
Biochemia: Ćw. – Metoda RT-PCR
1
M
ETODA
RT-PCR
Wyciąg z kart charakterystyki substancji niebezpiecznych:
bromek etydyny
– T+
EDTA
– Xi
etanol, 96%
– F
kwas octowy, 96%
– C
-merkaptoetanol
– N, T
Tris
– Xi
U
WAGI WSTĘPNE
Praca z kwasami nukleinowymi (np. izolacja, reakcja PCR) wymaga zachowania szczególnej czystości, ponieważ kwasy nukleinowe łatwo ulegają degradacji. Dotyczy to głównie
RNA, ze względu na powszechną obecność rybonukleaz (RNaz) – enzymów katalizujących niespecyficzne rozrywanie łańcuchów rybonukleinowych. Poniżej zamieszczono kilka
uwag istotnych podczas eksperymentów, w których używamy kwasów nukleinowych.
Wszystkie czynności wykonujemy w czystych rękawiczkach, które przemywamy 70% etanolem.
Wszystkie końcówki, probówki i szkło muszą być autoklawowanie.
Izolację kwasów nukleinowych oraz przygotowanie reakcji PCR przeprowadzamy w komorze laminarnej wysterylizowanym lampą UV. Miejsce do izolacji i aparat do
rozdziału elektroforetycznego przemywamy 70% etanolem.
Bufory z zestawu do preparatyki kwasów nukleinowych, składniki zestawu do reakcji PCR, polimerazy pobieramy używając końcówek z filtrem.
PBS sterylizujemy przez filtrowanie (filtr o średnicy porów 0.2
m); bufory z gotowych zestawów do izolacji i reakcji PCR są sterylne.
Metoda łańcuchowej reakcji polimerazy poprzedzona odwrotną transkrypcją, RT-PCR (ang. reverse transcription polymerase chain reaction) służy m.in. do
analizy ekspresji badanego genu. Bezpośrednim i najbardziej miarodajnym sposobem sprawdzenia, czy gen ulega ekspresji w danej komórce jest wykrycie
obecności transkryptu danego genu – cząsteczek mRNA. Technika RT-PCR składa się z dwóch etapów:
- reakcji odwrotnej transkrypcji, w której mRNA przepisywane jest na cDNA,
- amplifikacji (wielokrotnego powielenia) cząsteczek cDNA metodą klasycznej reakcji PCR.
Ostatnim etapem badania ekspresji genu jest elektroforeza produktów (amplifikatów) reakcji PCR wykonywana najczęściej w żelu agarozowym.
1.
P
RZYGOTOWANIE
RNA
1.1
I
ZOLACJA
RNA
(
ZESTAW FIRMY
Q
IAGEN
)
Materiał do izolacji kwasów nukleinowych stanowią skrawki tkankowe, całe narządy pobierane od zwierząt, komórki ustalonych linii komórkowych lub linii
pierwotnych hodowane in vitro w naczyniach hodowlanych (szalkach Petriego, płytkach wielodołkowych lub butelkach). Zamieszczona poniżej procedura w
punktach 1 – 4 odnosi się do komórek adherentnych hodowanych in vitro.
1. Po osiągnięciu 70 – 90% zarośnięcia naczynia hodowlanego (szalki Petriego o średnicy 6 cm) komórki przemyć zimną pożywką nie
zawierającą surowicy a następnie zimnym sterylnym PBS*.
2. Po dokładnym odciągnięciu roztworu PBS komórki zebrać sterylnym skrobakiem do probówki typu Eppendorf o poj. 1.5 ml w 350
l
buforu lizującego RLT zawierającego
-merkaptoetanol. Tak zebrane komórki można przechowywać w temp. -70
C przez okres 1
miesiąca.
3. Do ekstraktów komórkowych dodać 350
l 70% etanolu, zamieszać pipetą.
4. 700
l mieszaniny nałożyć na minikolumnę do izolacji RNA. Po zwirowaniu (8000 RPM**, 15 s, RT***) kolumnę przełożyć do nowej
probówki wirowniczej o poj. 2 ml.
5. Dodać 700
l buforu RW1, zwirować (8000 RPM, 15 s, RT).
6. Kolumnę umieścić w nowej probówce wirowniczej o poj. 2 ml i dodać 500
l buforu RPE, zwirować (8000 RPM, 15 s, RT).
7. Kolumnę umieścić w nowej probówce wirowniczej o poj. 2 ml i dodać 500
l buforu RPE, zwirować (8000 RPM, 2 min, RT).
8. Kolumnę umieścić w nowej probówce wirowniczej o poj. 2 ml i zwirować w celu wysuszenia membrany (15000 RPM, 1 min, RT).
9. Kolumnę przenieść do probówki wirowniczej o poj. 1.5 ml i dodać na membranę 100
l wody wolnej od RNaz, zwirować (8000 RPM,
1 min, RT). Czynność powtórzyć nakładając na kolumnę eluat otrzymany z pierwszego wirowania.
*PBS (ang. Phosphate Buffered Saline) – zbuforowany roztwór soli fizjologicznej
**RPM (ang. Revolutions Per Minute) – liczba obrotów na minutę
***RT (ang. Room Temperature) – temperatura pokojowa
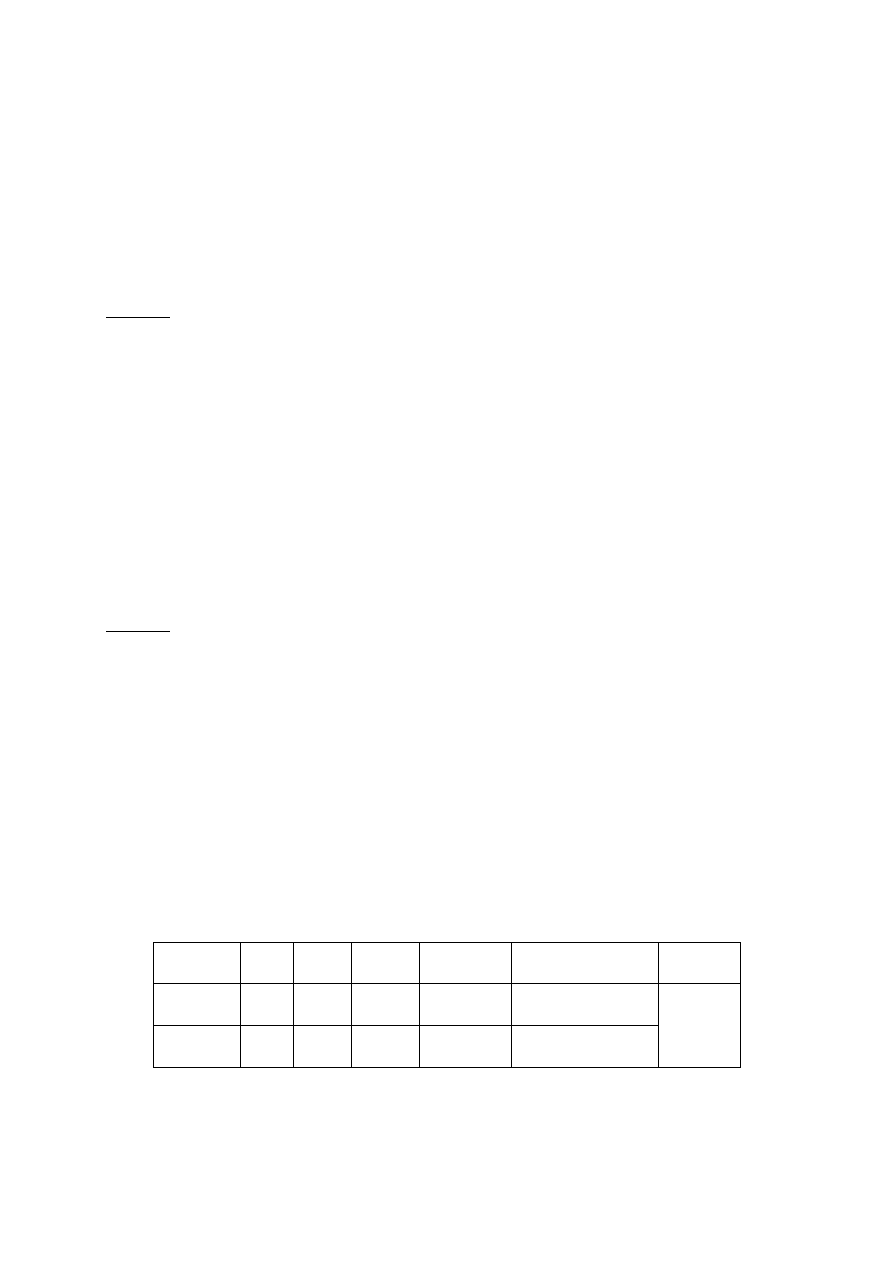
Biochemia: Ćw. – Metoda RT-PCR
2
1.2.
O
KREŚLENIE STĘŻENIA ORAZ CZYSTOŚCI
RNA
METODĄ SPEKTROFOTOMETRYCZNĄ
Kwasy nukleinowe selektywnie pochłaniają światło ultrafioletowe (UV) wykazując maksimum absorbancji przy długości fali 260 nm.
Zjawisko to znajduje zastosowanie w oznaczaniu stężenia kwasów nukleinowych, np. w preparatach kwasów nukleinowych po izolacji.
Ekstrakty kwasów nukleinowych mogą być zanieczyszczone np. białkami lub odczynnikami używanymi do izolacji. W celu określenia
czystości przygotowanego preparatu mierzy się OD również przy dł. fali 280 nm, stanowiącą maksimum absorbancji dla białek. Preparat
uznaje się za czysty wówczas, kiedy stosunek wartości absorbancji A
260
/A
280
dla RNA jest równy lub zbliżony do 2.0. W przypadku, gdy
stosunek wartości A
260
/A
280
jest niższy, roztwór RNA jest zanieczyszczony białkami.
Wykonanie: W spektrofotometrze do mikroobjętości (Thermo Scientific Nano-Drop 2000) zmierzyć absorbancję przy dł. fali
260 i 280 nm względem próby ślepej (wody wolnej of RNaz). Spektrofotometr dla warstwy absorbującej równej 10 mm
w oparciu o zadaną wartość współczynnika absorbancji (dla RNA 1 jednostka A
260
równa jest 40
g/ml) przelicza zmierzoną
wartość absorbancji na stężenie wyrażone w ng/µl. Wyliczona przez spektrofotometr wartość ilorazu A
260
/A
280
świadczy
o czystości preparatu.
1.3.
P
RECYPITACJA
RNA
W przypadku uzyskania preparatu RNA o bardzo małym stężeniu (często uniemożliwiającym pomiar metodą spektrofotometryczną)
roztwór możemy zagęścić przez precypitację (strącanie) z użyciem środków odwadniających. Najczęściej do precypitacji używa się
alkoholi: etanolu lub izopropanolu. Wydajność precypitacji kwasu nukleinowego zwiększa użycie schłodzonego alkoholu; samą
precypitację też wykonuje się w niskiej temperaturze (4
C lub -20
C).
Wykonanie: Przygotować probówkę typu eppendorf o poj. 0.5 ml. Do 100
l roztworu wyjściowego RNA dodać
10
l 3M CH
3
COONa, pH 5.2 (RT) oraz 250
l 96% etanolu (-20
C). Wymieszać dokładnie i inkubować przez
30 min w temp. -20
C. Wytrącone RNA wirować przez 10 min, 13000 RCF, RT (wirówka miniSpin plus, Eppendorf). Zebrać
dokładnie nadsącz a peletę przemyć 250
l 70% etanolu (RT) i zworteksować. Ponownie zwirować przez 10 min, 13000
RCF, RT (wirówka miniSpin plus, Eppendorf). Po dokładnym zebraniu nadsączu peletę wysuszyć pod lampką (ok.10 min)
a następnie rozpuścić w 10
l wody dejonizowanej.
W celu określenia odzysku RNA po precypitacji zmierzyć jego stężenie w roztworze wyjściowym oraz precypitacie (pkt. 1.2).
Określić ilość RNA w roztworze wyjściowym i precypitacie oraz procent RNA odzyskany po precypitacji. Wyniki zamieścić
w tabeli 1. Skomentować czystość roztworów RNA oraz wydajność precypitacji RNA.
Tab.1. Bilans precypitacji RNA.
roztwór RNA
A
260
A
280
A
260
/A
280
stężenie RNA
[ng/µl]
ilość RNA
w danej objętości [µg]
odzysk RNA
[%]
wyjściowy
precypitat
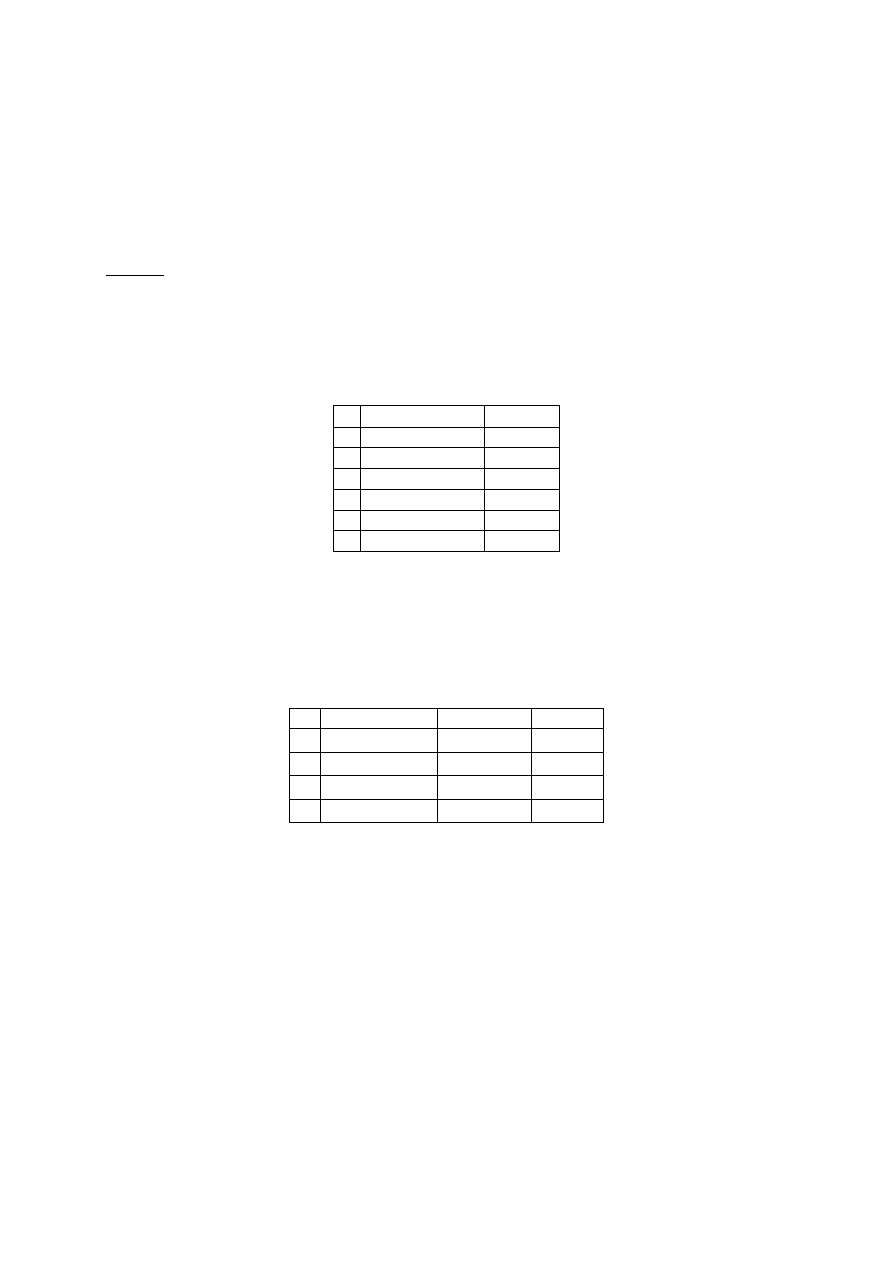
Biochemia: Ćw. – Metoda RT-PCR
3
2.
O
DWROTNA TRANSKRYPCJA
Cząsteczki kwasu mRNA przed amplifikacją w reakcji PCR należy przepisać na cDNA (ang. complementary DNA) w procesie odwrotnej
transkrypcji z użyciem enzymu – odwrotnej transkryptazy (RT, ang. reverse transcriptase).
Wykonanie: W probówce typu eppendorf o poj. 0.5 ml 1
g RNA rozcieńczyć wodą wolną od RNaz do obj. 5
l. W kolejnej probówce
o poj. 0.5 ml przygotować mieszaninę reakcyjną, składającą się z: buforu 10-krotnie stężonego, roztworu czterech
deoksyrybonukleozydotrifosforanów (dNTP), krótkich fragmentów oligodeoksyrybonukleotydowych (dT23), wody wolnej od RNaz oraz
enzymu – odwrotnej transkryptazy. Zamieszczona poniżej tabela 2 zawiera skład mieszaniny reakcyjnej na jedną próbkę.
Tab.2. Skład mieszaniny reakcyjnej do reakcji odwrotnej transkrypcji.
składniki mieszaniny
objętość [
l]
1.
bufor
10x stężony
2
2.
5 mM dNTP
2
3.
70
M oligo dT23
0,3
4.
woda wolna od RNaz
9,7
5.
odwrotna transkryptaza
1
.
objętość całkowita
15
Równocześnie z próbką badaną przygotować kontrolę negatywną: mieszanina reakcyjna z wodą wolną od RNaz w miejsce RNA.
Odwrotną transkryptazę dodać na końcu, tuż przed dodaniem mieszaniny do probówki zawierającej RNA. Po zwirowaniu, probówki
umieścić w bloku grzejnym i prowadzić reakcję zgodnie ze schematem podanym w tabeli 3.
Tab.3. Etapy odwrotnej transkrypcji.
nazwa cyklu
temperatura
czas [min]
1.
wstępna denaturacja
65ºC
5
2.
odwrotna transkrypcja
37ºC
60
3.
przerwanie reakcji
95ºC
5
4.
koniec reakcji
4ºC
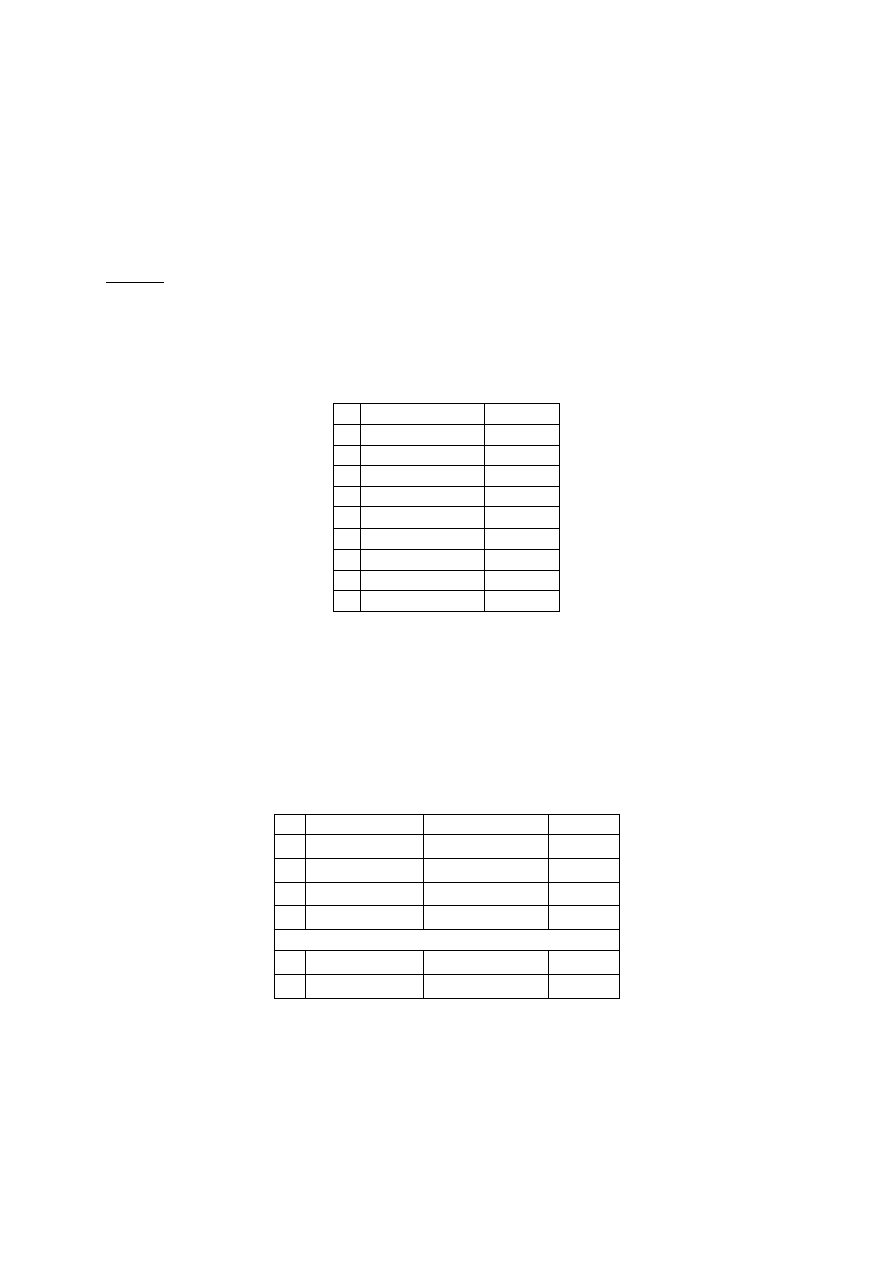
Biochemia: Ćw. – Metoda RT-PCR
4
3.
A
MPLIFIKACJA C
DNA
METODĄ
PCR
Technika PCR (reakcja łańcuchowa polimerazy lub reakcja łańcuchowa polimeryzacji) zapewnia uzyskanie w przeciągu krótkiego czasu
milionów – miliardów kopii badanego fragmentu DNA. Miejsce syntezy w cząsteczce cDNA wskazują startery – krótkie sekwencje
nukleotydowe komplementarne do początku i końca powielanego odcinka cDNA. Proces syntezy katalizowany jest przez termostabilny
enzym – polimerazę DNA (najczęściej stosowana jest polimeraza Taq wyizolowana z termofilnych bakterii Thermus aquaticus).
Wykonanie: Do probówki o poj. 0.2 ml dodać 2
l (300 ng) cDNA. Przygotować mieszaninę odczynników do reakcji PCR – jedną wspólną
dla wszystkich próbek dla danego genu. W skład mieszaniny reakcyjnej wchodzi: bufor (10-krotnie stężony), Q-solution, roztwór czterech
deoksyrybonukleozydotrifosforanów (dNTP), MgCl
2
(jony magnezu są kofaktorami polimerazy Taq), para starterów (R – ang. reverse
i F – ang. forward), woda wolna od RNaz oraz polimeraza Taq. Zamieszczona poniżej tabela 4 zawiera skład mieszaniny na jedną próbkę.
Tab.4. Skład mieszaniny reakcyjnej do reakcji PCR
składniki mieszaniny
objętość [
l]
1.
bufor
10x stężony
2.5
2.
Q-solution
5
3.
10 mM dNTP
1.6
4.
MgCl
2
1
5.
20
M starterów R
1
6.
20
M starterów F
1
7.
woda wolna od RNaz
7.65
8.
polimeraza Taq
0.25
objętość całkowita
20
Równocześnie z próbką badaną przygotować kontrolę negatywną (mieszanina reakcyjna z wodą wolną od RNaz w miejsce cDNA).
Polimerazę Taq dodać na końcu, tuż przed przeniesieniem mieszaniny do probówki zawierającej matrycę. Po krótkim zwirowaniu probówki
umieścić w termocyklerze PCR i prowadzić reakcję zgodnie ze schematem podanym w tabeli 5.
Tab.5. Etapy reakcji PCR.
nazwa cyklu
temperatura
czas [min]
1.
wstępna denaturacja
94ºC
3
2.
denaturacja
94ºC
1
3.
hybrydyzacja
temp. zależy od starterów
1
4.
wydłużanie
72ºC
2
30 - 35 cykli (pkty: 2 – 4)
5.
końcowe wydłużanie
72ºC
10
6.
koniec reakcji
4ºC
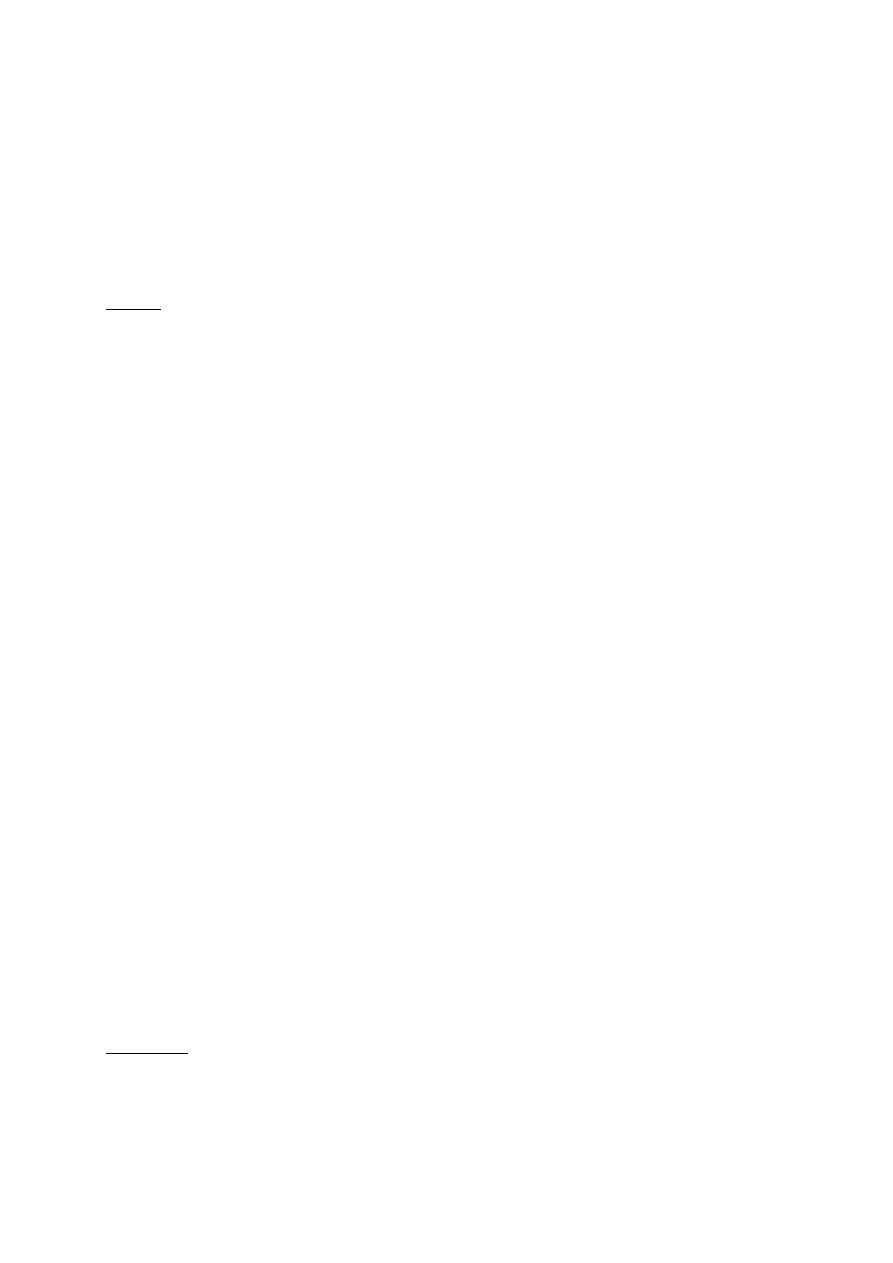
Biochemia: Ćw. – Metoda RT-PCR
5
4.
E
LEKTROFOREZA AGAROZO
WA PRODUKTÓW REAKCJI
PCR
Produkt reakcji PCR identyfikujemy rozdzielając uzyskany materiał w żelu agarozowym metodą elektroforezy. W celu określenia wielkości
badanego genu równocześnie rozdzielmy w żelu standardy masowe – fragmenty DNA o znanej wielkości wyrażonej w ilości par zasad (pz,
ang. base pair, bp). Na podstawie położenia badanego genu w stosunku do standardów masowych określamy jego wielkość. Rozdzielone
elektroforetycznie produkty reakcji PCR wizualizujemy przy użyciu fluorochromu bromku etydyny (EtBr), który interkalując z dsDNA
(dwuniciowym kwasem deoksyrybonukleinowym, ang. double stranded deoxyribonucleic acid) wykazuje intensywną fluorescencję
w świetle UV.
Wykonanie
Przygotowanie 2% żelu agarozowego
Do kolby stożkowej o poj. 250 ml dodać 0.8 g agarozy oraz 40 ml buforu TAE (40mM Tris-kwas octowy, pH 8.3, 1mM EDTA). Dokładnie
rozpuszczać agarozę ogrzewając w mikrofalówce przez 3 – 5 min. Zamieszać zawartość kolby; jeśli agaroza nie uległa rozpuszczeniu
kolbę umieścić ponownie na kilka minut w mikrofalówce. Złożyć podstawę na żel z płytkami zamykającymi oraz grzebieniem do
formowania studzienek. Do roztworu agarozy schłodzonego pod wodą bieżącą do temp. 50 – 60ºC dodać 2.5 μl bromku etydyny (uwaga!
bromek etydyny ma działanie karcynogenne!). Dokładnie wymieszać a następnie wylać żel na podstawę i odczekać do zastygnięcia
(30 – 40 min, RT). Wyjąć płytki zamykające podstawę z żelem oraz grzebień. Umieścić żel w aparacie horyzontalnym (poziomym)
wypełnionym buforem TAE (ok. 300 ml).
Przygotowanie próbek
Do probówki typu eppendorf o poj. 0.5 ml lub na kawałkach parafilmu przygotować próbki do elektroforezy dodając:
5 μl cDNA (amplifikatu uzyskanego w reakcji PCR),
1 μl buforu do próbek, 6-krotnie stężonego (Loading Dye Solution, Fermentas).
Wymieszać i krótko zwirować.
Rozdział elektroforetyczny
Próbki nałożyć do studzienek (kieszonek) wykonanych w żelu grzebieniem. Podłączyć elektrody do zasilacza i prowadzić rozdział przez
30 min przy napięciu 100 V. Pod wpływem pola elektrycznego ujemnie naładowane kwasy nukleinowe przemieszczają się w żelu
agarozowym od katody do dodatnio naładowanej anody z prędkością zależną od ich wielkości.
Wizualizacja kwasów nukleinowych
Po zakończeniu elektroforezy żel przenieść na folię. Wynik rozdziału elektroforetycznego
analizować w transiluminatorze Uvitec w świetle
UV.
Zalecana literatura:
"Biochemia" J.M. Berg, J.L.Tymoczko, L. Stryer, PWN; W-wa 2005.
"Biochemia Harpera" R.J. Murray, D.K. Granner, V.W. Rodwell, PZWL, W-wa 2008
„Biologia molekularna. Krótkie wykłady" PC Hames, AG McLennan, AD Bates, MRH White, PWN, W-wa 2007
„Biochemia. Krótkie wykłady" BD Hames, NM Hooper, PWN, W-wa 2005
Wyszukiwarka
Podobne podstrony:
Metoda RT-PCR, 3. rok, genetyka kliniczna
Metoda RT PCR
W 5 Izolacja RNA, RT PCR, hybrydyzacja 1
cw 12 pcr
BMiGO Wykład 5 Izolacja RNA, hybrydyzacja, RT PCR
ODWROTNA TRANSKRYPCJA RT PCR
genetyka, ćw 6 geny, 6 Techniki oparte na PCR do diagnozowania chorów genetycznych i uchwycenia zmie
PCR RAPD Genetyka molekularna ćw koło 3
Ćw 4 Elektroforeza wyników PCR
Ćw 3 Identyfikacja płci u ptakow metoda PCR
Ćw 3 Identyfikacja płci u ptakow metoda PCR
ćw 5 PCR
Wyniki PCR ćw. gr. I 2012, Studia, I semestr III rok, Biologia molekularna
ZIA Ćw 12 Badanie przekaźnika czasowego RT 431 doc
ćw 4 Profil podłużny cieku
biofiza cw 31
Kinezyterapia ćw synergistyczne
więcej podobnych podstron