Komputerowe wspomaganie
projektowania leków

Komputerowe wspomaganie projektowania
CAD ( Computer Aided Design)
– projektowanie wspomagane
komputerowo, czyli
zastosowanie sprzętu i
oprogramowania
komputerowego w
projektowaniu technicznym.
Metodologia CAD znajduje
zastosowanie między innymi
w inżynierii mechanicznej,
elektrycznej, budowlanej oraz
w farmacji. Znamienne dla
CAD jest cyfrowe
modelowanie geometryczne
mające na celu opracowanie
zapisu konstrukcji wyrobu
(jednego obiektu
technicznego lub ich układu).

Komputerowe wspomaganie
projektowania leków (CADD)
Opracowanie nowego leku,
wykazującego pożądane działanie
terapeutyczne, jest jednym z
najtrudniejszych i najbardziej
złożonych procesów w przemyśle
farmaceutycznym. Jest to proces
niezwykle kosztowny, czasochłonny
i nie zawsze dający zadawalające
wyniki. Przyczyną trudności w
odkryciu nowego leku jest fakt, że
jego aktywność zależy od wielu
czynników tj: biodostępność,
toksyczność, metabolizmu.
Komputerowe wspomaganie
projektowania leków (CADD) jest
jedną z technik, która pozwala na
zwiększenie efektywności odkrycia
nowego leku.


Komputerowe wspomaganie projektowania leków
wykorzystuje chemię obliczeniową , aby odkryć, ulepszyć
lub badać leki i związane biologicznie aktywne cząsteczki.
Podstawowym celem CADD jest przewidzieć, czy dana
cząsteczka będzie wiązać się z ,,celem’’ i jeśli tak to jak
mocno. Dzięki komputerowemu przetwarzaniu ogromnej
liczby informacji o przestrzennej strukturze cząstek, do
centrum aktywnego enzymu zaangażowanego w proces
chorobowy można dopasować blokujący go związek.

Krokiem milowym w rozwoju
metod CADD był rozwój technik
rentgenowskiej analizy
strukturalnej. Umożliwił on
poznanie pierwszych sekwencji
białkowych. Obecnie
międzynarodowa baza białek
PDB.org zawiera prawie 58
tys. rekordów i stanowi
podstawowe źródło wiedzy o
budowie enzymów białkowych.
Znajomość budowy receptorów
uwarunkowała powstanie i
rozwój metod obliczeniowych
uwzględniających ich strukturę
RD (ang. receptor dependent).
Najważniejszą grupę stanowią
tu metody RD-QSAR.

Metoda przeszukiwania farmakoforycznego.
Przeszukiwanie baz molekularnych w celu znalezienia
potencjalnie nowych leków możliwe jest dzięki farmakoforom,
które pełnią rolę wzorców molekularnych.
Umożliwia wstępną selekcję potencjalnych leków, które należy
w dalszej kolejności zsyntezować i przebadać klinicznie.
Farmakofor definiuje przestrzenne ulokowanie wybranych cech
budowy cząsteczki jak: ładunki, pierścienie aromatyczne,
ugrupowania hydrofobowe, miejsca donorowe i akceptorowe
wiązań wodorowych i koordynacyjnych.
Aby wygenerować model farmakofora należy dysponować
zbiorem aktywnych ligandów, najlepiej należących do wspólnej
klasy chemicznej. Można również nakładać związki o całkiem
odmiennych strukturach na molekułą wzorcową. Następnie
program znajduje powtarzające się motywy strukturalne istotne
dla oddziaływań lek-receptor i oblicza ich rozmieszczenie.
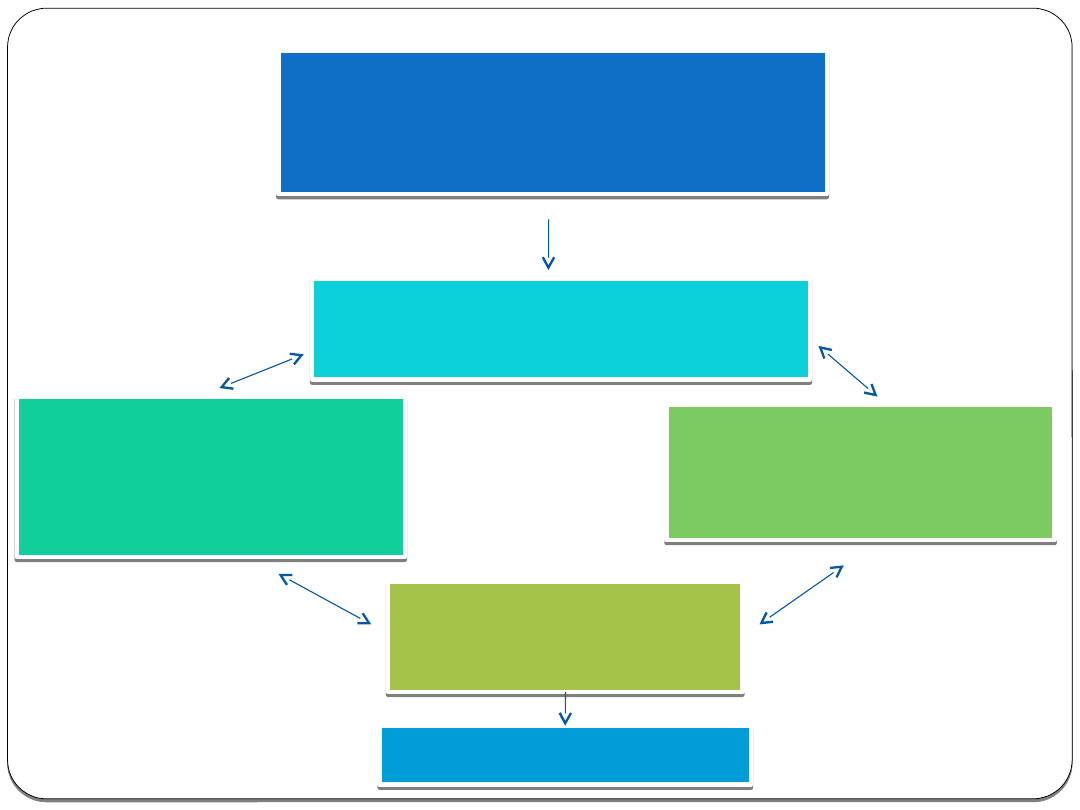
Identyfikacja celu biologicznego
• Genetyka
• Biologia molekularna
• Bioinformatyka
Identyfikacja celu biologicznego
• Genetyka
• Biologia molekularna
• Bioinformatyka
Określenie struktury
• Krystalografia rentgenowska
• Spektroskopia NMR
Określenie struktury
• Krystalografia rentgenowska
• Spektroskopia NMR
Komputerowe
wspomaganie
projektowania
• Modelowanie
molekularne
• Grafika komputerowa
Komputerowe
wspomaganie
projektowania
• Modelowanie
molekularne
• Grafika komputerowa
Testy biologiczne
•High-Throughput
Screening
•Computer-Based
Screening
Testy biologiczne
•High-Throughput
Screening
•Computer-Based
Screening
Synteza chemiczna
• Peptidomimetics
• Kombinatoryka
chemiczna
Synteza chemiczna
• Peptidomimetics
• Kombinatoryka
chemiczna
Badania kliniczne
Badania kliniczne

Oprogramowania do modelowania
molekularnego
Ogólne modelowanie molekularne (duże i małe cząsteczki): mechanika molekularna, dynamiczna, wielofunkcyjne programy

Jeszcze na początku obecnego stulecia moc powszechnie
dostępnych komputerów (klasy PC) była zbyt mała aby
swobodnie operować strukturami białkowymi. W związku
z tym w obliczeniach CADD wykorzystywano jedynie
metody niezależne od struktury receptora . W metodach
tych poszukuje się podobieństw strukturalnych dla zbioru
cząsteczek wywołujących podobny efekt biologiczny.
Następnie wyznacza się fragmenty strukturalne
odpowiedzialne za określone działanie (np. toksyczność).
Modyfikacja struktury cząsteczki ma na celu wzmożenie
określonego działania na organizm lub wyeliminowanie
działań niepożądanych. Dokonuje się tego najczęściej za
pomocą badań QSAR wraz z metodami: analizy
porównawczej pól molekularnych CoMFA, analizy
porównawczej powierzchni molekularnych CoMSA.
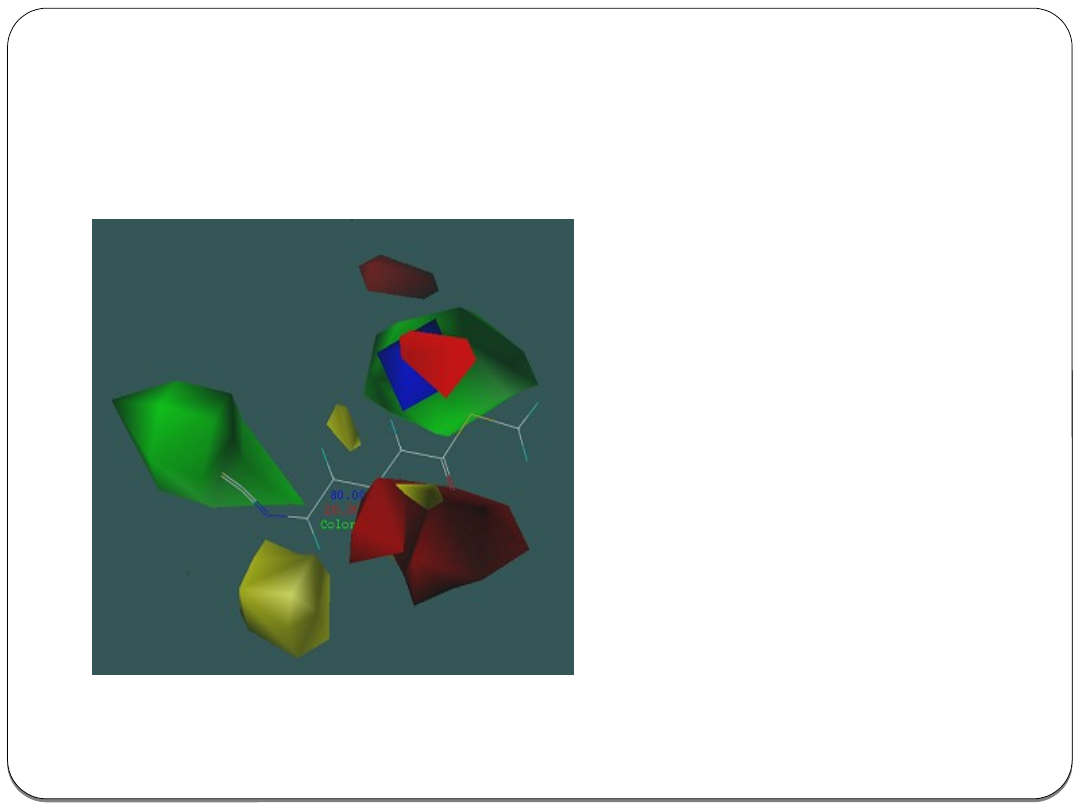
Analiza porównawcza pól
molekularnych CoMFA
(ang. Comparative Molecular Field
Analysis). Dysponując grupą leków o
podobnym działaniu biologicznym
możliwe jest wykonanie analizy
wpływu poszczególnych
podstawników na aktywność. W tym
celu nakłada się na siebie ich
trójwymiarowe konformacje
względem głównego motywu
strukturalnego. Analiza CoMFA
wyznacza w przestrzeni obszary,
które mają największy wpływ na
aktywność .
Użyteczność analizy CoMFA polega
na możliwości dowolnej modyfikacji
struktury badanej cząsteczki poprzez
wprowadzanie lub usuwanie różnych
podstawników w różnych miejscach.
Następnie możliwe jest szacowanie
aktywności zmodyfikowanego
związku.
QSAR
(ilościowa zależność pomiędzy strukturą, a reaktywnością)
Szereg technik obliczeniowych i
statystycznych wykorzystywanych
do przewidywania aktywności
biologicznej związków w oparciu o
ich strukturę bez odwoływania się
do metod modelowania
molekularnego.
Modelowanie QSAR z
zastosowaniem narzędzi
statystycznych korelacji
aktywności biologicznej , z
uwzględnieniem pożądanych
efektów terapeutycznych i
niepożądanych efektów ubocznych
pozwala na uzyskanie modeli
predykcyjnych w zakresie
chemikaliów (leków, toksyn,
zanieczyszczeń środowiska) .
Uzyskanie dobrego modelu
QSAR zależy od wielu
czynników, takich jak aktywność
biologiczna , wybór metod
statystycznych. Jakiekolwiek
modelowanie QSAR powinno
ostatecznie doprowadzić do
statystycznie solidnych modeli,
dających dokładne i
wiarygodne prognozy dotyczące
aktywności biologicznej nowych
związków.
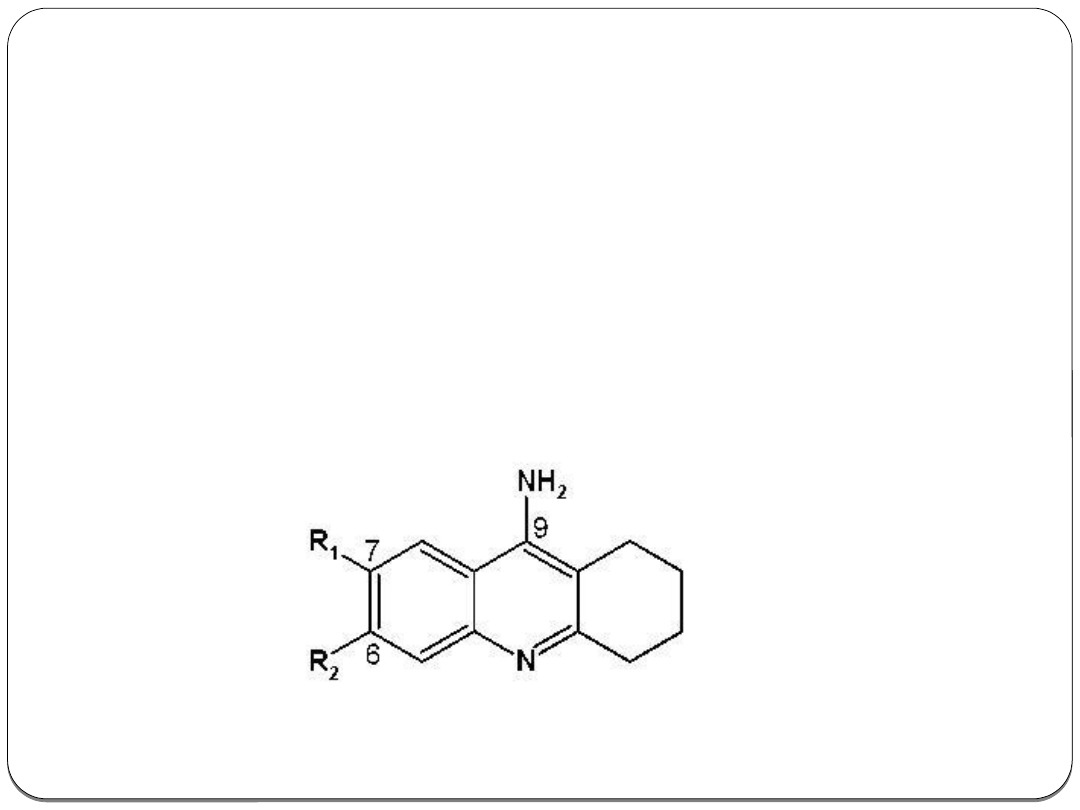
Przykład zastosowania QSAR
Analizę QSAR można zastosować do modyfikacji
pochodnych takryny Pochodne takryny są
inhibitorami acetylocholinosterazy i znajdują
zastosowanie jako leki przeciwko chorobie
Alzheimera.
Zsyntezowano 12 pochodnych, na podstawie
właściwości których wyprowadzono równanie QSAR.
Obliczenia te pozwoliły na otrzymanie
bromopochodnej o wyższej aktywności biologicznej.

Grafika komputerowa w
CADD
Komputery są niezbędnym
narzędziem w nowoczesnej chemii
mechanicznej i są ważne zarówno w
odkrywanie nowych leków .
Rozwój grafiki komputerowej
pozwala chemikowi przewidzieć
strukturę i właściwości znanych,
nieznanych, stabilnych i
niestabilnych molekuł poprzez
wykorzystanie odpowiednich
równań matematycznych, których
rozwiązanie pozwala na uzyskanie
odpowiednich informacji.
W chemii medycznej grafika
komputerowa pozwala na
wizualizację trójwymiarowego
kształtu ligandów i ich miejsc
docelowych.
Modelowanie
molekularne
Modelowanie molekularne nierozłącznie jest
związane z komputerami, których moc
obliczeniowa decyduje o dokładności
wykonywanych symulacji rozmaitych zjawisk na
poziomie pojedynczych cząsteczek. W układach o
dużej złożoności stosuje się uproszczone założenia
lub wychodzi się z pewnych założeń początkowych,
wynikających z wcześniejszych danych
eksperymentalnych.
Molekularne modelowanie jest rutynowo
stosowane do poznawania struktury dynamiki i
termodynamiki rozmaitych związków chemicznych.

Mechanika molekularna
Wykorzystuje mechanikę
klasyczną do modelowania
układów molekularnych.
Energia potencjalna
każdego rozpatrywanego
systemu jest wyznaczana za
pomocą odpowiedniego pola
siłowego. Mechanika
molekularna może zostać
użyta zarówno do badania
prostych cząsteczek jak i
złożonych biomolekuł oraz
układów
nanotechnologicznych
zbudowanych z milionów
atomów.
Mechanika molekularna
Podstawowym zastosowaniem
mechaniki molekularnej jest
minimalizacja energii cząsteczki
poprzez wykorzystanie odpowiedniego
pola siłowego oraz algorytmu
służącego do znajdowania lokalnych
minimów minimum na
hiperpowierzchni energii potencjalnej.
Globalne minimum może zostać
odnalezione przy pomocy metod takich
jak symulowane wyżarzanie oraz różne
implementacje metody Monte Carlo
wykorzystującej algorytm Metropolisa.
Niezależnie od przyjętej metody
zasadniczym celem optymalizacji jest
znalezienie zestawu konformacji
cząsteczki o najniższej energii. Pole
siłowe reprezentuje tutaj jedynie człon
entalpowy energii swobodnej Gibbsa.

Dynamika molekularna
Dynamika molekularna jest metodą
pozwalającą na badanie ewolucji
układu oddziałujących atomów
(molekuł) zgodnie za znanymi
prawami fizyki. Inaczej mówiąc jest
to metoda obliczeniowa używana
do śledzenia pozycji i prędkości
oddziałujących ze sobą atomów
(cząstek) w materiałach przez
integrację ich równań ruchu.
Położenie każdego atomu i w
systemie złożonym z N atomów jest
wyliczane zgodnie z równaniem
ruchu Newtona.
Dynamika molekularna jest metodą
deterministyczną (podane
początkowe pozycje i prędkości
oraz czas późniejszej ewolucji są
dokładnie określone).
Dynamika molekularna znajduje
zastosowanie między innymi w
biochemii jako narzędzie do
poznawania struktury i oddziaływań
w białkach, kwasach nukleinowych
i innych biomolekułach.

Analiza konformacyjna
Sprowadza się do zidentyfikowania
"uprzywilejowanych" konformacji cząsteczki
(okolice minimum energetycznego).
Niezbędne posiadanie oddzielnego algorytmu
do generowania początkowych konformacji
cząsteczki (do późniejszej minimalizacji).
W przypadku bardzo wielu minimów znajduje
się wszystkie "dostępne" minima (bariery
kinetyczne lub termodynamiczne).
Względne populacje cząsteczek dla każdego z
minimów wyznacza się z rozkładu Boltzmanna.

Model bezpośredni
Strateg
ie
CADD
Strateg
ie
CADD
Idealna metoda obliczeniowa
powinna być w stanie przewidzieć
prawidłową strukturę leku przed
jego zsyntezowaniem. Niestety
obecne metody obliczeniowe są
niedoskonałe i pozwalają na
prawidłowe oszacowanie jedynie
jakościowego powinowactwa
leku do celu. Dlatego w praktyce
nadal kilka razy powtarza się
operację projektowania, syntezy i
testowania przed odkryciem
optymalnej cząsteczki.
Z drugiej strony, metody
obliczeniowe pozwalają na
przyspieszenie odkrycia poprzez
zmniejszenie liczby potrzebnych
interakcji, a ponadto często
pozwalają na opracowanie
większej ilości nowych małych
struktur cząsteczek, które mogą
posłużyć jako lek.

Podsumowanie:
Niektóre związki pochodzenia naturalnego
wykorzystuje się jako prekursory nowych leków.
Modyfikacja struktury chemicznej ma na celu
poprawę określonych właściwości leczniczych i
osłabienie lub nawet wyeliminowanie działań
niepożądanych. Obecnie możliwe jest także
zaprojektowanie leku od podstaw.
Niektóre związki pochodzenia naturalnego
wykorzystuje się jako prekursory nowych leków.
Modyfikacja struktury chemicznej ma na celu
poprawę określonych właściwości leczniczych i
osłabienie lub nawet wyeliminowanie działań
niepożądanych. Obecnie możliwe jest także
zaprojektowanie leku od podstaw.
Przemysł farmaceutyczny każdego roku wprowadza
na rynek kilkanaście, kilkadziesiąt nowych leków.
Liczba ta nie wydaje się oszałamiająca. Należy jednak
zwrócić uwagę, iż koszt opracowania leku liczony od
początku badań do zakończenia prób klinicznych
szacowany jest na kilkaset milionów dolarów, przy
czym proces ten zajmuje czasem kilkanaście lat.
Przemysł farmaceutyczny każdego roku wprowadza
na rynek kilkanaście, kilkadziesiąt nowych leków.
Liczba ta nie wydaje się oszałamiająca. Należy jednak
zwrócić uwagę, iż koszt opracowania leku liczony od
początku badań do zakończenia prób klinicznych
szacowany jest na kilkaset milionów dolarów, przy
czym proces ten zajmuje czasem kilkanaście lat.

Dziękuje
my za
uwagę !!!
Document Outline
- Komputerowe wspomaganie projektowania leków
- Slide 2
- Komputerowe wspomaganie projektowania leków (CADD)
- Slide 4
- Slide 5
- Slide 6
- Metoda przeszukiwania farmakoforycznego.
- Slide 8
- Oprogramowania do modelowania molekularnego
- Slide 10
- Analiza porównawcza pól molekularnych CoMFA
- QSAR (ilościowa zależność pomiędzy strukturą, a reaktywnością)
- Przykład zastosowania QSAR
- Grafika komputerowa w CADD
- Modelowanie molekularne
- Mechanika molekularna
- Mechanika molekularna
- Dynamika molekularna
- Analiza konformacyjna
- Slide 20
- Slide 21
- Podsumowanie:
- Slide 23
Wyszukiwarka
Podobne podstrony:
komputerowe wspomaganie projekt Nieznany
Labolatorium projektowania układów i systemów sterowania, Narzędzia komputerowego wspomagania projek
komputerowe wspomaganie projektowania lekcja 8
sciaga ze wspomagania, Politechnika Lubelska, Studia, Semestr 6, sem VI, Komputerowe wspomaganie pro
Komputerowe wspomaganie projektowania w3
komputerowe wspomaganie projekt Nieznany (6)
komputerowe wspomaganie projektowania, Politechnika Lubelska, Studia, Semestr 6, sem VI, Komputerowe
komputerowe wspomaganie projektowania godz2255, Politechnika Lubelska, Studia, Semestr 6, sem VI, Ko
komputerowe wspomaganie projektowania lekcja 4
Politechnika Lubelska mathcad, Politechnika Lubelska, Studia, Semestr 6, sem VI, semestr 6, komputer
komputerowe wspomaganie projektowania lekcja 2
Wzor opisu do projektu - sem 6 a, IŚ Tokarzewski 27.06.2016, VI semestr COWiG, Komputerowe Wspomagan
komputerowe wspomaganie projekt Nieznany (2)
Wzor opisu do projektu - sem 5, IŚ Tokarzewski 27.06.2016, V semestr COWiG, KWP (Komputerowe wspomag
Wzor opisu do projektu - sem 6, IŚ Tokarzewski 27.06.2016, VI semestr COWiG, Komputerowe Wspomaganie
Wzor opisu do projektu - sem 6 pw mw, IŚ Tokarzewski 27.06.2016, VI semestr COWiG, Komputerowe Wspom
KWP 6 sem Andruszkiewicz Biedrzycka Płochocki Pyra, IŚ Tokarzewski 27.06.2016, VI semestr COWiG, Kom
komputerowe wspomaganie projekt Nieznany (5)
komputerowe wspomaganie projekt Nieznany (3)
więcej podobnych podstron