
Choroby jednogenowe
autosomalne dominujące
Mateusz Skowronek
Gr 46

Kryteria dziedziczenia
autosomalnego dominującego
●
Cecha (choroba) ujawnia się już u heterozygot (Aa),
natomiast homozygoty AA spotyka się rzadko, gdyż
są najczęściej letalne
●
Ze związku Aa + aa = 50% dzieci obarczonych
chorobą (Aa), a 50% to osobnicy zdrowi (aa)
●
Ze związku Aa + Aa = 50% dzieci to chore
heterozygoty (Aa), 25% to chore homozygoty (AA),
25% to zdrowe homozygoty recesywne (aa)

Kryteria dziedziczenia
autosomalnego dominującego
●
Cecha (choroba) występuje z jednakową częstością u obu
płci
●
Pionowe przekazywanie cechy- z pokolenia na pokolenie
●
Obraz kliniczny choroby zależy od:
✓
Zmiennej ekspresji genu
✓
Stopnia penetracji genu
✓
jeżeli geny dominujące wykazują niepełną penetracje
występuje zjawisko ,,wyciszenia” typowych objawów
chorobowych, aż do pełnego ich zaniku
✓
Stopień penetracji zależy od wieku

Kryteria dziedziczenia
autosomalnego dominującego
●
Nasilenie objawów choroby może zależeć od płci
chorego rodzica przekazującego zmutowany gen
●
Niektóre choroby monogenowe ujawniają się w
późnym wieku np. choroba Huntingtona

Dziedziczenie autosomalne
dominujące
Dziedziczenie autosomalne recesywne
Chorują homozygoty (AA) i
heterozygoty (Aa), homozygoty są
najczęściej letalne
Chorują homozygoty (aa)
Ok. 50% potomstwa jest chorych , jeżeli
jedno z rodziców jest heterozygotą
W przypadku rodziców
heterozygotycznych tylko 25%
potomstwa ma określoną cechę
Ryzyko wystąpienia cechy u potomstwa
zwiększa się z wiekiem ojca
Nie ma zależności miedzy wiekiem
rodziców a wystąpieniem choroby u
dzieci
Zmienna ekspresja
Stała ekspresja
Pionowy wzór rodowodu
Poziomy wzór rodowodu
Nie zaobserwowano wpływu
pokrewieństwa na częstość
występowania choroby u potomstwa
Pokrewieństwo rodziców zwiększa
ryzyko wystąpienia cechy u potomstwa

Nerwiakowłókniakowatość
(Neurofibromatosis- NF ,
choroba Recklinghausena
Choroba heterogenna, występująca
w dwóch postaciach : NF-1 i NF-2

Nerwikowłókniakowatość
●
Gen NF-1, odpowiedzialny za chorobę Recklinghausena,
zlokalizowany na 17 chromosomie jest genem
autosomalnym dominującym
●
W obrębie genu mutacje typu delecje występują z tą samą
częstością u kobiet i u mężczyzn
●
Mutacje typu de novo , najczęściej pochodzenia ojcowskiego
są przyczyną 50% nowych zachorowań
●
Obniżony poziom produktu tego genu- białka
neurofibrominy , sprzyja rozwojowi nowotworów
●
Częstość choroby wynosi 1: 3500 urodzeń

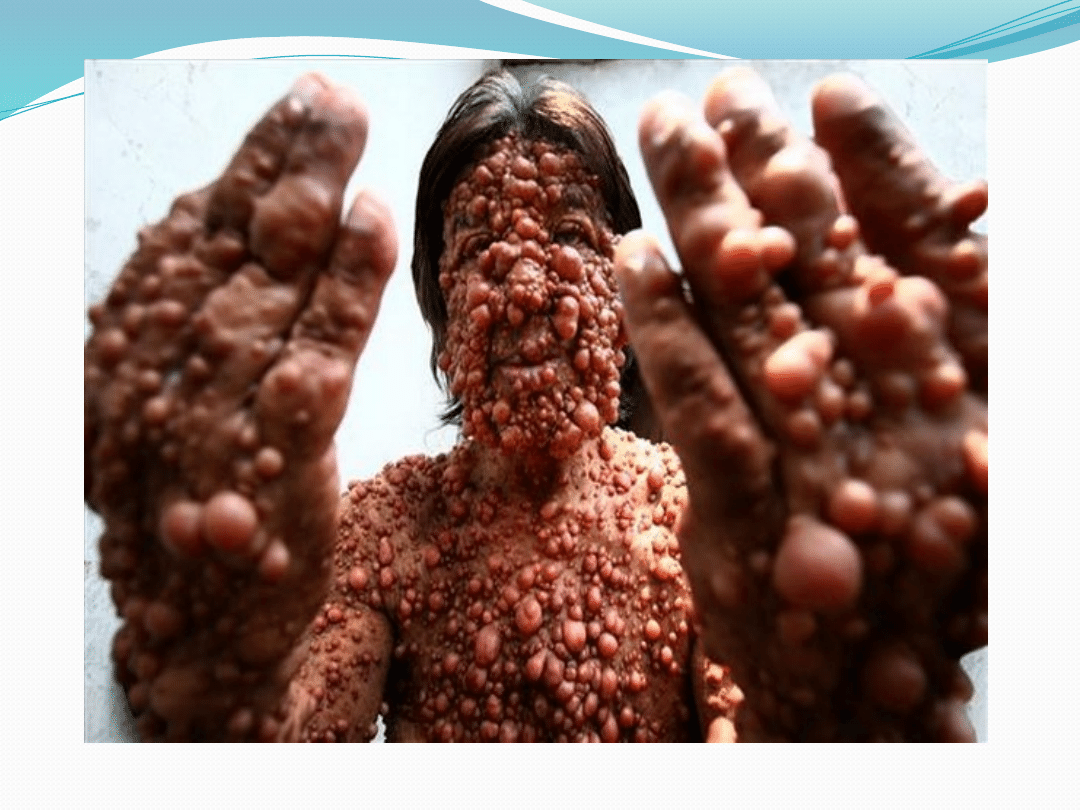
Objawy
●
We wczesnym dzieciństwie pojawiają się zmiany
barwnikowe koloru ,, kawy z mlekiem’’
●
W okresie dojrzewania pojawiają się liczne guzki
wywodzące się z nerwów obwodowych, wyrózniamy:
włókniaki, nerwiakowłókniaki oraz glejaki nerwu
wzrokowego
●
Guzy te są zwykle łagodne ale w 3-13% ulegają
zezłośliwieniu
●
Często występuje również niedorozwój umysłowy,
padaczka i skrzywienie kręgosłupa

Nerwikowłókniakowatość
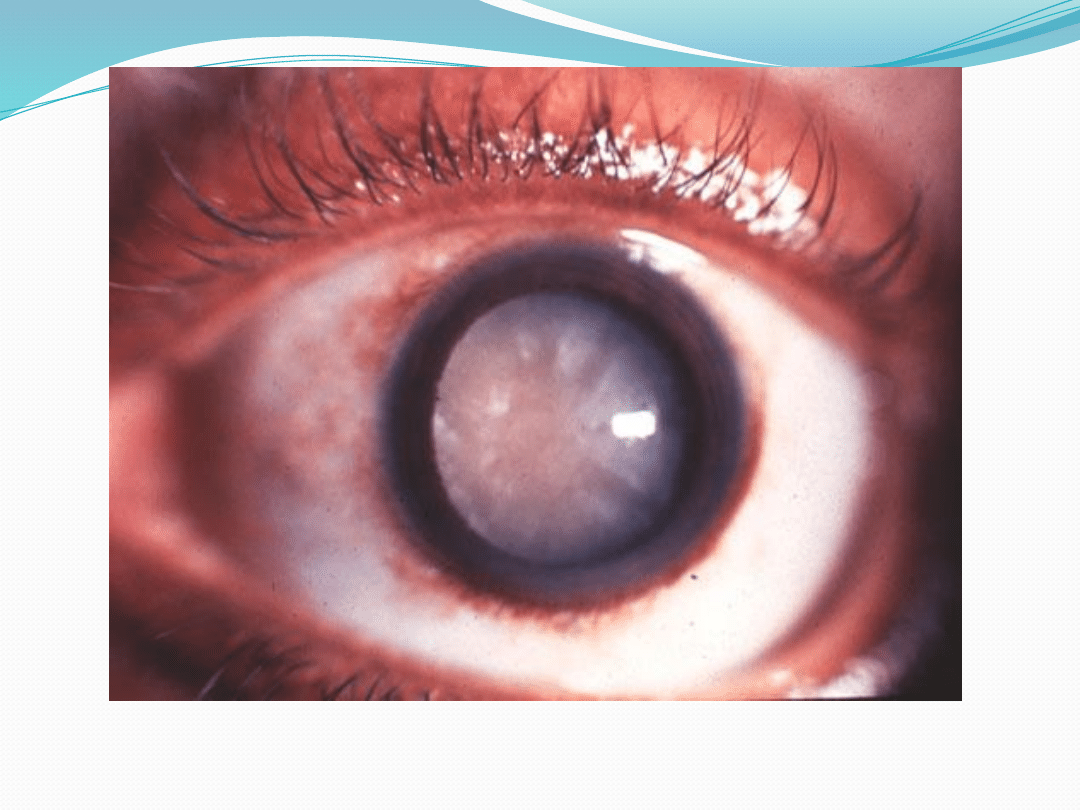
●
Gen NF-2 znajduje się na chromosomie 22 i
dziedziczy się autosomalnie dominująco
●
Częstość występowania to 1: 3500-40000 urodzeń
●
Produktem genu jest merlina, będąca białkiem
cytoszkieletu
●
Mutacja tego genu powoduje powstanie
nerwiaków osłonkowych a także zmętnienie
soczewki

Achondroplazja
( chondrodystrofia,
karłowatość chondrodystroficzna)
●
Jedna z najczęsciej występujących karłowatości u
ludzi
●
Dziedziczona jako cecha autosomalna dominująca
●
Częstość występowania 1: 15000- 77000
●
W ponad 80% jest wynikiem powstania nowej
mutacji ( rodzice są zdrowi)
●
Częstość mutacji de novo rośnie wraz z wiekiem ojca

Achondroplazja
●
Gen wywołujący chorobę jest zlokalizowany na
ramieniu krótkim chromosomu 4 (4p16.3)
●
Jest to gen receptora czynnika wzrostu fibroblastów
( FGFR3)
●
W nukleotydzie 1138 tego genu może wystąpić:
➢
Tranzycja G A
➢
Transwersja G C

Achondroplazja
●
Konsekwencją tych mutacji punktowych jest zmiana
glicyny (G) na argininę (R) w 380 pozycji w łańcuchu
białka – G380R
●
Obie mutacje w 1138 nukleotydzie genu FGFR3 mogą
być wykryte za pomocą technik biologii molekularnej

Objawy
●
Skrócenie kończyn, zwłaszcza odcinków
proksymalnych
●
Mikromelia (małe dłonie)
●
Szpotowate kolana
●
Ograniczenie prostowania stawu łokciowego
●
Nadmierna lordoza lędźwiowa
●
Charakterystyczna twarz z wypukłym czołem i
zapadniętą nasadą nosa

Objawy
●
Wysokość ciała u mężczyzn to ok. 132cm, a u
kobiet ok. 123cm
●
Radiologicznie stwierdza się m.in.: małe sześcienne trzony
kręgów, lordozę lędźwiową, kifozę piersiowo-lędźwiową,
wąskim kanał kręgowy
●
Rozwój umysłowy jest prawidłowy
●
Śmiertelność wzrasta od urodzenia do 4 roku życia i
poźniej w 4-5 dekadzie życia
●
Homozygoty (AA) mają tak liczne wady , że większość
umiera w 1 roku życia



Zespół Marfana- arachnodaktylia
●
Częstość występowania to 1:10000 urodzeń
●
Gen odpowiedzialny za chorobę nazwano FBN1 ( gen
fibriliny) i znajduje się on na ramieniu długim
chromosomu 15 (15q21.1)
●
W 25% przypadków choroby są to nowe mutacje
●
Gen koduje fibrylinę, białko które jest głównym
składnikiem zewnątrzkomórkowych mikrofibryli

Zespół Marfana- arachnodaktylia
●
Skutkiem mutacji genu FBN1 jest defekt tkanki
mezenchymalnej, uszkodzenie włókien sprężystych i
zaburzenie w tworzeniu łańcuchów alfa kolagenu w
układzie kostno- stawowym, krążenia i w gałkach
ocznych.

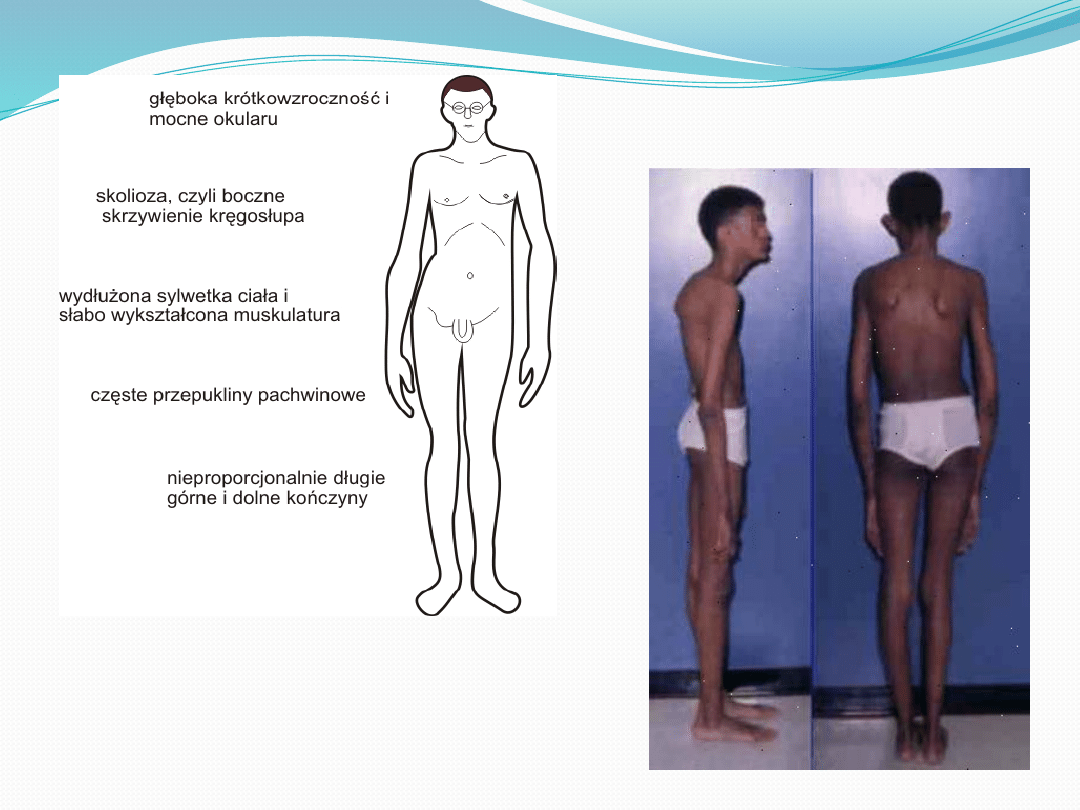
Objawy
●
Nadmierny wzrost kości długich i zaburzenie
stosunku długości tułowia do długości kończyn
dolnych
●
Smukła sylwetka, wysoki wzrost, nadmiernie długie
palce rąk i stóp
●
Klatka piersiowa o kształcie kurzym lub lejkowatym
●
Nadmiernie elastyczna skóra
●
Podwichnięcie soczewki, krotkowzroczność, wady
serca, przepukliny

Choroba Huntingtona
●
Częstość występowania 4-7: 100000
●
Gen HD odpowiedzialny za chorobe zlokalizowany
jest na ramieniu krótkim chromosomu 4 w regionie
4p16.3
●
Gen koduje białko o masie 348 kD o nazwie
huntingtina
●
Choroba dziedziczy się autosomalnie dominująco

Choroba Huntingtona
●
Jest wynikiem niestabilnej liczby powtórzeń
sekwencji nukleotydowej CAG na końcu 5’ genu
kodującego huntingtynę
Osoby zdrowe 10-29 powtórzeń CAG
Stan przedmutacyjny 30-35 powtórzeń
Osoby chore 36 i powyżej powtórzeń
●
Istnieje korelacja miedzy większą niż 50 liczbą
powtórzeń i rozwojem choroby we wczesnym okresie
życia

Choroba Huntingtona
●
Choroba wykazuje antycypację tzn. ze choroba
występuje w coraz młodszym wieku i z coraz
cięższym przebiegiem w kolejnych pokoleniach
●
Antycypacja jest mocniej wyrażona jeśli zmutowany
gen pochodzi od ojca
●
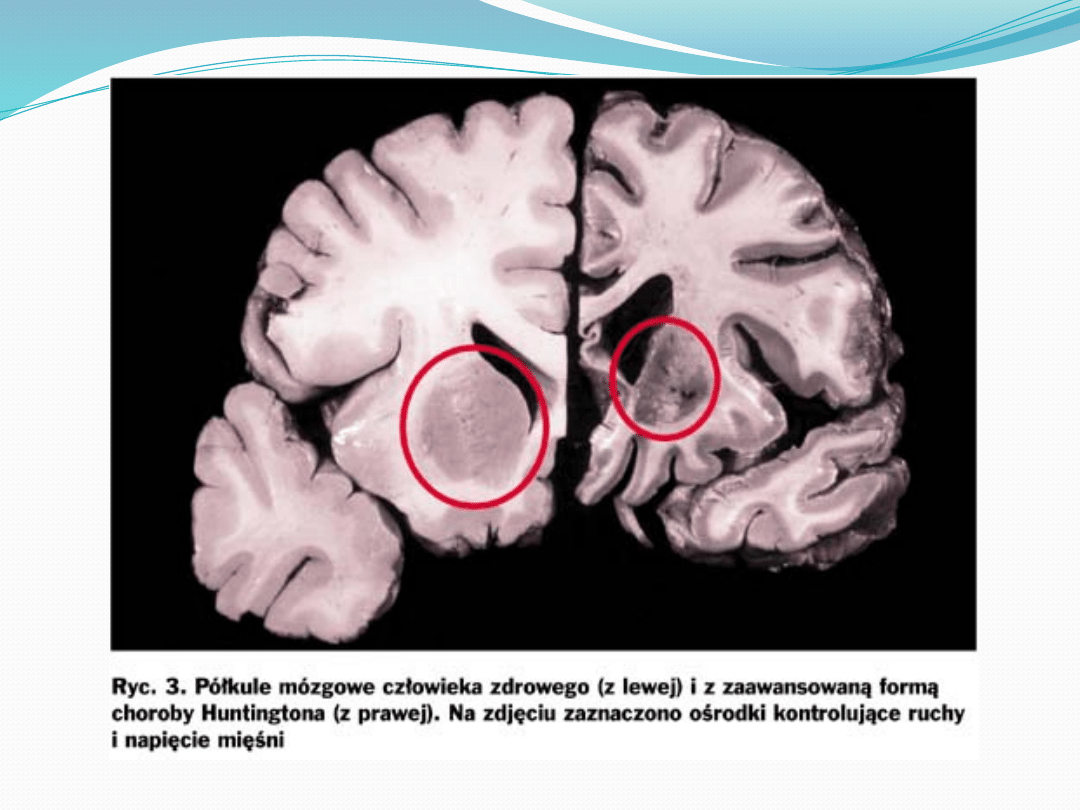
Początek choroby następuje zwykle w 4 dekadzie
życia- zanik małych neuronów w jądrze ogoniastym i
skorupie oraz dużych neuronów gałki bladej

Objawy
●
Zaburzenia hiperkinezy- przypominające taniec
●
Zaburzenia mowy
●
Otępienie umysłowe
●
Charłactwo fizyczne
●
W czasie snu objawy pląsawicy ustępują
●
Śmierć następuje w ciągu 10-15 lat od momentu
wystąpienia pierwszych objawów

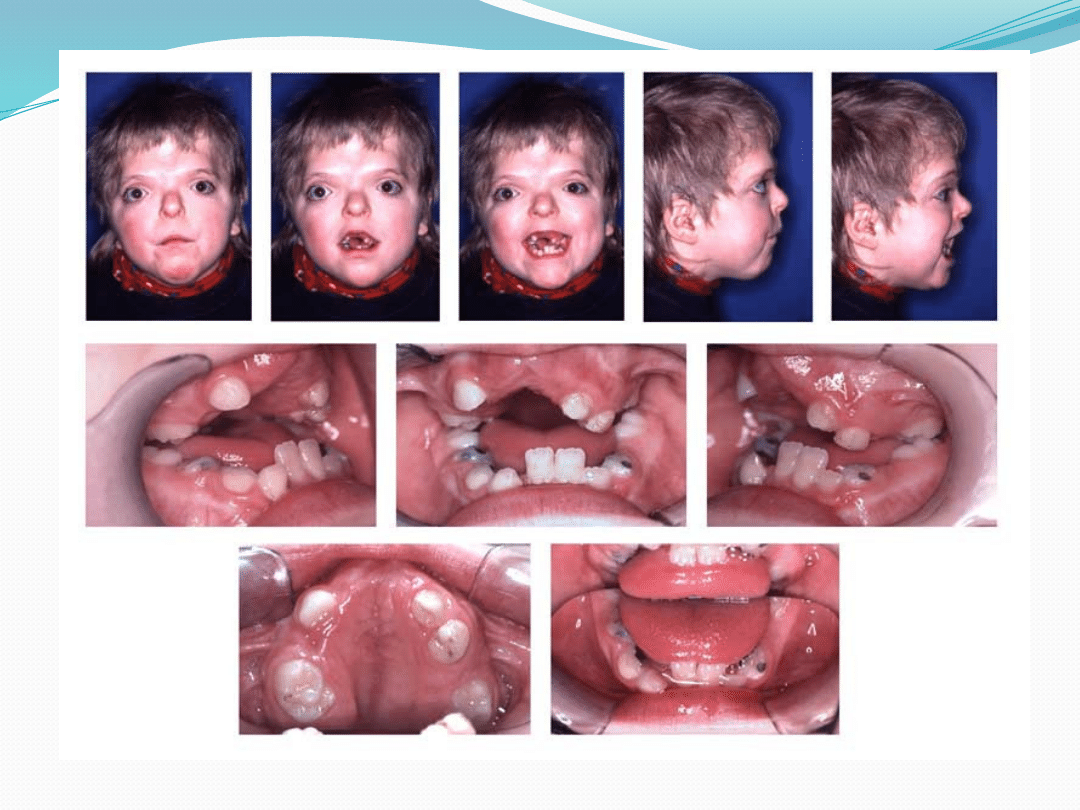
Zespół Aperta
●
Choroba wywołana mutacją w genie receptora 2
czynnika wzrostu fibroblastów -FGFR2
●
Gen odpowiedzialny za zespół występuje na
ramieniu długim chromosomu 10 w pozycji 10q26
●
Obrazem klinicznym jest wrodzone zarośnięcie
szwów czaszkowych oraz palcozrost

Objawy
●
Deformacje czaszki związane z przedwczesnym
zarośnięciem szwów czaszkowych
●
Zrośnięcie palców 2-5 (syndaktylia) rąk i stóp
●
Zmiany w obrębie twarzoczaszki prowadzą
do pseudoprognatyzmu ( nadmiernie wysuniętej
twarzoczaszki ) wskutek względnej hipoplazji szczęki i
innych struktur kostnych środkowej części twarzy
●
Hipertolaryzm oczny, rozszczep podniebienie,
niedorozwój umysłowy

Osteogenesis imperfecta-
wrodzona łamliwość kości
●
Choroba wywołana mutacjami w genie
odpowiadającym za prawidłową budowę
kolagenu
Typ
Gen
Locus
Białko
Typ I (OI1)
COL1A1
COL1A2
17q21.31-q22
7q22.1
Kolagen I
Typ II (OI2)
COL1A1
COL1A2
17q21.31-q22
7q22.1
Kolagen I
Typ III (OI3)
COL1A1
COL1A2
17q21.31-q22
7q22.1
Kolagen I
Typ IV (OI4)
COL1A1
COL1A2
17q21.31-q22
Kolagen I
Typ VII (OI7)
CRTAP
3p22, 3p24.1-p22
CRTAP
Typ VIII (OI8)
LEPRE1
1p34
Leprekan

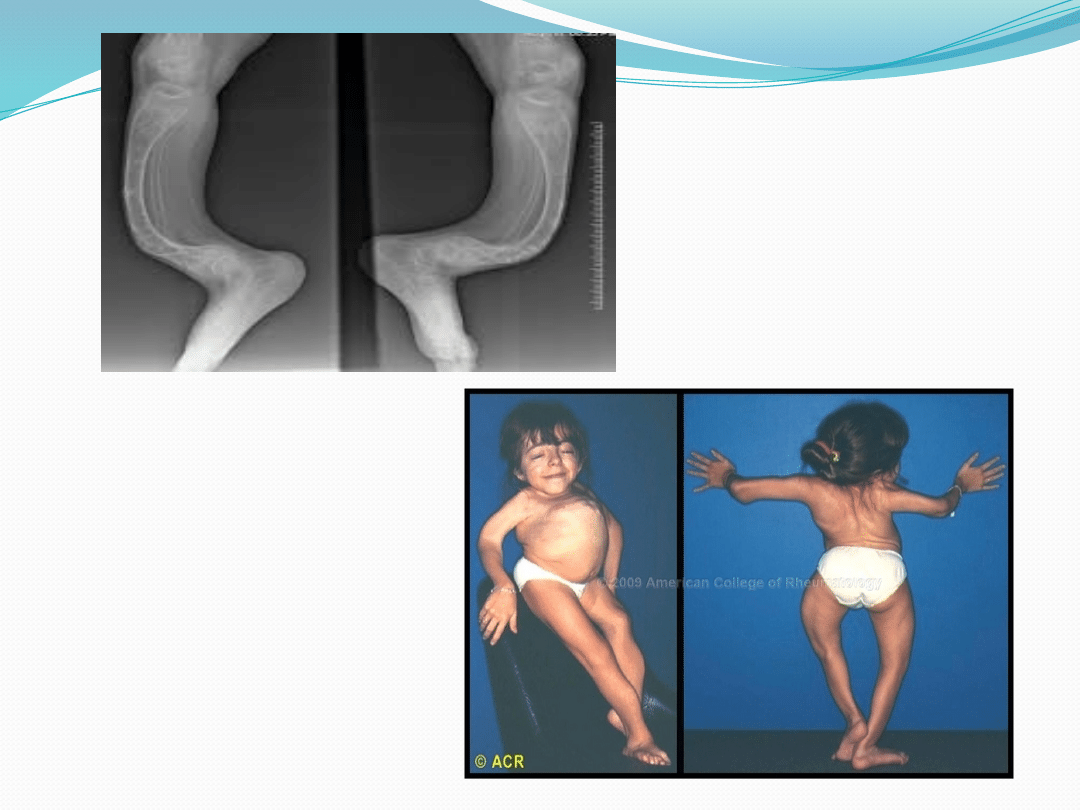
Osteogenesis imperfecta
●
Częstość występowania 1: 30000 urodzeń
●
Nagminne złamania kości, nawet podczas snu
●
Najczęściej złamaniom ulegają kości ramion i stóp
●
Występuje ogromna giętkość kości, przez co
przybierają one kształty łuków
●
Bardzo niski wzrost ( poniżej 1,5 m)
●
Może występować kamica nerkowa, astygmatyzm
oraz niebieskawe zabarwienie twardówki

Dziękuję za uwagę
☺
Wyszukiwarka
Podobne podstrony:
4 Ch jednogenowe autos dominujące Skowronek
5 Ch jednogenowe cz 2 Ch wieloczynnikowe Skrzypek
4 Ch jednogenowe cz 1 Ładosz
(Laudate Dominum ch 363r m
9 Ch organiczna WĘGLOWODANY
ch wrzodowa prof T Starzyńska
ch zwyrodnieniowa st
Ch 28 Pelites
11 Ch organiczna AMINOKWASY I BIAŁKAid 12388 ppt
3 ch org zwiazki funkcyjne
dziedziczenie chorob jednogenowych
WYKúAD 4 MASA» J CH cd
Reh amb w ch Parkinsona
Wykład Ch F konduktometria
więcej podobnych podstron