

Kryteria określania
płci
Budowa anatomiczna i histologiczna
gonad
›
- jest podstawą określenia płci gonadalnej;
osobnik posiadający jądra ma płeć męską,
osobnik posiadający jajniki ma płeć gonadalną
żeńską
Budowa anatomiczna zewnętrznych
i wewnętrznych narządów płciowych
›
określa płeć somatyczną człowieka

Kryteria określania
płci
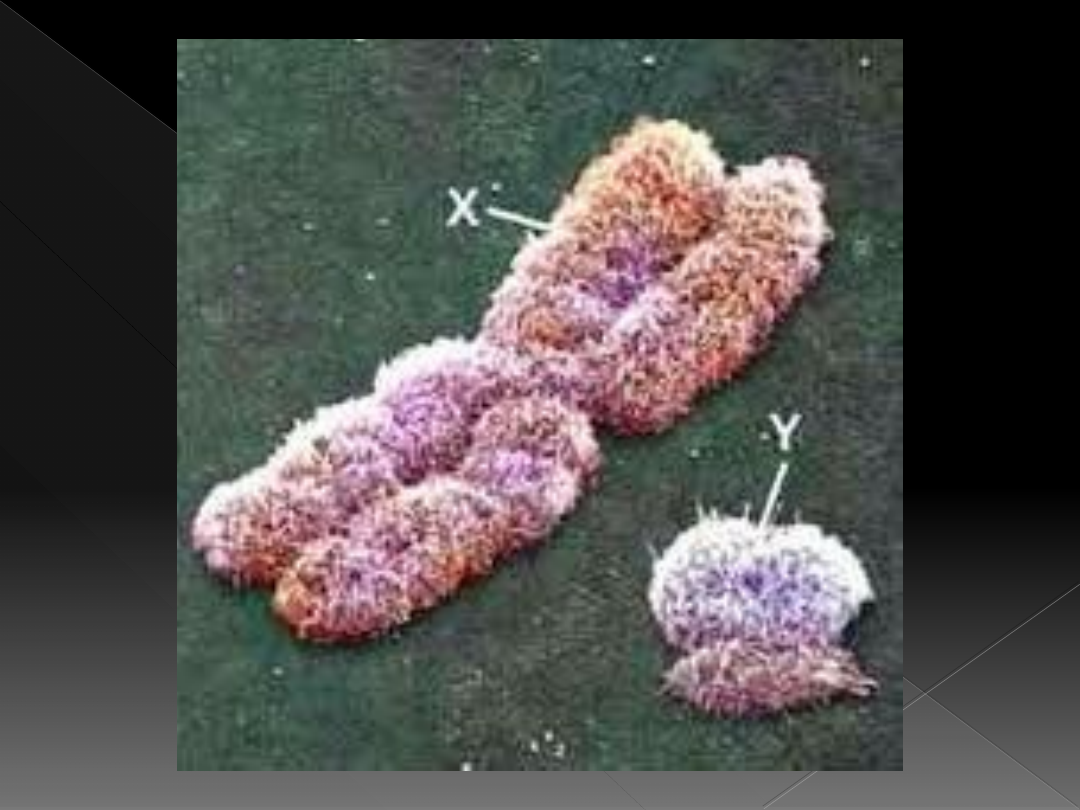
Rodzaj chromosomów płciowych
›
jest kryterium rozpoznania płci chromosomalnej:
obecność chromosomu Y jest podstawą
rozpoznania płci chromosomalnej męskiej, brak
chromosomu Y- płci chromosomalnej żeńskiej
Występowanie w genomie regionu
determinującego płeć (SRY)
›
stanowi kryterium rozpoznania płci genetycznej;
obecność SRY – płeć genetycznie męska, brak SRY-
płeć genetycznie żeńska

Kryteria określania
płci
Płeć chromatynowa
›
- określają obecność 1 ciałka Barra w jądrach
prawidłowych komórek u kobiet i obecność 1
ciałka Y w jądrach prawidłowych komórek u
mężczyzn
Płeć hormonalna
›
- jest uwarunkowana rodzajem i ilością
wydzielanych hormonów płciowych oraz
wzajemną proporcją androgenów i estrogenów
determinujących rozwój cech płciowych

Kryteria określania
płci
Płeć psychiczna
›
- określana przez psychiczne poczucie bycia mężczyzną
lub kobietą: osobę, której płeć psychiczna jest inna niż
gonadalna, nazywamy transwestytą
Określenie płci w dokumentach
danej
osoby (metryce urodzenia, dowodzie tożsamości)
nazywamy płcią metrykalną:
›
niezgodności między płcią metrykalną a innymi
określeniami płci mogą powstawać w wyniku
zdarzających się trudności jednoznacznego określania
płci somatycznej u noworodka

Determinacja płci
chromosomalnej i
genetycznej
Rozwój zarodka i płodu, a następnie dziecka w kierunku
męskim determinuje obecność chromosomu Y, natomiast
rozwój w kierunku żeńskim- brak chromosomu Y
Zygota o kariotypie 46,XY rozwija się w kierunku –
męskim
Zygota o kariotypie 46,XX rozwija się w kierunku-
żeńskim
Determinacja płci (chromosomalnej i genetycznej)
zachodzi na etapie zapłodnienia

Chromosomy płci
Powstały w drodze ewolucji z pary homologicznych
autosomów
Chromosom X podobny jest do chromosomów z
grupy C, chromosom Y do grupy G
Na chromosomie X zlokalizowano wiele genów
( znanych jest 286 cech recesywnych i 9
dominujących sprzężonych z X)chromosom Y
posiada niewiele genów, większą jego część
stanowi region heterochromatynowy „genetycznie
pusty”


Chromosomy płci
Opisano przypadki osób o kariotypie 46,XX
i fenotypie męskim, jak też przypadki
kariotypu 46,XY z fenotypem żeńskim
Paradoksy te wyjaśnia fakt, że dla
determinacji płci istotny jest nie cały
chromosom Y, lecz jego część zwana
„regionem determinującym płeć”
(SRY, ang. Sex Determining Region of Y)

Chromosomy płci
Regin determinujący płeć zawiera hipotetyczny
gen kodujący tzw. czynnik determinujący
jądro, TDF (ang. Testis Determining Factor)
Osoby o kariotypie 46,XY i fenotypie żeńskim
nie mają SRY w chromosomie (delecja)
Osoby o kariotypie 46,XX i fenotypie męskim
mają w chromosomie X odcinek SRY
(translokacja z Y)

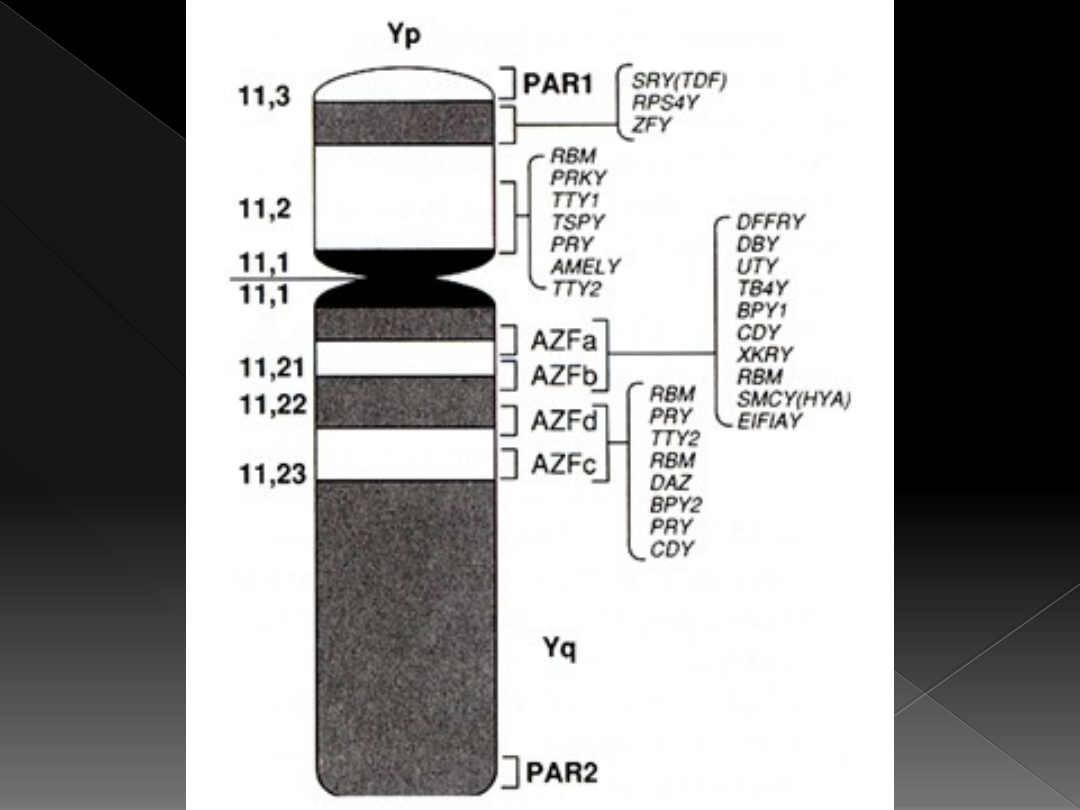
Chromosomy płci
SRY stanowi część krótkiego ramienia
chromosomu Y, nie zawiera intronów i
koduje białko o wielkości 204
aminokwasów (białko SRY należy do
czynników transkrypcyjnych)
miejsce transkrypcji białka SRY to
prekursory komórek Sertoliego
Białko SRY może być aktywatorem
transkrypcji genu kodującego wytwarzanie
czynnika anty-müllerowskiego

Mechanizmy
różnicowania płci
Genotyp zygoty determinuje rodzaj
gonad (jądra lub jajniki)
Gonady, dzięki produkowanym przez
nie hormonom, kierują różnicowaniem
narządów płciowych wewnętrznych i
zewnętrznych
Pod nieobecność czynników kierujących
rozwój cech płciowych w kierunku
męskim, płód rozwija się jako żeński

Mechanizmy
różnicowania płci
Różnicowanie płci w kierunku męskim
wymaga wysokiego stężenia
testosteronu oraz tzw. hormonu anty-
müllerowskiego
Hormon anty-müllerowski (AMH)
jest glikoproteiną wytwarzaną przez
komórki podporowe Sertoliego jądra
płodu oraz jądra osobnika dorosłego

Mechanizmy
różnicowania płci
Hormon ten wywołuje zanik przewodów
przyśródnerczowych Müllera, które są
zawiązkami jajowodów, macicy i
górnego odcinka pochwy
Kluczowym enzymem odpowiedzialnym
za różnicowanie płci jest aromataza-
enzym katalizujący konwersję
testosteronu do estradiolu

Mechanizmy
różnicowania płci
Wysoka aktywność aromatazy u zarodków
genetycznie żeńskich powoduje konwersję
produkowanego w gonadach testosteronu do
estradiolu
Niskie stężenie testosteronu przy wysokim
stężeniu estradiolu warunkuje:
›
Różnicowanie gonad niezróżnicowanych w jajnik
›
Zanik struktur prekursorowych dla narządów płciowych
męskich (przewodu śródnerczowego Wolffa)
›
Rozwój przewodów przyśródnerczowych (Müllera) i ich
pochodnych (jajowodów, macicy, pochwy)

Rozwój męskich i żeńskich
narządów płciowych
wewnętrznych
Przewód śródnerczowy (Wolffa
)
›
-parzysty, wykształca się w 4 tygodniu rozwoju;
powstają z niego: najądrza, nasieniowody,
pęcherzyki nasienne oraz nadjajnik, przyjajnik,
przewód Gartnera
Przewody przyśródnerczowe (Müllera)
›
-dają początek jajowodom, macicy, górnej części
pochwy oraz szczątkowym narządom układu
płciowego męskiego (przyjądrze, przyczepek jądra)

Różnicowanie narządów
płciowych zewnętrznych
Powstają z przedniej części steku
(kloaki) przez podział przez przegrodę
moczowo-odbytową
Do 7 tyg. życia zarodkowego narządy
płciowe zewnętrzne są jednakowe u
zarodków genetycznie męskich i
żeńskich

Różnicowanie narządów
płciowych zewnętrznych
Pod wpływem androgenów
niezróżnicowane narządy płciowe
zewnętrzne przekształcają się w
narządy o budowie męskiej
Przy braku androgenów- w narządy o
budowie żeńskiej

Różnicowanie narządów
płciowych zewnętrznych
W 6 tyg. rozwoju widoczne są struktury:
›
Guzek płciowy (powstaje z niego prącie i łechtaczka)
›
Bruzda moczowo-płciowa- u płodów męskich
zarasta, u żeńskich pozostają jako szpara prowadząca
do przedsionka pochwy
›
Fałdy moczowo-płciowe- po obu stronach bruzdy
moczowo-płciowej, u płodów męskich zrastają się
tworząc brzuszną część prącia, u żeńskich tworzą
wargi sromowe mniejsze
›
Guzki wargowo-mosznowe- u płodów męskich
tworzą mosznę, u żeńskich wargi sromowe większe

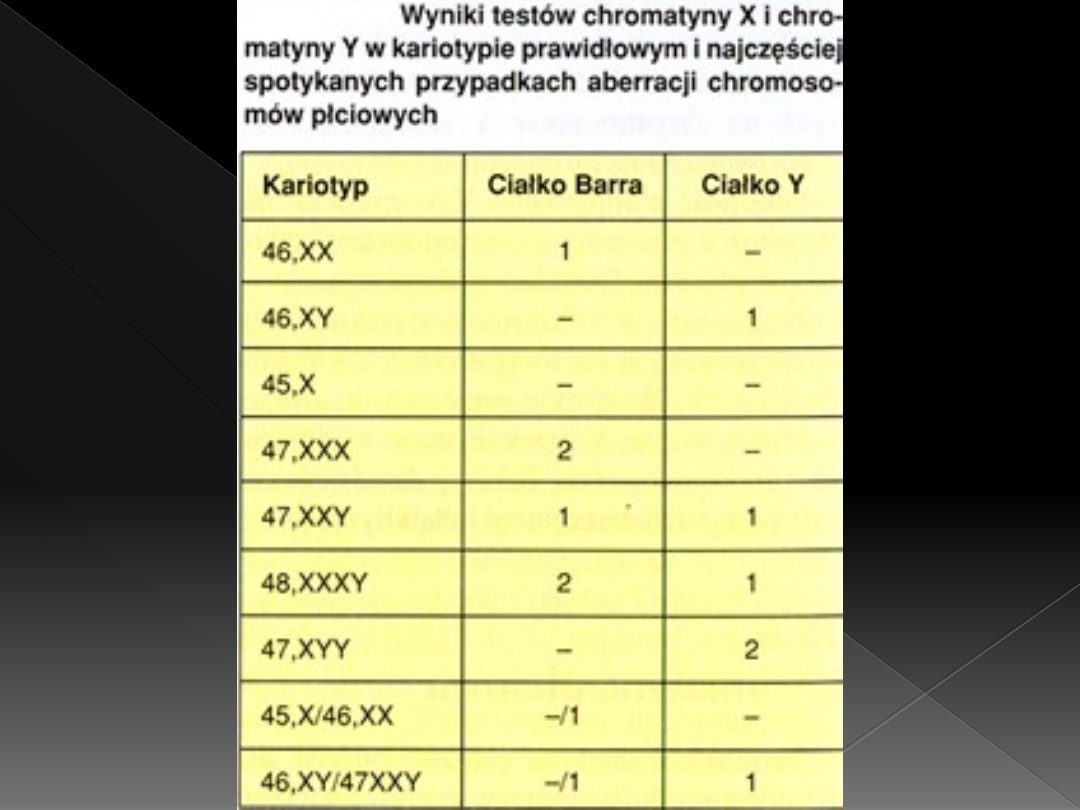
Chromatyna płciowa
Chromatyna płciowa to odpowiednio
wybarwione struktury chromatyny widoczne w
jądrach interfazowych, odpowiadające
chromosomom X i Y
Ciałko Barra (ciałko X) obecne jest w jądrach
interfazowych komórek osobników żeńskich
Chromatyna Y (ciałko Y) obecne jest w jądrach
interfazowych komórek osobników męskich

Ciałko Barra
Grudka zasadochłonnej chromatyny dyskowato
przylegająca do błony jądrowej
Najczęściej bada się je w rozmazach z nabłonka
jamy ustnej i w komórkach płynu owodniowego
W granulocytach obojętnochłonnych występuje
w postaci „pałeczek dobosza”-grudek
chromatyny wyrzuconych poza obręb jądra, ale
pozostających z nim w kontakcie

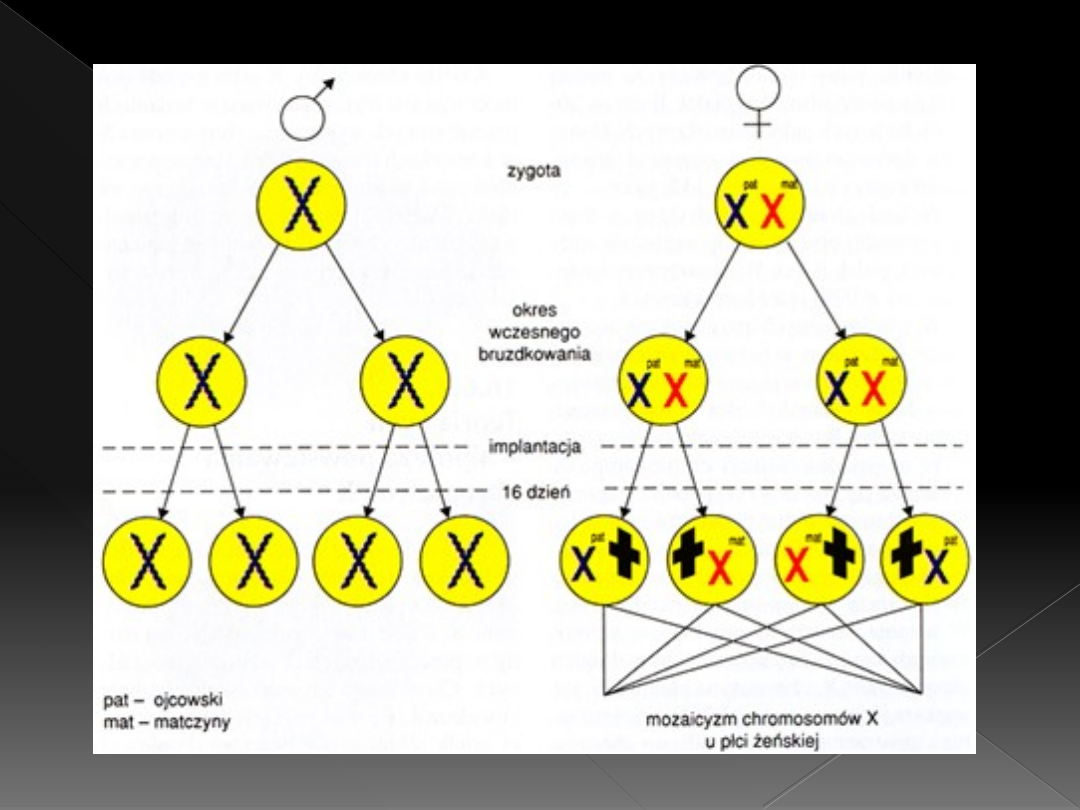
Teoria Lyon
W komórkach zarodka żeńskiego
człowieka około 16 dnia życia
płodowego dochodzi do inaktywacji
jednego z chromosomów X
Chromosom ten staje się nieaktywny i
widoczny jest jako grudka chromatyny
płciowej (ciałko Barra)

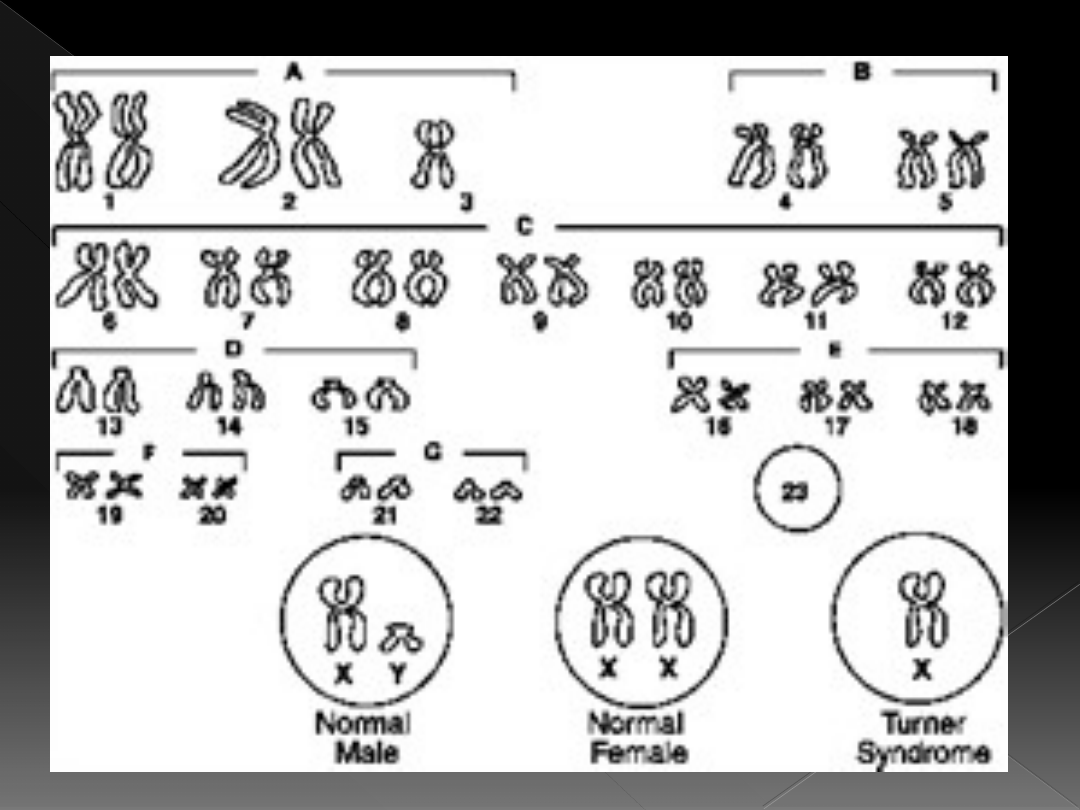
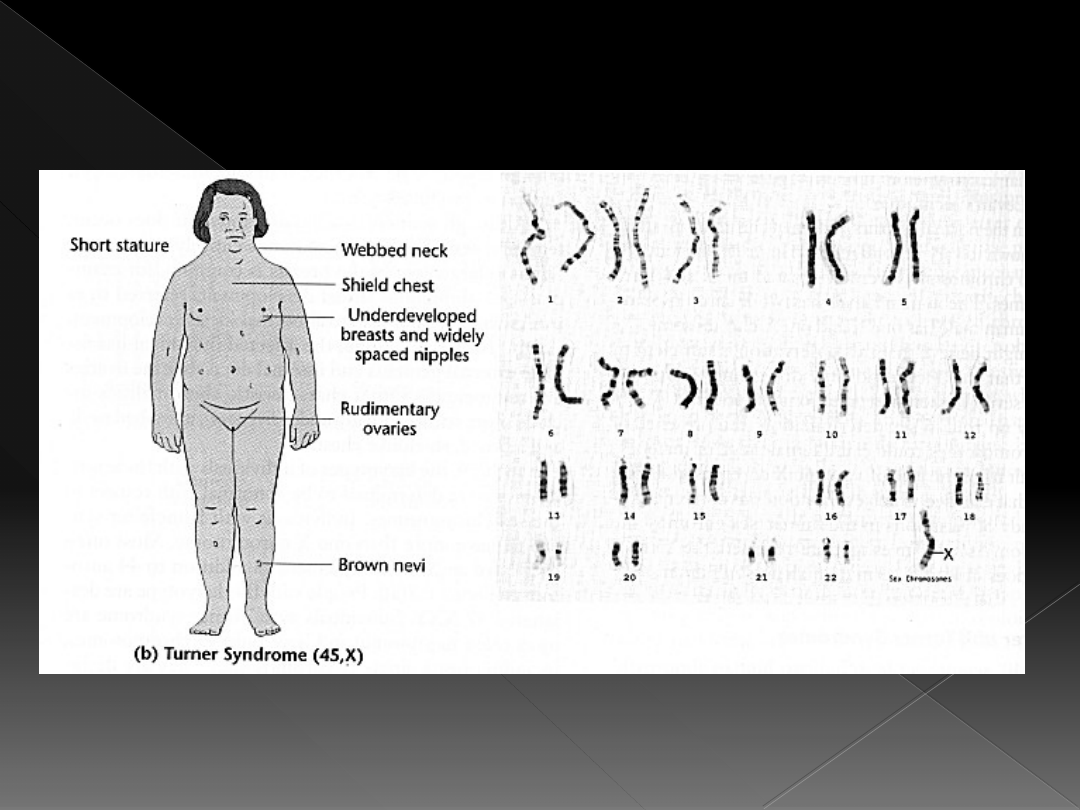
Zespół Turnera
częstość występowania około 1:3000
urodzonych dziewczynek
kariotyp:
›
45,X w około 60% przypadków
›
45,X/46,XX w około 20% przypadków
›
46,X,i (Xq) w 5-13% przypadków

Zespół Turnera
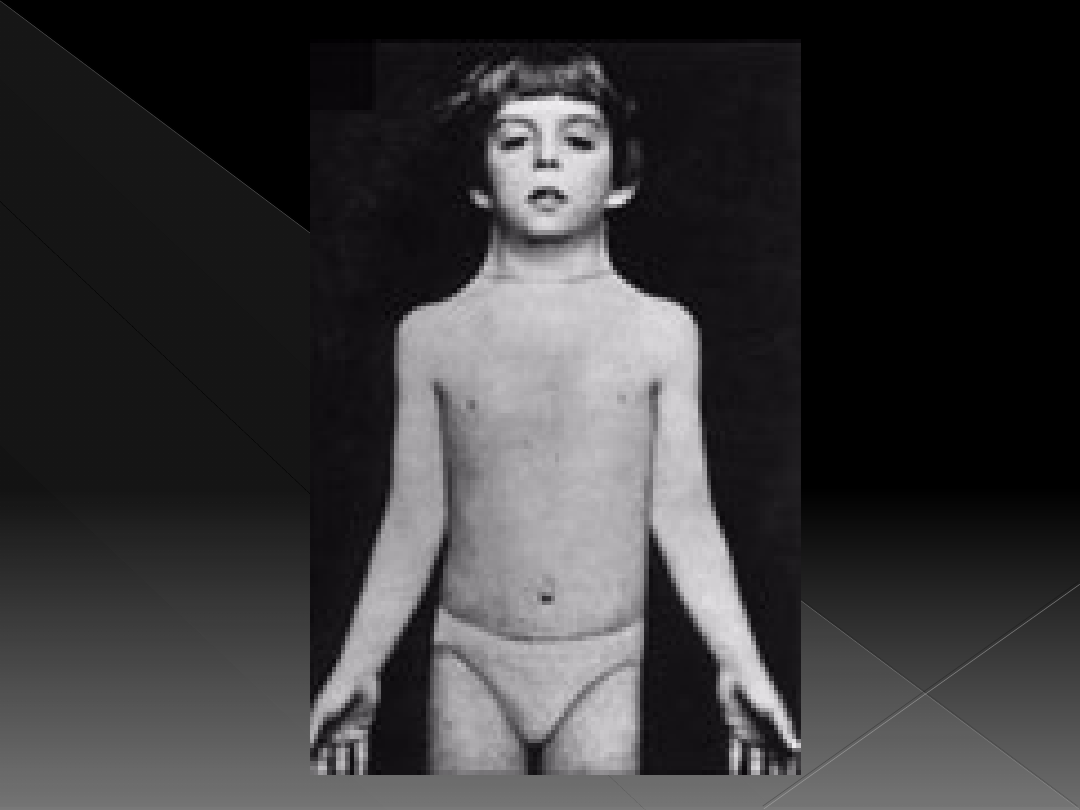
objawy klasycznego zespołu Turnera z
kariotypem 45,X:
zaburzenia wzrostu
cechy dysmorficzne twarzy
›
wysokie czoło
›
szerokie szpary powiekowe i nasada nosa,
›
Hiperteloryzm
›
zmarszczka nakątna
krótka szyja z widocznym parzystym fałdem skóry
co powoduje tzw. płetwistość szyi

Zespół Turnera
krępa budowa ciała, brak talii i
zaokrąglenia bioder, skłonność do nadwagi
klatka piersiowa szeroka, uwypuklona na
boki, brak rozwoju piersi
zmiany barwnikowe na skórze szyi i klatki
piersiowej
zewnętrzne narządy płciowe żeńskie
niedorozwinięte
skąpe owłosienie pachowe i łonowe

Zespół Turnera
wady narządów wewnętrznych dotyczą układu
krążenia, kośćca, uzębienia i nerek
pierwotna niewydolność jajników spowodowana
ich hipoplazją, mała spłaszczona macica
występuje pierwotny brak miesiączki
i pierwotna niepłodność
iloraz inteligencji w granicach normy

Zespół Turnera
u noworodków występują obrzęki
limfatyczne grzbietów rąk i
stóp
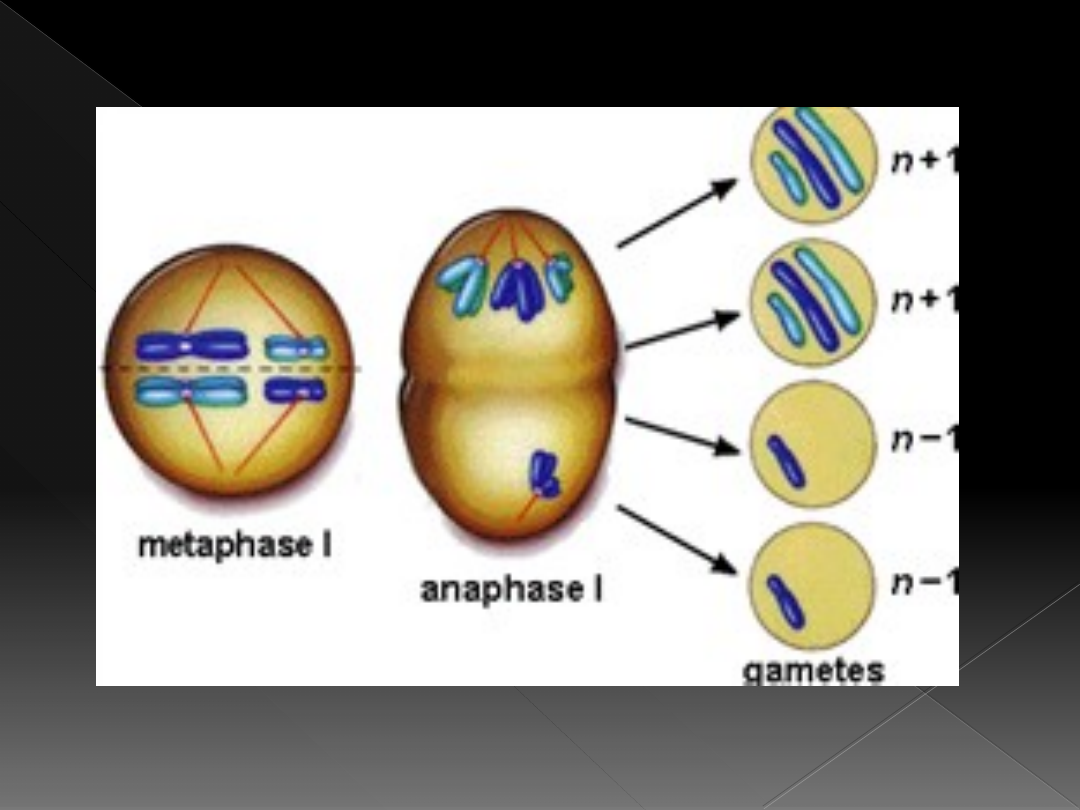
znaczny odsetek zarodków z
kariotypem 45,X ulega
samoistnemu poronieniu

Zespół kobiety 47,XXX
występuje z częstością 1:1000
urodzeń płci żeńskiej
przyczyną powstawania
kariotypu 47,XXX jest nondynsjunkcja
w I lub II podziale mejotycznym u
kobiety albo w drugim
podziale mejotycznym u mężczyzny

Zespół kobiety 47,XXX
u kobiet wzrost i budowa ciała jest
prawidłowa
15-25% pacjentek wykazuje upośledzenie
umysłowe w stopniu lekkim
pojawiają się zaburzenia
miesiączkowania i wcześniejsza
menopauza

Zespół kobiety 47,XXX
Kobiety mogą być płodne (ok.75%)
Wyraźny efekt wieku matki

Zespół Klinefeltera
Występuje 1/1000 urodzonych
chłopców
Kariotyp:
›
80% 47,XXY
›
15% 46,XY/47,XXY
›
48,XXXY lub 49,XXXXY (rzadko)
Dodatkowy chromosom X w ponad 50%
przypadków pochodzi od matki
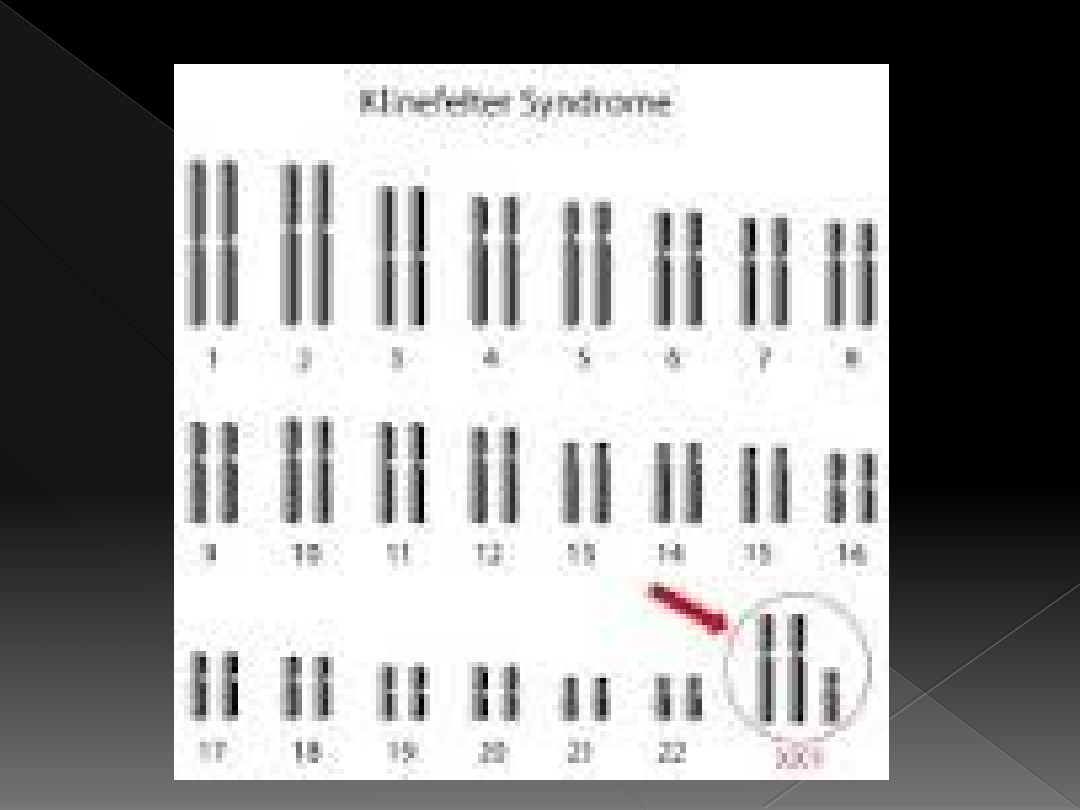

Zespół Klinefeltera
Cechy fenotypowe
Wysoki wzrost powyżej 180cm
Twarz o słabym zaroście mająca często
chłopięcy wygląd
Półkolista linia włosów na czole bez
typowego łysienia skroniowego

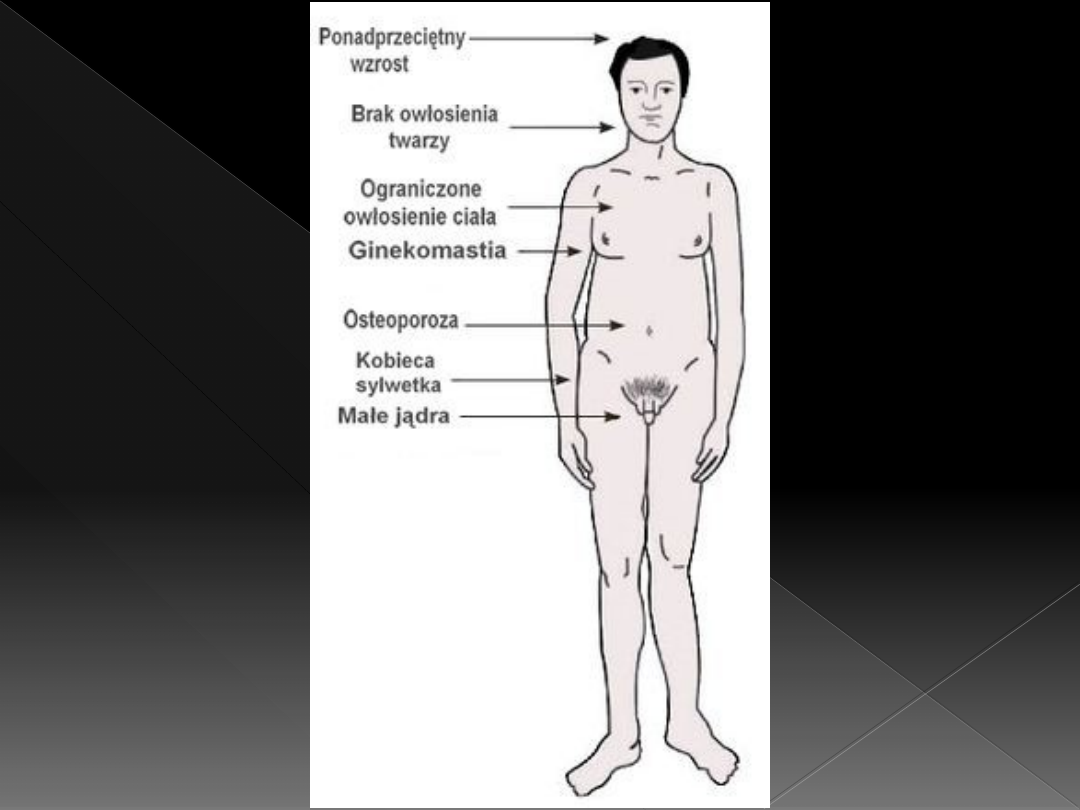
Zespół Klinefeltera
Sylwetka typu kobiecego
Odkładanie tkanki tłuszczowej
Ginekomastia
Skąpe owłosienie pachowe i łonowe
W uzębieniu brak zębów 8

Zespół Klinefeltera
Niedorozwój narządów płciowych
zewnętrznych i wewnętrznych:
›
Prącie – od małych do prawie normalnych
›
Słabo rozwinięta moszna
›
Małe jądra
›
Degeneracja kanalików nasiennych
Nieznaczne upośledzenie umysłowe i
obniżenie IQ

Zespół Klinefeltera
Zmiany rtg w obrębie kości czaszki
›
Spłaszczenie obu okolic skroniowych
›
Powiększone zatoki czołowe
›
Przedwczesne zrastanie szwów
wieńcowych
›
Małe siodełko tureckie
Wydłużone kończyny dolne

Mężczyźni z kariotypem
46,XX
Częstość 1:20 000 urodzeń
Hipotezy rozwoju fenotypu męskiego z
kariotypem 46,XX:
›
Translokacja części lub całego chromosomu Y na
ramię krótkie chromosomu X
›
Utrata chromosomu Y w komórkach zarodka o
kariotypie 47,XXY we wczesnym okresie rozwoju
›
Mutacja genu związanego z różnicowaniem
płciowym umiejscowionego na chromosomie X
lub autosomach

Mężczyźni z kariotypem
46,XX
Objawy kliniczne podobne do zespołu Klinefeltera,
ale:
›
Średnia wzrostu jest niższa (ok. 170cm)
›
Długość kończyn dolnych i proporcje ciała są prawidłowe
W 10% hipogonadyzm
Ginekomastia
Azoospermia
Bezpłodność

Zespół mężczyzny
47,XYY
1:1000 urodzonych chłopców
Objawy kliniczne:
Wysoki wzrost, typowo męska budowa ciała
Częste powikłania potrądzikowe w postaci blizn na
skórze twarzy i pleców
Rozwój zewnętrznych narządów płciowych-
prawidłowy

Zespół mężczyzny
47,XYY
Mężczyźni są płodni i mają zdrowe
potomstwo
W badaniu rtg:
›
przewaga długości kości śródręcza nad
paliczkami i skrócenie paliczków dystalnych
›
Progenia
Czasami zaburzenia zachowania w postaci
nadmiernej agresywności

Zespoły dysgenezji
gonad
Dysgenezja gonad- nieprawidłowa
budowa i czynność gonad spowodowana
najczęściej aberracjami chromosowymi lub
mutacjami genowymi
Charakteryzuje się:
›
żeńskim fenotypem
›
Pierwotnym brakiem miesiączki
›
Brakiem rozwoju drugo- i trzeciorzędowych
cech płciowych
›
obecnością szczątkowych gonad

Zespoły dysgenezji
gonad
Czysta dysgenezja gonad z kariotypem
46,XY (zespół Swyera)
Czysta dysgenezja gonad z kariotypem
46,XX
Mieszana dysgenezja gonad z
kariotypem 45,X/46,XY

Obojnactwo rzekomo
męskie
Niepełny rozwój narządów płciowych w
kierunku męskim u osobnika płci
genetycznie męskiej (46,XY)
Obecność różnie rozwiniętych jąder z
narządami płciowymi zewnętrznymi
obojnaczymi lub żeńskimi

Obojnactwo rzekomo
żeńskie
Maskulinizacja w okresie życia
płodowego narządów płciowych
zewnętrznych u płci genetycznie
żeńskiej (46,XX)
Wewnętrzne narządy płciowe
rozwinięte w kierunku żeńskim

Obojnactwo prawdziwe
U jednego osobnika stwierdza się obecność zarówno
tkanki jajnikowej, jak i jądrowej
Wady rozwojowe dotyczą głównie narządów
płciowych
Grupy obojnactwa prawdziwego:
›
Obustronne (tzw. „ovotestis”)
›
Jednostronne
›
Naprzemienne
Kariotypy: 46,XX: 46,XY oraz kariotyp mozaikowy
46,XX/46,XY (chimeryzm)
Document Outline
- Slide 1
- Kryteria określania płci
- Kryteria określania płci
- Slide 4
- Kryteria określania płci
- Kryteria określania płci
- Determinacja płci chromosomalnej i genetycznej
- Chromosomy płci
- Slide 9
- Slide 10
- Chromosomy płci
- Chromosomy płci
- Chromosomy płci
- Mechanizmy różnicowania płci
- Mechanizmy różnicowania płci
- Mechanizmy różnicowania płci
- Mechanizmy różnicowania płci
- Rozwój męskich i żeńskich narządów płciowych wewnętrznych
- Różnicowanie narządów płciowych zewnętrznych
- Różnicowanie narządów płciowych zewnętrznych
- Różnicowanie narządów płciowych zewnętrznych
- Chromatyna płciowa
- Slide 23
- Ciałko Barra
- Teoria Lyon
- Slide 26
- Zespół Turnera
- Slide 28
- Slide 29
- Zespół Turnera
- Zespół Turnera
- Zespół Turnera
- Zespół Turnera
- Slide 34
- Slide 35
- Zespół kobiety 47,XXX
- Zespół kobiety 47,XXX
- Zespół kobiety 47,XXX
- Zespół Klinefeltera
- Slide 40
- Zespół Klinefeltera
- Zespół Klinefeltera
- Zespół Klinefeltera
- Zespół Klinefeltera
- Slide 45
- Slide 46
- Mężczyźni z kariotypem 46,XX
- Mężczyźni z kariotypem 46,XX
- Zespół mężczyzny 47,XYY
- Zespół mężczyzny 47,XYY
- Zespoły dysgenezji gonad
- Zespoły dysgenezji gonad
- Obojnactwo rzekomo męskie
- Obojnactwo rzekomo żeńskie
- Obojnactwo prawdziwe
Wyszukiwarka
Podobne podstrony:
ANOMALIE HETEROCHROMOSOMÓW
ANOMALIE HETEROCHROMOSOMÓW
4 Prawidłowy kariotyp człowieka Anomalie auto i heterochromosomów
Wrodzone anomalie nerwu wzrokowego
Homo i heteroglikany 2012 2013
Związki heterocykliczne, Chemia
ANOMALNE WŁAŚCIWOŚCI LITU I JEGO DIAGONALNE PODOBIRŃSTWO DO MAGNEZU
Lekcja 7 ?presja inbredowa i heterozja
heteroazeotrop
Analysis of soil fertility and its anomalies using an objective model
Cząsteczki heterojądrowe
modelowanie DFT w katalizie heterogenicznej
Dziwna anomalia odmierzania czasu na Sycylii, W ஜ DZIEJE ZIEMI I ŚWIATA, ●txt RZECZY DZIWNE
20 ODŻYWIANIE HETEROTROFICZNE ORGANIZMÓW
I heterofobi dla stud pedag, Kulturoznawstwo, III rok, Etyka
Kinetyka reakcji heterogenicznych
więcej podobnych podstron