II zasada termodynamiki
II zasada termodynamiki wprowadza
ENTROPIĘ (S)
termodynamiczną funkcję stanu, która wskazuje kierunek przemian samorzutnych
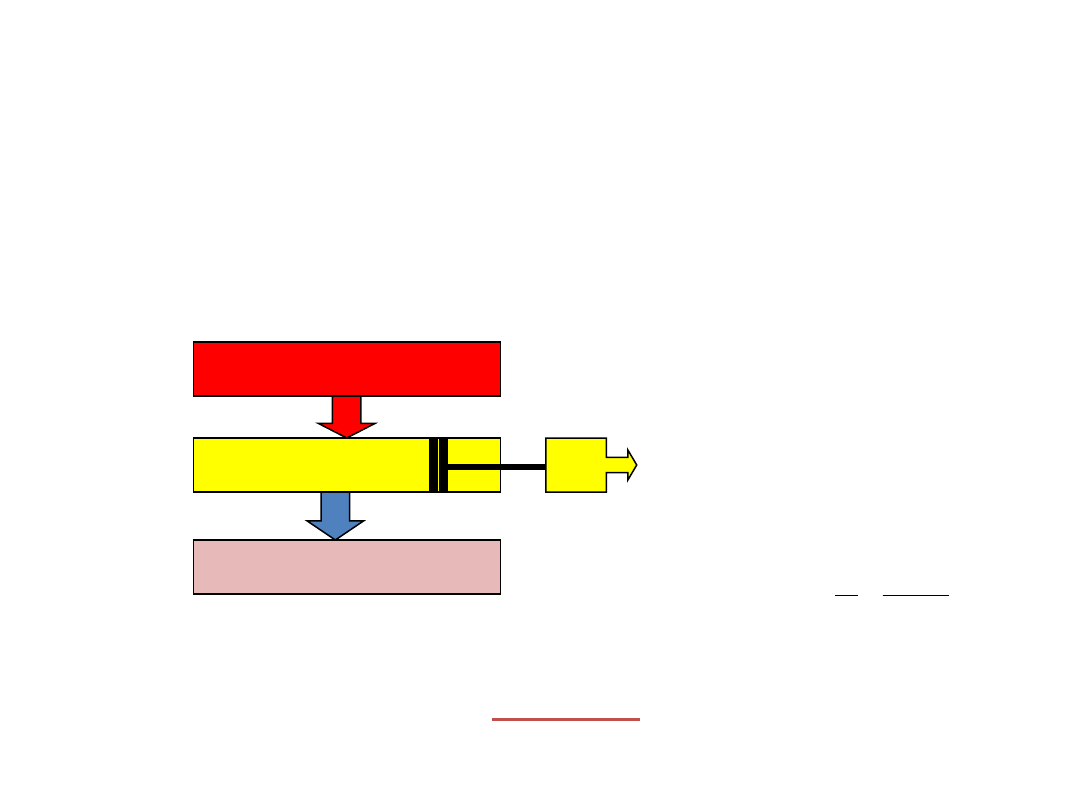
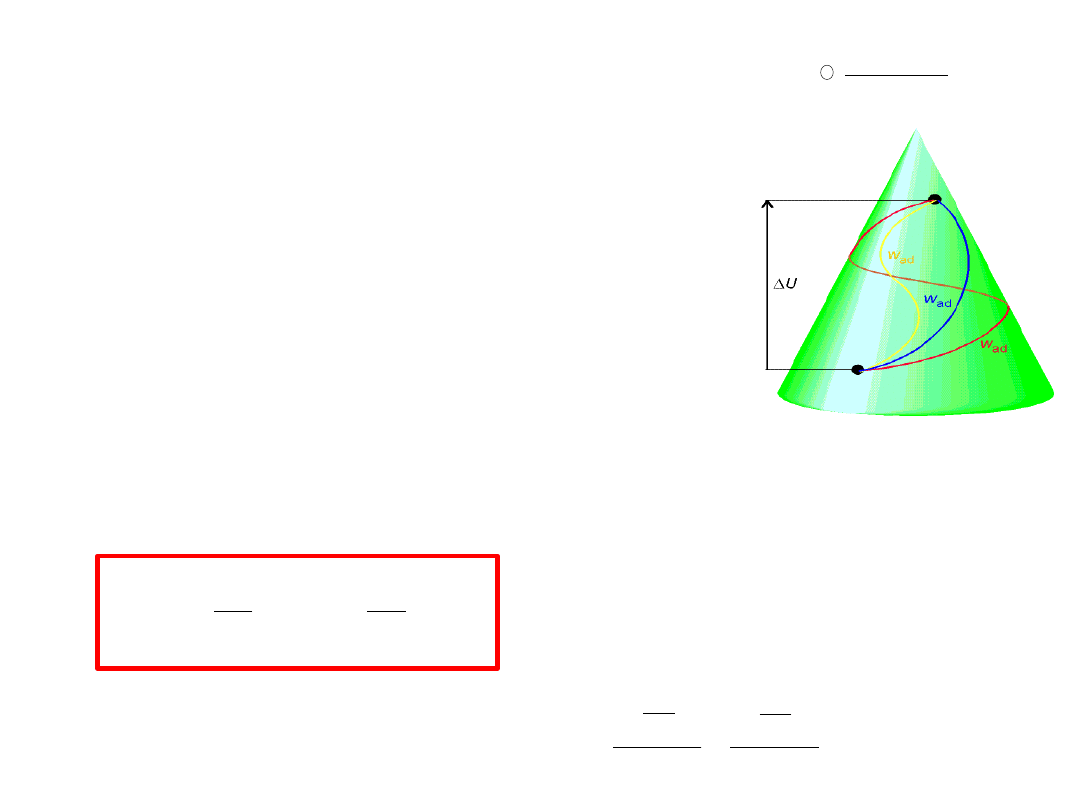
Zbiornik ciepła
Ciało robocze
(maszyna)
chłodnica
T
1
T
2
q
1
q
2
w
T
1
> T
2
Zamknięty cykl roboczy:
ciało robocze wraca do stanu początkowego
∆U = 0
∆U = q + w
- w = q = q
1
+ q
2
Wydajnośd maszyny cieplnej
1
2
1
1
q
q
q
q
w
η
(q
2
< 0)
zajmuje się
kierunkiem przepływu ciepła
,
tym jaka częśd ciepła może byd zamieniona na pracę
oraz kiedy proces przebiega samorzutnie
Maszyna cieplna

Clausius 1850: „Ciepło nie może przechodzid samorzutnie
od ciała chłodniejszego do ciała cieplejszego”
Thomson 1851: „Ciepło najzimniejszego z ciał biorących
udział w procesie kołowym nie może byd źródłem pracy
Rudolf Julius Emanuel Clausius (1822-1888)
II zasada termodynamiki
William Thomson. Lord Kelvin (1892)
1824-1907
Nicolas Léonard Sadi Carnot
1796-1832
Fizyk, inżynier wojskowy i matematyk francuski
autor teorii silników cieplnych, w tym cyklu idealnego silnika
cieplnego – tzw. cyklu Carnota
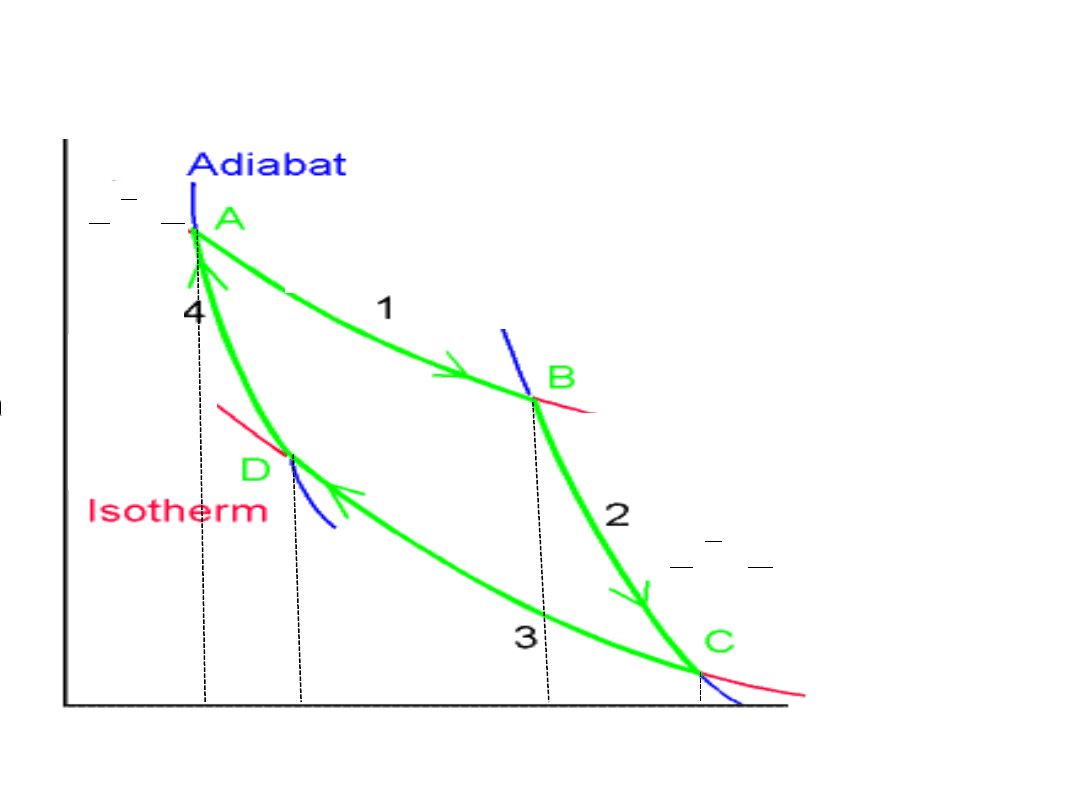
Cykl Carnota
:
zamknięty cykl
czterech odwracalnych
przemian gazu doskonałego
w
3
= -nRT
2
ln (V
4
/ V
3
)
w
2
= nc
V
(T
2
–T
1
)
w
4
=nc
V
(T
1
–T
2
)
4
1
R
c
1
2
V
V
T
T
V
T
1
=const
T
2
=const
w
1
= - nRT
1
ln (V
2
/ V
1
)
q
2
= 0
3
2
R
c
1
2
V
V
T
T
V
q
4
= 0
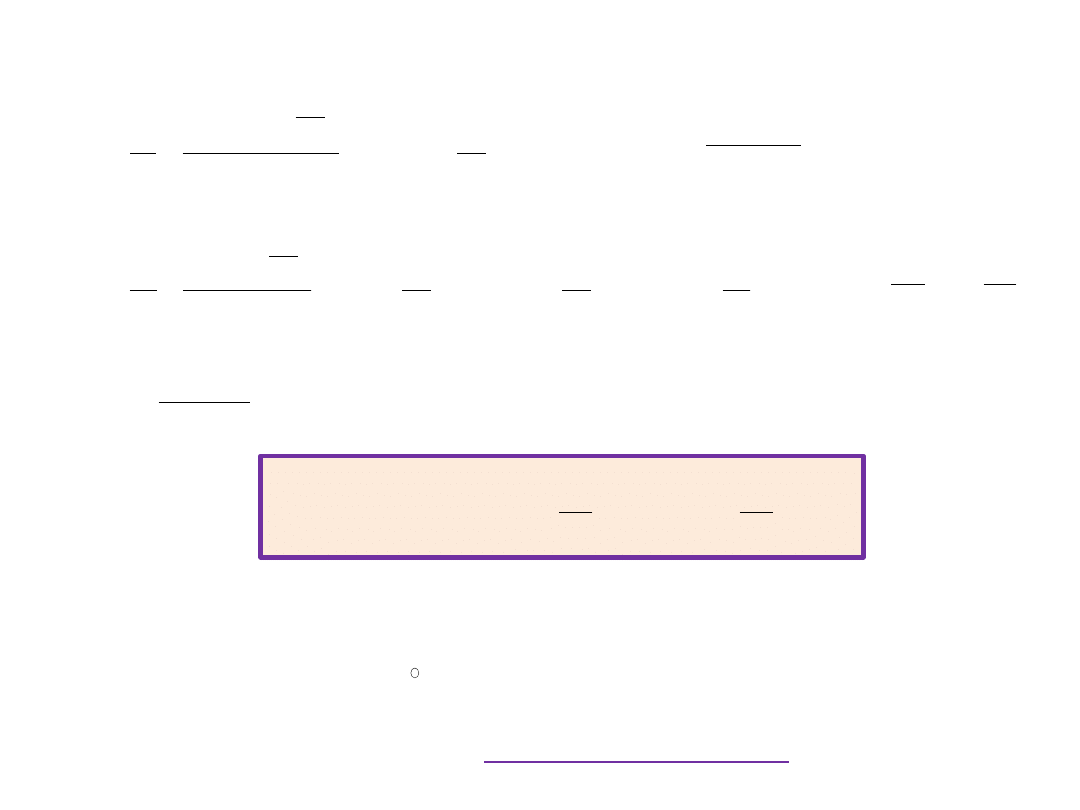
P
V
1
V
4
V
V
2
V
3
1 = A
B
Ciepło q
1
przepływa od źródła ciepła do maszyny
(gazu) Gaz rozpręża się V
1
→ V
2
w odwracalnym procesie
izotermicznym:
T
1
= const ∆ U
1
= 0,
q
1
= - w
1
2 = B
C
Gaz rozpręża się V
2
→ V
3
w
odwracalnym procesie adiabatycznym:
q
2
= 0
,
T
1
↘ T
2
,
∆U
2
= w
2
3 = C
D
Tłok spręża gaz V
3
→ V
4
w
odwracalnym procesie izoterm.: T
2
= const
ciepło q
3
przepływa z gazu do chłodnicy,
∆ U
3
= 0
q
3
= - w
3
4 = D
A
Tłok spręża
gaz V
4
→ V
1
w
odwracalnym procesie
adiabatycznym:
q
4
= 0
,
T
2
↗ T
1
,
∆U
4
= w
4
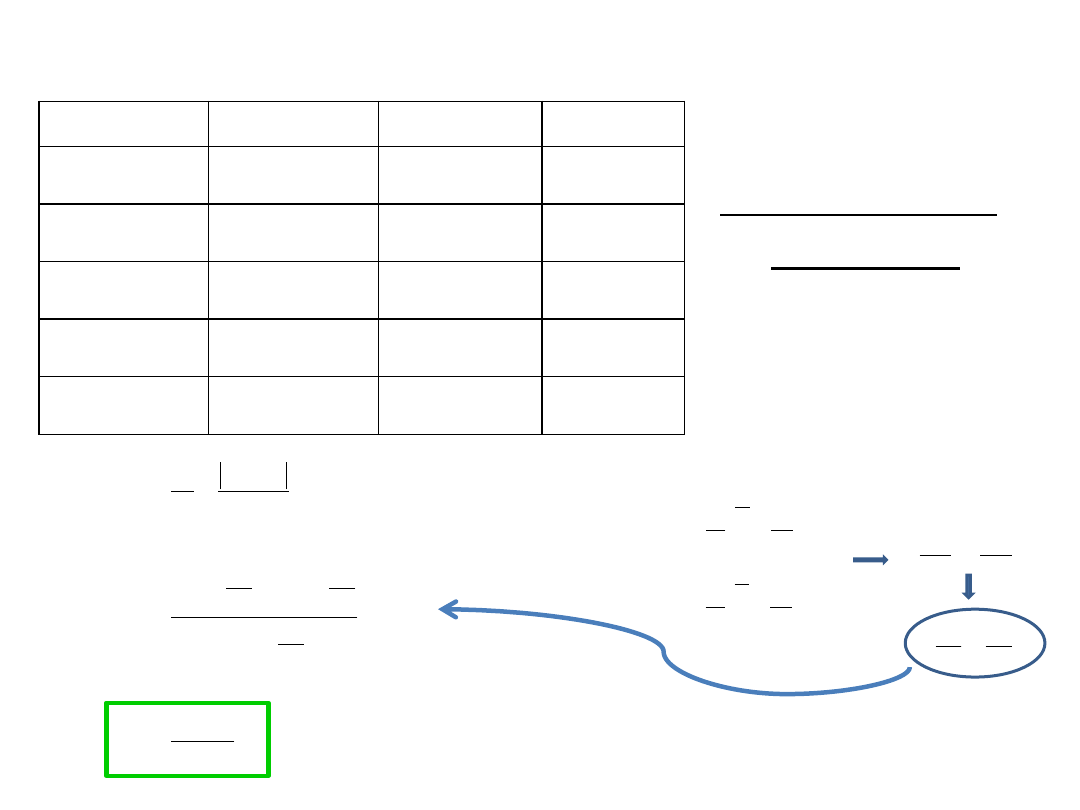
Bilans energetyczny w odwracalnym cyklu Carnota
przemiana
Ciepło, q
Praca, w
U
1. Izotermiczna
T
1
= const
nRT
1
ln(V
2
/V
1
)
-nRT
1
ln(V
2
/V
1
)
0
2. Adiabatyczna
q
2
= 0
0
nc
V
(T
2
-T
1
)
nc
V
(T
2
-T
1
)
3. Izotermiczna
T
2
= const.
nRT
2
ln(V
4
/V
3
)
-nRT
2
ln(V
4
/V
3
)
0
4.Adiabatyczna
q
4
= 0
0
nc
V
(T
1
-T
2
)
nc
V
(T
1
-T
2
)
Cały cykl
Carnota
nRT
1
ln(V
2
/V
1
)
+nRT
2
ln(V
4
/V
3
)
-nRT
1
ln(V
2
/V
1
)
-nRT
2
ln(V
4
/V
3
)
0
Sprawnośd silnika cieplnego
= wykonana praca
ciepło pochłonięte
1
2
1
1
2
1
4
3
2
1
2
1
1
3
1
1
T
T
T
η
V
V
ln
RT
V
V
ln
RT
V
V
ln
RT
η
q
q
q
q
w
η
3
2
R
c
1
2
V
V
T
T
V
4
1
R
c
1
2
V
V
T
T
V
4
3
1
2
V
V
V
V
Z równao adiabat (2) i (4)
4
1
3
2
V
V
V
V
(2)
(4)
max (100%) gdy T
2
0
1
2
1
1
3
1
T
T
T
η
q
q
q
η
Analizując sprawnośd maszyny cieplnej Carnota
otrzymujemy:
1
2
1
1
3
1
T
T
T
q
q
q
q
3
< 0 oddane chłodnicy
W procesie cyklicznym nie można zamienid ciepła na pracę bez
równoczesnego przeniesienia części tego ciepła ze zbiornika ciepła
(T
1
) do chłodnicy (T
2
)
1
2
1
3
T
T
1
q
q
1
1
1
2
3
T
q
T
q
0
T
q
T
q
1
1
2
3
0
T
q
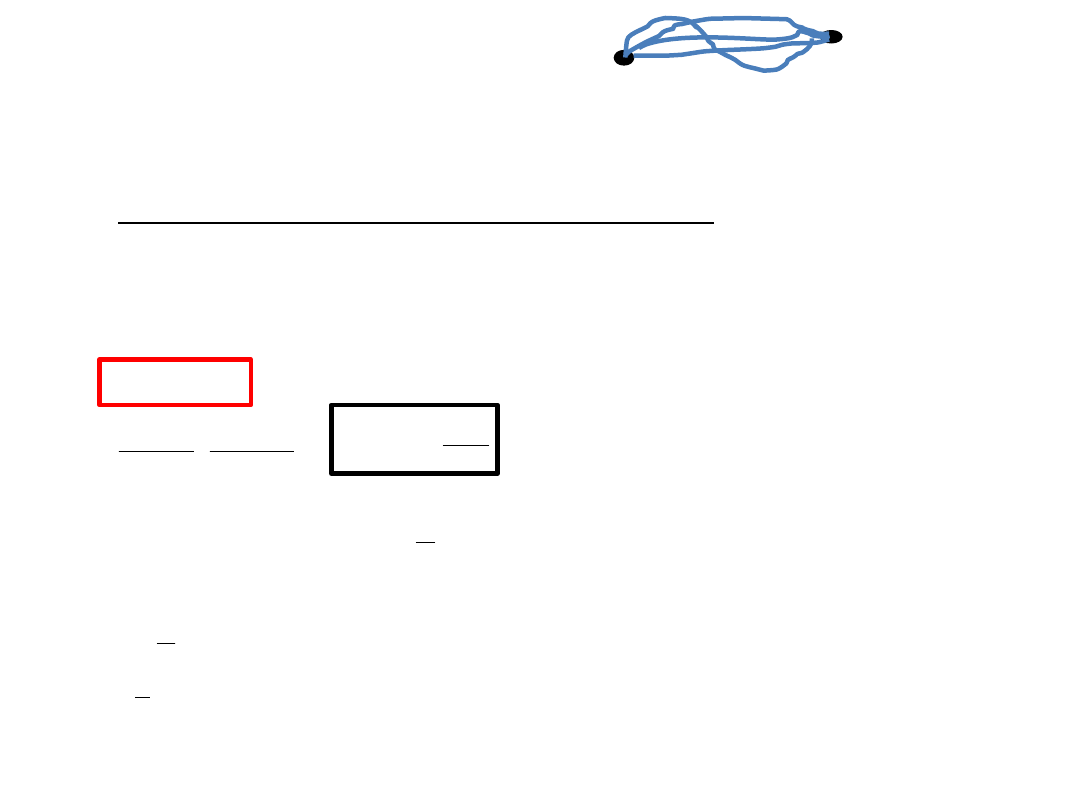
Dowolny
cykl odwracalny
dla gazu doskonałego można
uważad za sumę wielkiej liczby cykli Carnota
0
T
q
)
(
i
odw
i
q
i
– ciepło wymienione w temperaturze T
i
w i-tym odwracalnym procesie cyklu zamkniętego
0
T
dq
odw
Wyrażenie pod całką jest różniczką funkcji stanu
ENTROPII S
T
dq
dS
odw
Gdy układ zamknięty o temperaturze T wymienia z otoczeniem ciepło q w procesie
odwracalnym to ENTROPIA tego układu zmienia się o wartośd dS
PRZYPOMNIENIE
1. Funkcja stanu jest niezależna od drogi zależy jedynie od stanu
początkowego i koocowego układu.
2. Zmiana funkcji stanu w cyklu zamkniętym (takim, który wraca do
stanu początkowego) wynosi 0
Jeśli całka krzywoliniowa po drodze zamkniętej ∮ dF = 0,
to dF jest różniczką zupełną
dy
y
F
dx
x
F
dF
x
y
x
y
F
y
x
F
x
y
3. Drugie pochodne cząstkowe funkcji stanu
są sobie równe
WŁAŚCIWOŚCI
funkcji stanu
F= f (x,y)
PRZYPOMNIENIE
0
T
dq
odw
Zmiana entropii w cyklu Carnota:
1
2
1
1
2
1
1
1
1
V
V
ln
R
n
T
V
V
ln
T
R
n
T
q
ΔS
0
T
0)
dq(
ΔS
2
1
T
T
2
1
2
4
3
3
4
2
3
4
2
2
3
3
V
V
nRTln
V
V
nRTln
V
V
nRln
T
V
V
ln
nRT
T
q
ΔS
0
T
0)
dq(
ΔS
1
2
T
T
4
0
V
V
ln
R
n
V
V
ln
R
n
ΔS
ΔS
1
2
1
2
i
i
1
2
4
3
V
V
V
V
0
dS
bo
.
Entropia w zamkniętym cyklu przemian odwracalnych nie uległa zmianie,
S = 0,
co można zapisad:
czyli entropia jest funkcją stanu.
ENTROPIA
Ciepło i praca zależą od sposobu przeprowadzenia procesu
Ciepło wymienione w izotermicznym procesie odwracalnym:
q
odw
= -w
odw
(max) = - nRT ln(V
2
/ V
1
)
jest większe od ciepła wymienionego w nieodwracalnym procesie izotermicznym
q
nod
=-w
nod
= -p
2
(V
2
– V
1
)
q
odw
> q
nod
T
dq
T
dq
dS
nieod
odw
jest funkcją stanu
Zmiana entropii układu wywołana przebiegiem jakiegoś procesu zależy od stanu początkowego i koocowego
układu ∆S = S
2
– S
1
, nie zależy od tego czy proces był odwracalny czy nieodwracalny.
T
dq
dS
ENTROPII nie potrafimy zmierzyd możemy ją tylko obliczyd.
Zmianę entropii możemy obliczyd gdy przemiany zachodzą
odwracalnie.
Aby obliczyd ∆S dla procesu nieodwracalnego trzeba go
zastąpid odwracalnymi etapami przejśd ze stanu 1 do 2.
S
1
(T
1
, p
1
,V
1
)
S
2
(T
2
, p
2
,V
2
)
ENTROPIA jest kryterium samorzutności i stanu równowagi procesu zachodzącego
w układzie izolowanym.
0
T
q
dS
0
T
q
dS
Jeżeli proces przebiega samorzutnie
Jeżeli układ znajduje się w stanie równowagi
T
q
dS
W procesach nieodwracalnych tj. samorzutnych
występuje nadmiar entropii.
∮dS = 0
ENTROPIA jako kryterium samorzutności procesu
Analiza cyklu Carnota pozwoliła na zdefiniowanie entropii w ujęciu makroskopowym i wyrażenie
jej za pomocą wielkości fizycznych (q,T)
T
dq
dS
W cyklu Carnota wymiana ciepła następuje między
układem
i
otoczeniem, które dostarcza (zb. ciepła)
lub odbiera (chłodnica) wymienione ciepło
.
Połączenie układu i otoczenia stanowi UKŁAD IZOLOWANY
.
ENTROPIA układu izolowanego
U = const. dU=0 (I zasada termodynamiki)
Niech T
A
> T
B
q ≅ 0
T
A
= const
T
B
= const
A
B
T
A
T
B
∆S = ∆S
A
+ ∆S
B
∆S
A
=q
A
/ T
A
∆S
B
=q
B
/ T
B
q
A
= - q q
B
= q
0
T
T
T
T
q
T
1
T
1
q
T
q
T
q
T
q
T
q
ΔS
B
A
B
A
A
B
B
A
B
B
A
A
Jeżeli w układzie izolowanym zachodzą nieodwracalne przemiany to
zachodzą tak, że entropia układu rośnie: ∆S > 0
Dla procesu zachodzącego odwracalnie, tzn. pod wpływem nieskooczenie
małej siły napędowej T
A
≅ T
B
∆S = 0
Części A i B układu izolowanego
można potraktowad jak układ (A) i
otoczenie (B)
We wszystkich procesach nieodwracalnych tj SAMORZUTNYCH,
łączna entropia układu i otoczenia wzrasta.
q
đw
obj
= 0, V = const dV = 0
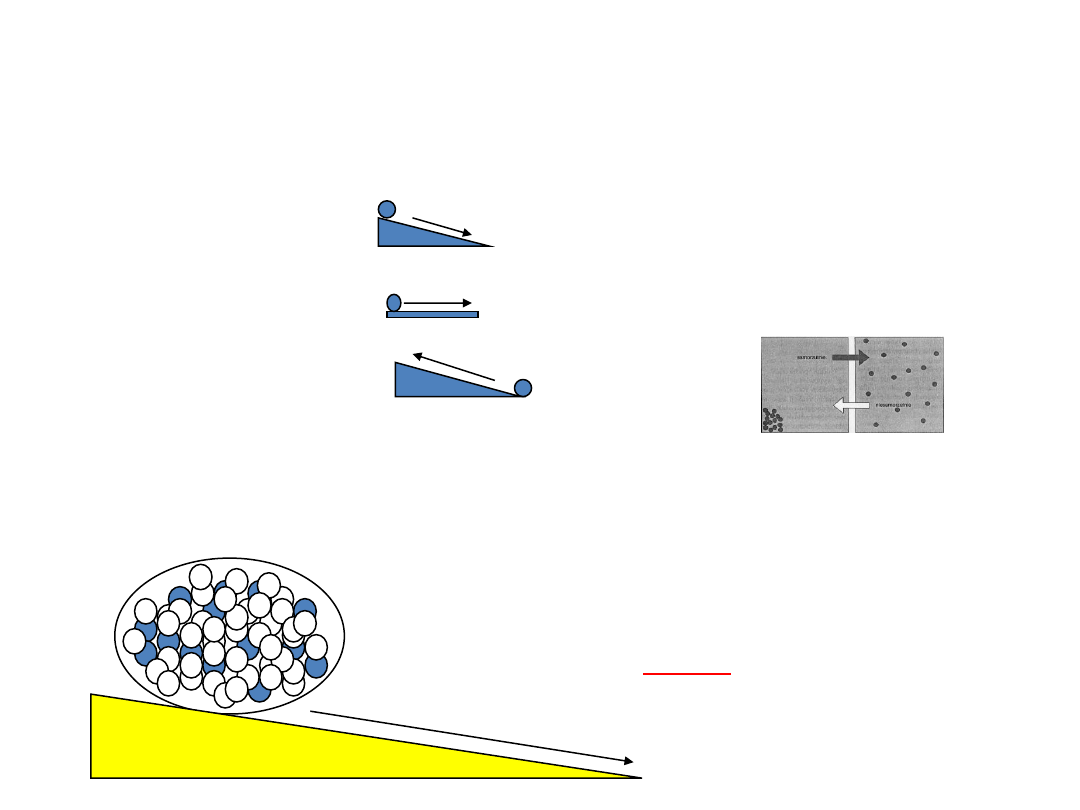
PROCESY SAMORZUTNE
zachodzą w przyrodzie i biegną do momentu osiągnięcia przez UKŁAD STANU RÓWNOWAGI np.
przechodzenie ciepła z ciała o wyższej temp. do ciała o niższej temp. czy dyfuzja substancji
rozpuszczonej z roztworu stężonego do rozcieoczonego
Podział procesów zachodzących w przyrodzie (wg kryt. możliwości wykonania pracy):
SAMORZUTNE
w < 0 (rozprężanie gazu, reakcje chem.)
układ wykonuje pracę
RÓWNOWAGOWE
w = 0 (reakcja w stanie równowgi A + B ⇄ C)
NIESAMORZUTNE
w > 0
pracę trzeba wykonad na układzie ( sprężanie gazu )
Siłą napędową procesów samorzutnych jest różnica wielkości intensywnych:
∆p – rozprężanie gazu, ∆T – przepływ energii cieplnej, ∆
,
c - przepływ masy (dyfuzja), ∆E - przepływ ładunków
elektr.( E potencjał elektryczny)
Gdy różnica wielkości intensywnych = 0 proces samorzutny ustaje a zachodzi proces równowagowy.
Naturalne procesy w przyrodzie są samorzutne i przebiegają w
sposób nieodwracalny, a więc ich entropia wzrasta.
Clausius:
„ENERGIA wszechświata jest stała
( I zasada termodynamiki),
a jego ENTROPIA dąży stale do max.
( II zasada termodynamiki)
Obliczanie zmian entropii
1.
Przemiana adiabatyczna
q
el
= 0
dS = 0 ∆S = 0
2.
Przemiana izochoryczna
V = const q
el
= dU
T
dq
dS
q
el
= dU - w
el
jeśli w = w
obj
q
el
= dU + pdV
q
el
= dH - Vdp
T
pdV
dU
dS
1
2
V
T
T
V
S
S
T
T
V
V
T
T
ln
nc
T
dT
c
n
ΔS
T
dT
nc
dS
T
dT
nc
dS
T
dU
dS
2
1
2
1
2
1
Jeśli c
V
= f (T)
dlnT
c
n
ΔS
dlnT
T
dT
2
1
lnT
lnT
V
Proces nie musi byd odwracalny
T
Vdp
dH
dS
T
dq
dS
T
pdV
dU
dS
4. Przemiana izobaryczna
p= const
q
el
= dU +pdV
q
el
= dH – Vdp = dH
1
2
p
T
T
p
S
S
T
T
p
p
T
T
ln
nc
T
dT
nc
ΔS
T
dT
nc
dS
T
dT
nc
dS
T
dH
dS
2
1
2
1
2
1
dlnT
c
n
ΔS
2
1
lnT
lnT
p
Jeśli c
p
= f (T)
Proces nie musi byd odwracalny
Obliczanie zmian entropii
3. Przemiana izotermiczna
T = const q
el
=- w
el
= nRT ln(V
2
/ V
1
)
∆S = n R ln(V
2
/ V
1
)
T
Vdp
dH
dS
5. Obliczanie zmian entropii
w dowolnej przemianie
q
el
= dU + pdV
q
el
= dH - Vdp
q
el
= nc
V
dT + (nRT/V)dV
q
el
= nc
p
dT – (nRT/p)dp
1
2
1
2
V
V
V
T
T
V
S
S
V
V
V
nRln
T
T
ln
nc
ΔS
V
dV
nR
T
dT
nc
dS
V
dV
nR
T
dT
nc
dS
2
1
2
1
2
1
6. Przemiana fazowa:
f
p,
f
p,
T
H
Δ
ΔS
1
2
1
2
p
p
p
T
T
p
S
S
p
p
p
nRln
T
T
ln
nc
ΔS
p
dp
nR
T
dT
nc
dS
dp
p
nR
dT
T
nc
dS
2
1
2
1
2
1
T
dq
dS
T
pdV
dU
dS
T
Vdp
dH
dS

Statystyczna interpretacja ENTROPII
Do opisu entropii 1mola = 6,023 x 10
23
cząsteczek stosuje się
prawdopodobieostwo termodynamiczne W:
S = k
B
lnW
statystyczna definicja ENTROPII
podana przez Maxa Planck’a,
k
B
–stała Boltzmanna= 1,38 x 10
-23
J / K
W
( podstawowe pojęcie termodynamiki statystycznej)
-
określa liczbę położeo cząsteczki w dowolnym układzie zmiennych stanu (p,T,V, n
i
), czyli liczbę
sposobów podziału energii
- stanowi miarę uporządkowania układu w danym stanie ( dla 1 mola at. He w temp.298K W ~ 10
25
)
- jest tym mniejsze im wyższy jest stopieo uporządkowania
-
dla kryształu doskonałego w temp.0 K, w którym wszystkie cząsteczki mają ściśle określone
położenia w przestrzeni oraz pędy, W = 1 i jest to jego minimalna wartośd
.
W ujęciu molekularnym ENTROPIA jest miarą liczby sposobów podziału ENERGII WEWNĘTRZNEJ
między poszczególne cząsteczki i poszczególne rodzaje ich ruchów.
ENTROPIA jest miarą różnorodności stanów energetycznych cząsteczek układu - miarą
nieuporządkowania energetycznego cząsteczek.

Z równania
S = k
B
lnW
wynika, że
dla dowolnej substancji w postaci kryształu doskonałego standardowa entropia
w temperaturze 0 K równa jest 0
lim S = 0
teoremat cieplny Nernsta
T → 0
Gdy T→ 0, to S→ 0
III zasada termodynamiki S
0
0
= 0
wg Planck’a
„ W temperaturze zera bezwzględnego entropia ciała indywidualnego doskonale
krystalicznego jest równa 0”.
Statystyczna interpretacja ENTROPII
ENTROPIA jest jedyną funkcją termodynamiczną, dla której możemy policzyd jej
wartośd bezwzględną.
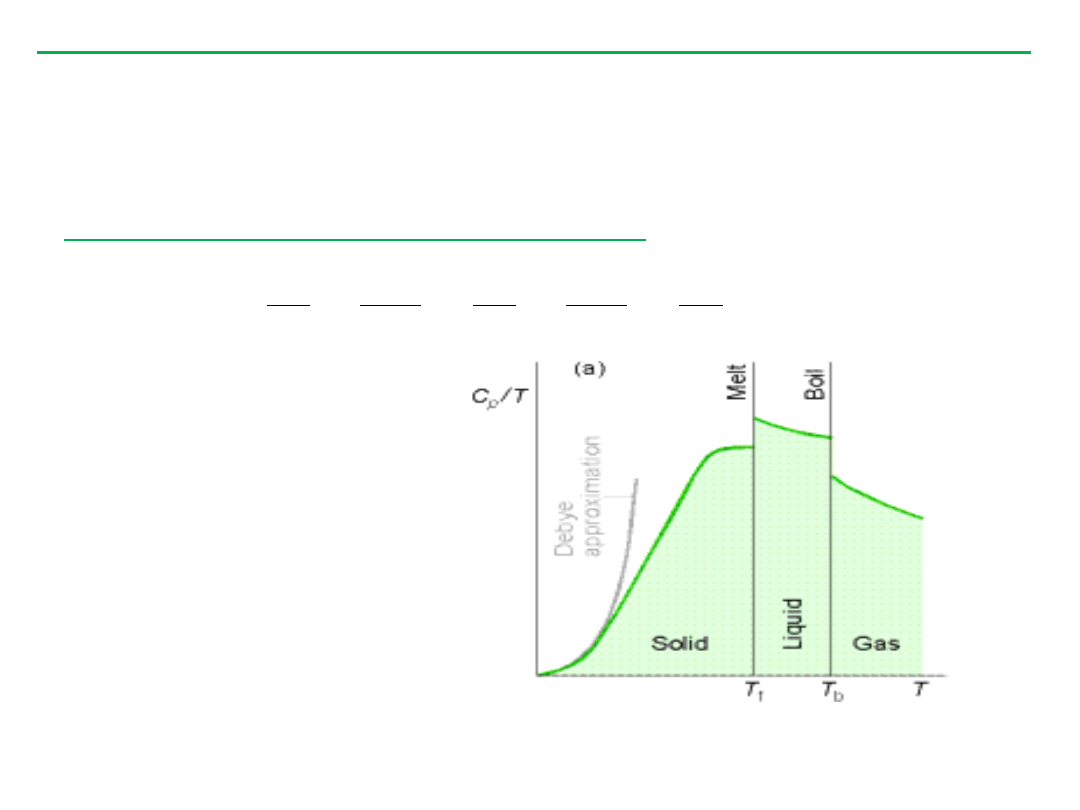
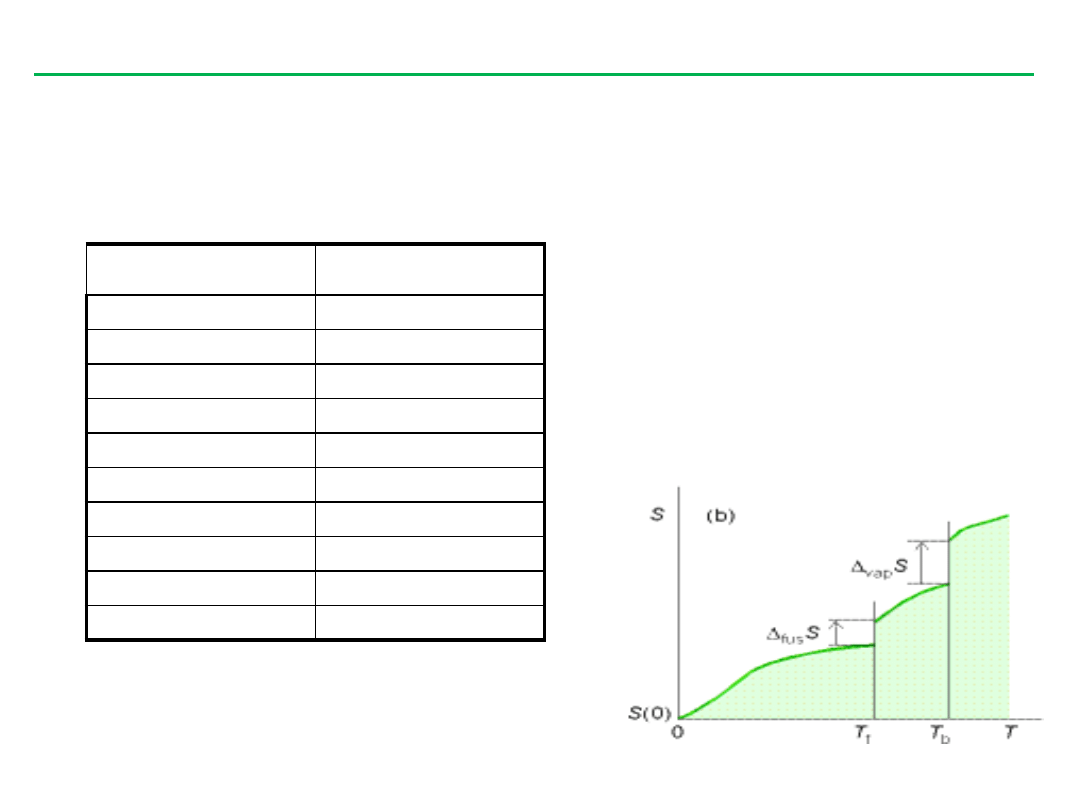
Standardowa entropia molowa S
0
pierwiastków i związków chemicznych
S
0
298
– standardowa entropia molowa w 298K
S
0
T
- standardowa entropia molowa w temp.T
Na podst. III zasady termodynamiki można obliczyd absolutne wartości entropii czystych pierwiastków
lub związków w dowolnym stanie skupienia i w dowolnej temperaturze.
ENTROPIA 1 mola związku (pierwiastka) w temperaturze T:
dT
T
c
T
ΔH
dT
T
c
T
ΔH
dT
T
c
S(0)
S
T
T
p(g)
wrz
par
T
T
p(c)
top
top
T
0
p(s)
T
wrz
wrz
top
top
gdzie:
• c
p
– izobaryczne ciepło molowe,
(s) – faza stała
(c) – faza ciekła,
(g) – faza gazowa
• H
top
– entalpia topnienia
• H
wrz
– entalpia parowania
• T
top
– temp. topnienia
• T
wrz
– temp. wrzenia
c
p
w zakresie temp. 0-10 K wyznacza się
na podst ekstrapolacji Debye’a
c
p
= aT
3
substancja
S
0
298
[J
mol
-1
K
-1
]
C (grafit)
5,73
C (diament)
2,50
NaCl
72,31
H
2
O (c)
69,93
Hg
75,95
H
2
130,45
O
2
204,86
H
2
O (g)
188,55
NH
3
192,32
CO
2
213,51
Standardowe entropie w 298K
Standardowa entropia molowa S
0
pierwiastków i związków chemicznych
Standardowa entropia reakcji chemicznej ∆
r
S
0
0
im
substraty
i
0
j m
produkty
j
0
S
ν
S
ν
S
r
to różnica entropii pomiędzy czystymi
rozdzielonymi produktami a czystymi
rozdzielonymi substratami przy czym
wszystkie reagenty znajdują się w stanie
standardowym dla danej temperatury.
ENTROPIA przemiany to stosunek ilości ciepła
wymienionego w procesie do temperatury tej wymiany.
f
p,
f
p,
f
p,
T
H
Δ
S
Δ
substancja
∆
par
H
0
[kJ
mol
-1
]
T
wrz
[
0
C]
∆
par
S
0
[J
K
-1
mol
-1
]
C
6
H
6
+30,8
80,1
+87,2
CCl
4
+30,0
76,7
+85,8
c-heksan
+30,1
80,7
+85,1
H
2
S
+18,7
-60,4
+87,9
CH
4
+8,18
-161,5
+73,2
H
2
O
+40,7
100,0
109,1
Standardowa entropia molowa ∆S
0
przemiany
Dla wielu cieczy
∆
par
S
0
~ 85 J / K mol
reguła Troutona
f
p,
0
f
p,
0
f
p,
T
H
Δ
S
Δ
Standardowe molowe entropie parowania
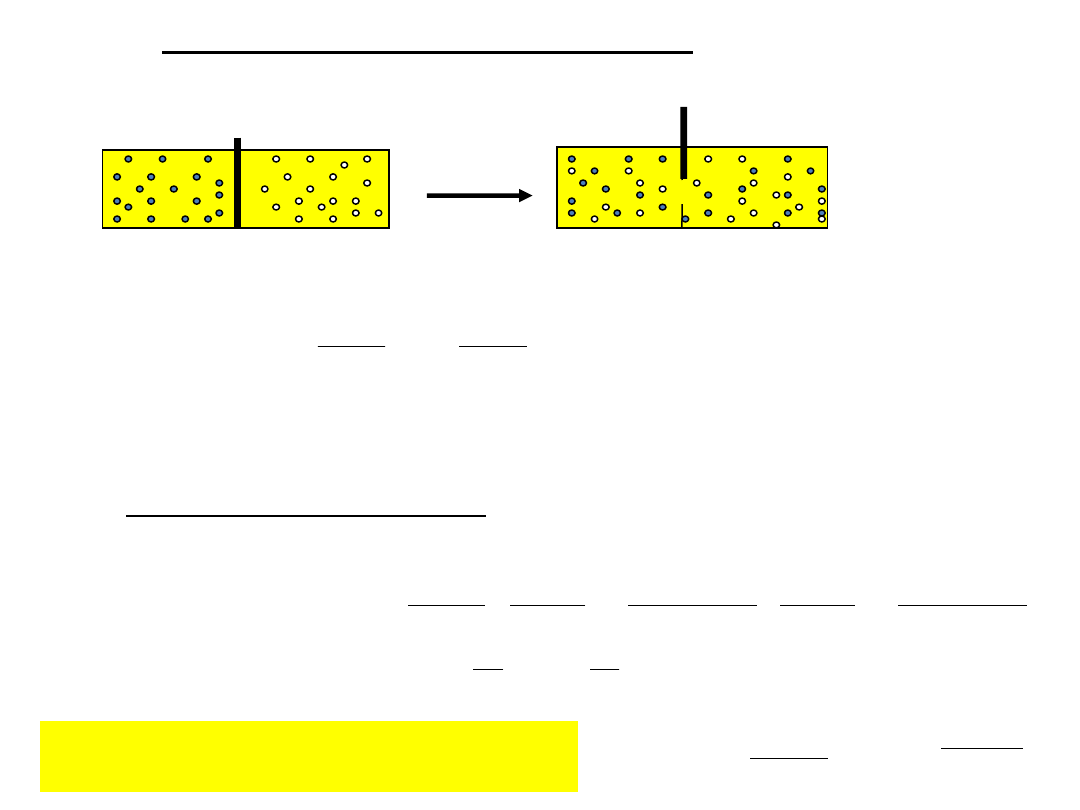
Entropia mieszania gazów doskonałych
Gaz A
n
A
Gaz B
n
B
∆S > 0
V
A
V
B
Stan przed mieszaniem (1)
Stan po zmieszaniu (2)
W warunkach T = const gazy A (n
A
) i B(n
b
) rozprężają się niezależnie od siebie do całego zbiornika.
∆S
miesz
= ∆S
A
+ ∆S
B
B
B
A
B
A
B
A
A
V
V
V
Rln
n
V
V
V
Rln
n
V
m
– objętośd 1 mola gazu doskonałego w danych warunkach p i T
V
A
= n
A
V
m
V
B
= n
B
V
m
W wyniku mieszania wzrasta objętośd przestrzeni dostępnej dla każdej cząsteczki, a więc także swoboda jej ruchu.
Wzrasta liczba sposobów podziału energii wewn. ( głównie translacyjnej) pomiędzy poszczególne cząsteczki, czyli
prawdopodobieostwo termodynamiczne W ↗, a S ↗ zgodnie z równaniem Boltzmanna: S = k
B
ln W
Średnia molowa entropia mieszania
B
B
A
A
miesz
m
B
m
B
m
A
B
A
B
m
A
m
B
m
A
B
A
A
B
A
miesz
miesz
x
1
Rln
x
x
1
Rln
x
ΔS
V
n
V
n
V
n
Rln
n
n
n
V
n
V
n
V
n
Rln
n
n
n
n
n
ΔS
ΔS
B
A
A
A
n
n
n
x
B
A
B
B
n
n
n
x
Proces samorzutny ∆S
miesz
> 0
)
lnx
x
lnx
R(x
S
Δ
B
B
A
A
miesz

Termodynamiczna skala temperatury
Z wyrażenia na wydajnośd maszyny cieplnej
1
2
1
T
T
T
η
wynika, że silnik cieplny ma maksymalną wydajnośd gdy T
2
= 0
Na tym wyniku Kelvin oparł swoją termodynamiczną skalę temperatury.
Jako jednostkę podstawową skali przyjął stopieo Celsjusza
.
Najniższa temperatura jaką udało się uzyskad to
20 nK = 20 x 10
-9
K
T[K] = 0[
0
C] + 273,15

Jak można osiągnąd bardzo niskie temperatury?
Z doświadczenia Joule’a – Thomsona wynika, że ekspansja gazu prowadzona poniżej jego
temperatury inwersji prowadzi do jego ochłodzenia.
W urządzeniu przeciwprądowym (Siemens 1860r) następuje wymiana ciepła między gazem
rozprężonym i ochłodzonym a gazem sprężonym, prowadząca do stopniowego ochładzania się gazu
aż do jego skroplenia.
Destylacja ciekłego powietrza daje
ciekły O
2
T
wrz
= 90 K
i
ciekły N
2
T
wrz
= 77 K.
Ekspansja
H
2
ochłodzonego za pomocą ciekłego azotu (poniżej temp.193 K) powoduje jego
skroplenie w temperaturze
22 K
.
Ciekły H
2
pozwala na schłodzenie i skroplenie
He T
wrz
= 4 K.
Zmniejszenie ciśnienia nad helem
pozwala uzyskad temp.
1 K.
Temp. < 1 K
uzyskuje się metodą rozmagnesowywania adiabatycznego ( 1926 r P.Debye i W.F. Giauque)
Kryształ soli paramagnetycznej ochładza się do temperatury ciekłego helu w obecności pola
magnetycznego.
Po odizolowaniu termicznym kryształu wyłącza się pole magnetyczne. Kryształ ulega samorzutnemu
adiabatycznemu rozmagnesowaniu. Entropia wzrasta kosztem energii oscylacyjnej sieci krystalicznej soli i
temperatura kryształu obniża się. W ten sposób uzyskano
temp. 10
-7
K.
Stosując metodę jądrowego rozmagnesowania adiabatycznego dla Cu ustanowiono światowy rekord
niskiej
temp. 2 x 10
-8
K.
Wyszukiwarka
Podobne podstrony:
7 I i II zas termodynamiki
4. Podstawy organizacji pracy redakcyjnej - 29.10.15 r, Rok1, Edytorstwo, Problemy organizacji pracy
2. Sprawozdanie 29.10.2014 - Statyczna próba ściskania, Studia ATH AIR stacjonarne, Rok II, Semestr
W5 29.10.03, Choroby wewnętrzne
zarz b9dzanie+finansami+ii+ +wyk b3ad+3+ 2824 10 2005 29 4IZCMOGXOJNMP5V74PSQ7X6DACOYTF47UQYPCEA
zarz b9dzanie+finansami+ii+ +wyk b3ad+2+ 2817 10 2005 29 WVDK6WE6KAD7RE4XECWU4H2KEA43EGQV32S6EVA
W3 I zas term (15 10 15)
plan 18.10-29.10, plany, scenariusze, Plany
29 10?rmakoterapia bólu
10 15
Elektrorafinacja 16 10 15
Wyklad II -psychopatologia i podzial w ICD 10, Psychopatologia
10 (15)
03 Immunologia prelekcja 10 15 2007id 4167 (2)
Koszykówka atak szybki 02 10 15
Koszykówka atak szybki 02 10 15
Wykład 8, 29.10.08
18 entropia i II zasada termodynamiki
więcej podobnych podstron