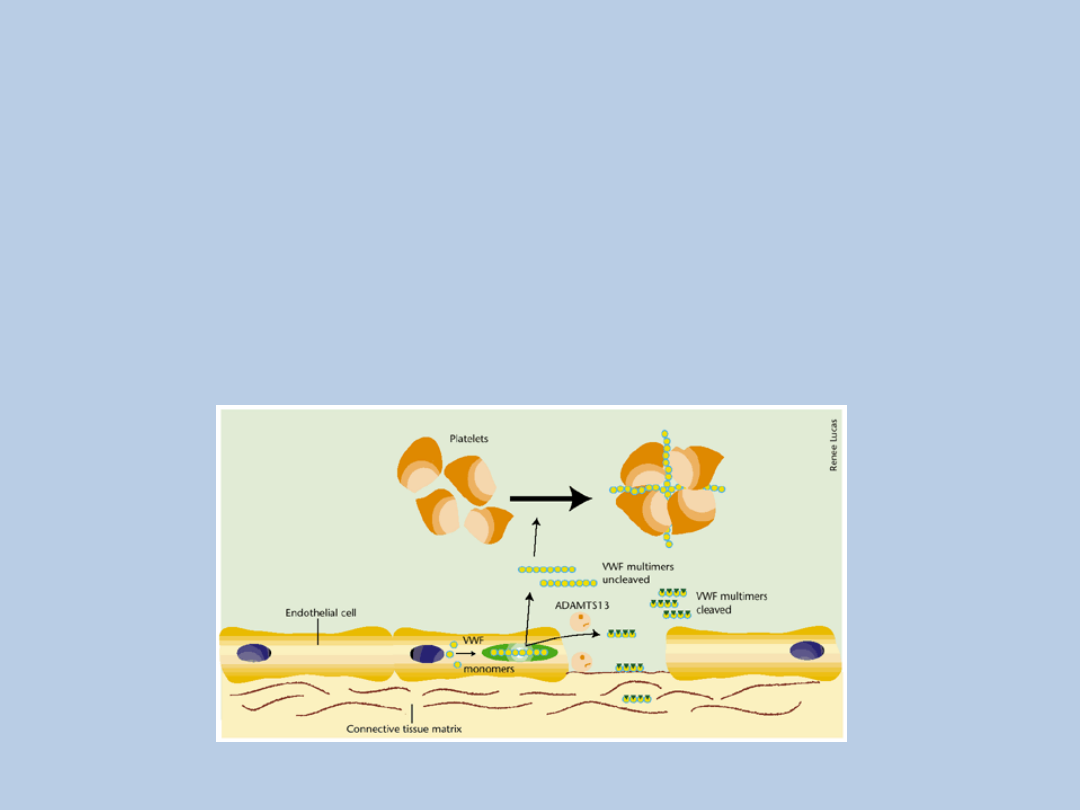
Niedobór proteazy vWF
wrodzony niedobór mieloproteinazy ADAMTS-13,
odpowiedzialnej za rozkład olbrzymich
multimerów czynnika von Willebranda (ULvWF - ultra
large von Willebrand factor multimers)
wiązanie ULvWF z antygenami
powierzchniowymi trombocytów
agregacja płytek krwi
zmiany zakrzepowe w drobnych naczyniach
tętniczych i włosowatych
Brass, L, 2001, Nature Med7:1177-1178.
Niedobór proteazy vWF
wrodzony niedobór ADAMTS-13 dziedziczony autosomalnie recesywnie –
Zespół Upshawa – Shulmana
ujawnia się po urodzeniu –
niedokrwistość hemolityczna i małopłytkowość
zajęcie nerek pojawia się w późniejszym okresie
przebieg przewlekły
nawracające epizody TTP
nabyty niedobór ADAMTS-13 - przeciwciało anty-ADAMTS-13
(59-100% chorych z nabytym niedoborem)
początek ostry, mogą towarzyszyć: silne bóle głowy, brzucha i mięśni,
zaburzenia
neurologiczne, hepato-,splenomegalia, żółtaczka
Brass, L, 2001, Nature Med7:1177-1178.
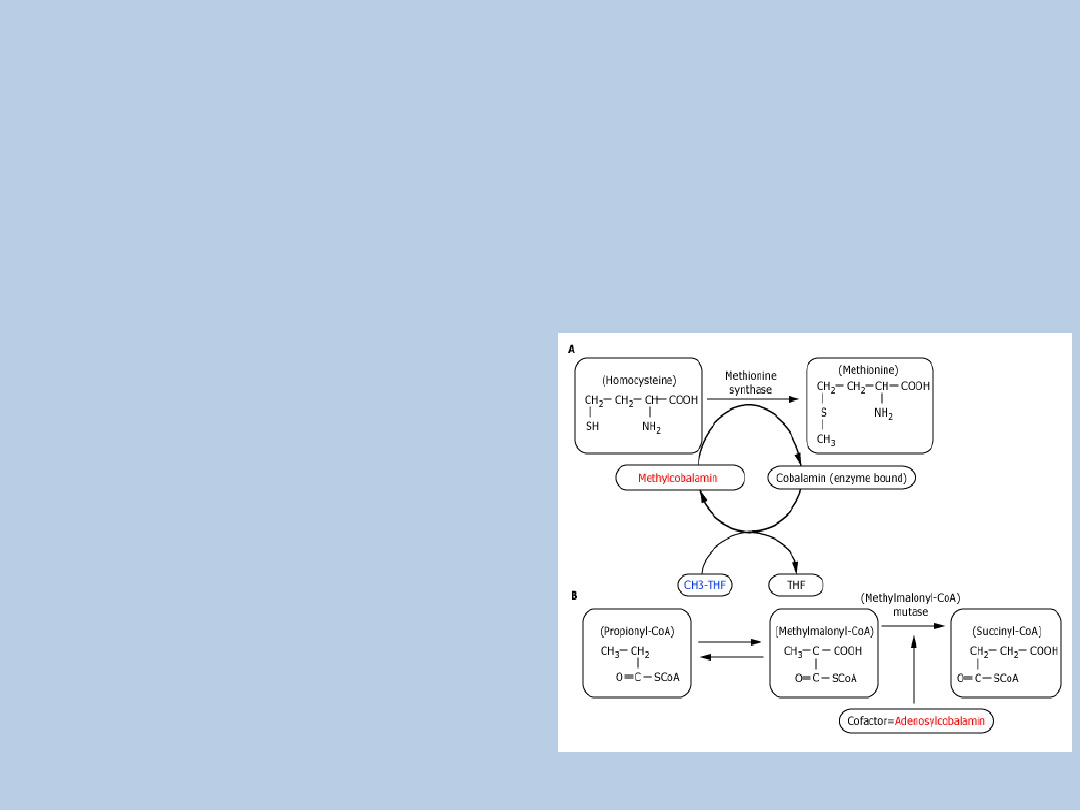
NIEDOBÓR KOBALAMINY-C
zaburzenie metabolizmu witaminyB12(kobalaminy)
hiperhomocysteiemia
kwasica metylomalonowa
klinicznie - atypowyHUS + objawy neurologiczne!!!
drgawki
hipotonia, zaburzenia karmienia, apatia
opóźnienie rozwoju
retinopatia
odchylenia w badaniach laboratoryjnych:
niedokrwistość makrocytarna
leukopenia, neutropenia
tylko krwiomocz!!!
• Tefferi, A, et al, 1994, Mayo Clin Proc69:181-186.
Etiologia
Choroby nowotworowe (prostat, żołądek,
trzustka)
Chemioterapia, radioterapia
Choroby autoimmunologiczne (np. SLE)
Ciąża, zespół HELLP
Doustne środki antykoncepcyjne
Glomerulopatie
Leki: cyklosporyna,
takrolimus, mitomycyna C
Heroina

• yklosporyna A, takrolimus, OKT3, IFN),
tiklopidyna, klopidogrel (przeciwciała
anty-ADAMTS 13), doustne środki
antykoncepcyjne, chinina, walcyklowir,
acyklowir, statyny, penicylina, chinolony,
sulfonamidy.cisplatyna, bleomycyna,
mitomycyna
• Hantawirus, gorączka Q, streptococcus
pneu, staphylococcus
Document Outline
- Slide 1
- Slide 2
- HUS – mikroangiopatia zakrzepowa
- Slide 4
- Podział
- Etiologia
- Patogenna E.coli
- Inne czynniki infekcyjne – atypowy HUS
- Patomechanizm
- HUS - D „+”
- Etiologia
- The European Paediatric Research Group For D(-) HUS
- .
- Aktywacja układu dopełniacza Droga lektynowa
- Regulacja układu dopełniacza
- Regulacja rozkładu
- Niedobór czynnika H
- Niedobór czynnika I
- Slide 20
- Regulacja układu dopełniacza
- BŁONOWY KOFAKTOR BIAŁKOWY (MCP = CD46)
- Patogeneza-atypowy HUS
- Patogeneza-atypowy HUS
- ZABURZENIA FUNKCJI DOPEŁNIACZA
- Niedobór proteazy vWF
- Niedobór proteazy vWF
- NIEDOBÓR KOBALAMINY-C
- Etiologia
- Slide 30
Wyszukiwarka
Podobne podstrony:
Zespół hemolityczno mocznicowy
ZESPÓŁ HEMOLITYCZNO- MOCZNICOWY, Nefrologia(1)
Zespół hemolityczno mocznicowy
Zespół nerczycowy
9 RF ZEspól 0 Środki trwałe
Choroba hemolityczna p odu na kurs
hemolityczne2006
Zespół kanału łokciowego i nerw pachowy (tryb edytowalny)
Zespoly paranowotworowe
Zespoly interdyscyplinarne
Teoria organizacji i kierowania w adm publ prezentacja czesc o konflikcie i zespolach dw1
zespoly otepienne
więcej podobnych podstron