
4. Wprowadzenie do enzymologii
4.1. Koncepcja biokatalizy
W ˝ywym organizmie zachodzà tysiàce ró˝nych reakcji chemicznych. Wi´kszoÊç z nich wy-
maga dostarczenia z zewnàtrz energii, w przeciwnym razie nawet si´ nie rozpocznà (mówimy,
˝e nie zostanà zainicjowane). W temperaturach, w których mo˝e funkcjonowaç ˝ywy orga-
nizm, tempo tych reakcji jest tak ma∏e, ˝e a˝ niezauwa˝alne. Wynika to z tego, ˝e energia we-
wn´trzna uk∏adu, który ma reagowaç, jest zbyt niska. Dlatego atomy i (lub) czàsteczki
zderzajà si´ ze sobà zbyt rzadko i niezbyt mocno. Nie wystarcza to do pokonania bariery pro-
gu energetycznego reakcji. Mo˝na powiedzieç (w ogromnym uproszczeniu), ˝e substraty sà
zbyt leniwe i powolne, aby reagowaç.
Tak wi´c, aby znaczàco przyspieszyç pr´dkoÊç reakcji, mo˝na:
1. Dostarczyç energii do uk∏adu przez podniesienie
temperatury – niewàtpliwie osiàgniemy sukces, bo-
wiem szybkoÊç reakcji zwi´kszy si´ w miar´ wzrostu
temperatury. B´dzie to jednak pyrrusowe zwyci´-
stwo – coÊ w rodzaju: „operacja si´ uda∏a, tylko pa-
cjent zmar∏”. Wynika to z tego, ˝e bia∏ka ustrojowe
denaturujà ju˝ w temperaturze ok. 45°C (por.
ROZDZ. 2.2). Tymczasem organizm zginie, zanim
osiàgnie po˝àdane tempo metabolizmu (por. te˝ ryc. 16). Nale˝y tutaj dodaç, ˝e energia
cieplna zaliczana jest do form nisko u˝ytecznych biologicznie – trudno jà magazynowaç,
przerabiaç i przesy∏aç. Ciep∏o jest wi´c z∏em koniecznym, a jego dostarczanie musi byç roz-
sàdnie ograniczane (por. PODR. KL. II).
2. Dostarczyç innej formy energii, na przyk∏ad Êwietl-
nej, o wysokiej u˝ytecznoÊci biologicznej. Niestety,
iloÊç Êwiat∏a, jaka by∏aby tu niezb´dna, to luksus
rzadko spotykany na naszej planecie (pomijam ju˝
problem, co np. robiç nocà?).
3. Zrezygnowaç z „pomys∏u” dostarczania du˝ych por-
cji energii i zadowoliç si´ ma∏ymi. W ten sposób po-
zostaniemy w zakresie temperatur dozwolonych
– tolerowalnych przez organizm. Rozwiàzania wymaga jedynie drobiazg – mniej wi´cej mi-
lion razy za wolne tempo reakcji biochemicznych w stosunku do potrzeb. Za∏amywaç ràk
nie trzeba, nale˝y obejÊç to ograniczenie przez zastosowanie enzymów (dawniej fermen-
tów). Nazwa tych zwiàzków pochodzi z greckiego enzyme, co oznacza czynnik obecny
w dro˝d˝ach, i zosta∏a wprowadzona przez Niemca Kuhnego, który bada∏ fermentacj´ al-
koholowà.
Enzymy sà katalizatorami, gdy˝ majà w∏aÊciwoÊç zwi´kszania szybkoÊci reakcji chemicz-
nych, same jednak nie ulegajà przemianom. Mówimy wi´c, ˝e enzymy nie zu˝ywajà si´
w przeprowadzanych przez siebie reakcjach (por. jednak ni˝ej).
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
31
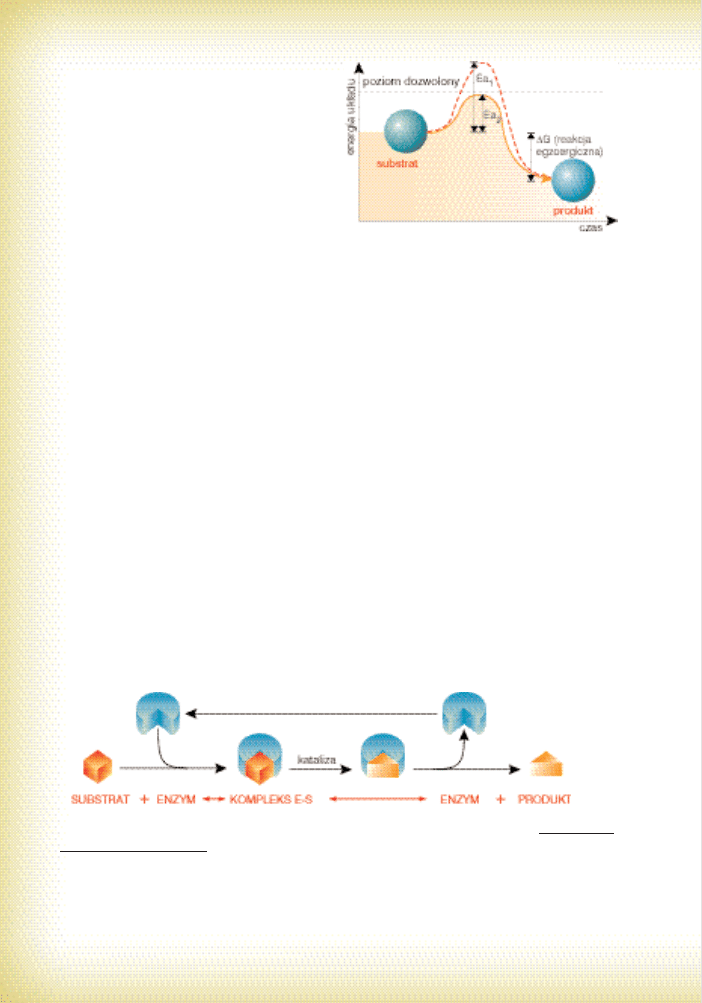
Ryc. 16.
Ogólne zasady energetyczne reakcji niekatali-
z o w a n y c h i k a t a l i z o w a n y c h u s t r o j u
(Ea
1
– energia aktywacji dla reakcji niekatali-
zowanej, Ea
2
– energia aktywacji dla reakcji
przyspieszanej przez enzym,
∆G – zmiana
energii swobodnej reakcji). Zwróç uwag´ na
poziom energetyczny substratu i produktu.
Uwaga:
Oprócz enzymów do biokatalizatorów zalicza si´ (skàdinàd nies∏usznie) hormony
i witaminy! W tej ksià˝ce poj´cie biokatalizatora b´dzie si´ jednak odnosi∏o tylko do
enzymów (w przeciwnym razie zostanie to wyraênie zaznaczone).
4.2. Budowa i dzia∏anie enzymów
WSZYSTKIE POZNANE DOTYCHCZAS ENZYMY SÑ BIA¸KAMI
Od czasu odkrycia i wyodr´bnienia w postaci krystalicznej pierwszego enzymu – ureazy
(dokona∏ tego Sumner w 1926 r.) wyizolowano, oczyszczono i zanalizowano setki ró˝nych bio-
katalizatorów. Zdecydowana wi´kszoÊç z nich nale˝y do bia∏ek z∏o˝onych. Jedynie cz´Êç hy-
drolaz i nieliczne izomerazy sà bia∏kami prostymi (por. ni˝ej). W zasadzie wi´c ca∏a
czàsteczka biokatalizatora, tak zwany holoenzym (gr. holo oznacza ca∏y), sk∏ada si´ z:
a) cz´Êci bia∏kowej – apoenzymu;
b) cz´Êci niebia∏kowej – grupy prostetycznej, przy∏àczonej do apoenzymu. JeÊli cz´Êç
niebia∏kowa po∏àczona jest nietrwale z apoenzymem, wówczas mówi si´ o koenzy-
mie. W celu unikni´cia zamieszania proponuj´ u˝ywaç nazwy kofaktor dla cz´Êci nie-
bia∏kowej (jest to jednak zabieg nieformalny!).
Nale˝y zadaç sobie trud znalezienia odpowiedzi na pytanie: dlaczego do przeprowadze-
nia reakcji enzym potrzebuje tylko niewielkiej porcji energii?
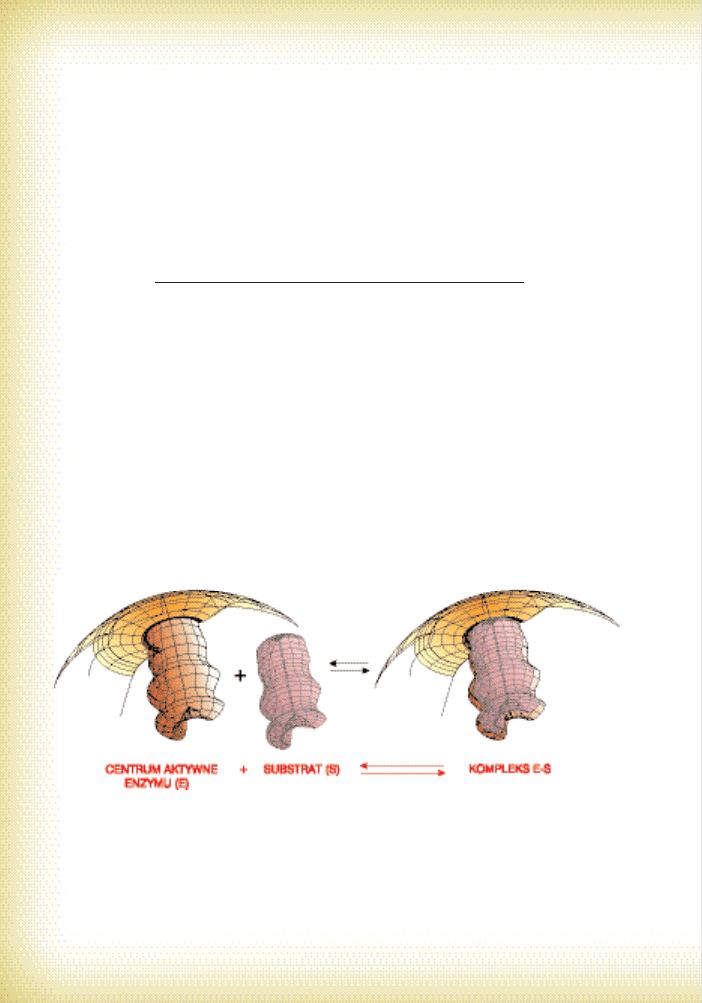
Ca∏e rozwiàzanie polega na tworzeniu przez enzym (E) przejÊciowego po∏àczenia z sub-
stratem (S) albo z substratami (zale˝y to od enzymu i rodzaju reakcji – znajdê kilka przyk∏a-
dów!). To po∏àczenie nazywa si´ kompleksem enzym-substrat (E-S). Ogólne równanie
reakcji katalizowanej przez enzym mo˝na zapisaç nast´pujàco:
W momencie wytworzenia kompleksu E-S w samym substracie dochodzi do przesuwania
okreÊlonych elektronów. Skutkiem jest powstawanie nowych wiàzaƒ lub rozrywanie ju˝ ist-
niejàcych. ÂciÊle mówiàc, obecnoÊç enzymu daje nast´pujàce korzyÊci:
1. Zwi´ksza si´ prawdopodobieƒstwo zderzeƒ pomi´dzy reagujàcymi czàsteczkami. To tak,
jak gdyby ktoÊ zbli˝y∏ do siebie zagubione osoby – bez tej interwencji szansa na spotkanie
by∏aby niewielka.
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
32

2. Substraty zostajà prawid∏owo zorientowane w przestrzeni trójwymiarowej – czàsteczki
w roztworze bez enzymu zderzajà si´ bez∏adnie, najcz´Êciej nie tymi „cz´Êciami” co trzeba.
Biokatalizator spe∏nia wi´c funkcj´ kogoÊ, kto ustawia je wzgl´dem siebie tak, ˝e reagujà-
ce wiàzania znajdà si´ w bezpoÊrednim sàsiedztwie.
3. Dochodzi do napr´˝ania wiàzaƒ w substracie – w momencie wpasowywania substratu do
enzymu (tworzenia kompleksu E-S) nast´puje nadwer´˝anie wiàzaƒ w samym substracie.
Takie naruszone wiàzanie doÊç ∏atwo mo˝na teraz zmieniç (por. tak˝e ni˝ej).
Nie oznacza to jednak, ˝e enzym mo˝e zrobiç z reakcjà, co chce. Przede wszystkim jego
obecnoÊç nie przesuwa stanu równowagi katalizowanej przemiany. Wyobraê sobie odwracal-
nà reakcj´, w której równowaga ustala si´ na nast´pujàcym poziomie: substratu jest 200 razy
wi´cej ni˝ produktu. I nie jest wa˝ne, czy enzym b´dzie obecny czy nie – wartoÊç ta nie ule-
gnie zmianie. KorzyÊci zastosowania biokatalizatora sà nast´pujàce:
a) znacznie szybsze osiàgni´cie stanu równowagi (w przybli˝eniu przyspieszenie reakcji –
por. ni˝ej);
b) zmniejszenie energii aktywacji – tej minimalnej porcji energii, która podniesie poziom
energetyczny substratu do progu umo˝liwiajàcego zainicjowanie i przeprowadzenie da-
nej reakcji (por. ryc. 16).
Istnieje tak˝e inne powa˝ne ograniczenie pracy biokatalizatorów. Jest nim problem
energetyczny – w warunkach ustrojowych enzymy mogà przyspieszaç jedynie reakcje egzo-
ergiczne (por. ryc. 16). Czy˝byÊmy mieli wi´c „biochemiczny pat” w wypadku reakcji endo-
ergicznych? Rozwiàzanie jest proste: trzeba w sensownym zakresie podnieÊç poziom
energetyczny substratu, ˝eby reakcja sta∏a si´ egzoergiczna. Jak „do∏adowaç” taki uk∏ad, wy-
jaÊni´ dok∏adniej w ROZDZ. 4.4. Tutaj musisz zadowoliç si´ stwierdzeniem, ˝e istniejà wy-
specjalizowane czàsteczki powstajàce w reakcjach silnie egzoergicznych, gromadzàce
w sobie energi´, którà potem mogà oddawaç na wspomniane do∏adowanie.
KA˚DY ENZYM MA CENTRUM AKTYWNE
Du˝a skàdinàd czàsteczka enzymu ma zawsze na swojej powierzchni ma∏e zag∏´bienie,
które nazwano centrum aktywnym (miejscem aktywnym; por. ryc. 17 i 18). Miejsce aktywne
jest przestrzennym wg∏´bieniem w kszta∏cie rowka lub do∏ka, zawierajàcym odpowiednie
aminokwasy. Ten „do∏ek” jest najwa˝niejszà cz´-
Êcià ca∏ej makroczàsteczki biokatalizatora.
Wchodzi tu bowiem substrat, tutaj tak˝e przy∏à-
cza si´ grupa prostetyczna (o ile wyst´puje).
¸aƒcuchy boczne (R) aminokwasów umo˝liwia-
jà rozpoznawanie, wpasowywanie i reagowanie
konkretnego substratu.
Ryc. 17. Prosty model budowy enzymu
Dlatego cz´sto nazywa si´ je grupami katalitycznymi enzymu. Liczba tych grup jest ró˝-
na (zale˝y od rodzaju enzymu), ale zawsze jest ich kilka do kilkunastu. Rodzaj i rozmieszcze-
nie przestrzenne aminokwasów w centrum aktywnym decyduje o w∏aÊciwoÊciach
konkretnego enzymu (por. ni˝ej klasyfikacja enzymów). Zwykle miejsca te zawierajà zarów-
no kilka niepolarnych, jak i polarnych reszt aminokwasowych, co zapewnia odpowiednie mi-
kroÊrodowisko.
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
33
ENZYMY SÑ SPECYFICZNE WZGL¢DEM SUBSTRATÓW
Budowanie bia∏kowego biokatalizatora kosztuje du˝o energii i materii, a jego trwa∏oÊç
jest ograniczona (por. ni˝ej). Dlatego nale˝y wykorzystaç do maksimum jego mo˝liwoÊci.
Przede wszystkim za∏ó˝my, ˝e komórka musi oszcz´dzaç energi´ i surowce. Oznacza to
mi´dzy innymi, ˝e nie staç jej na przeprowadzanie niepotrzebnych reakcji. Rozwiàzaniem jest
„specjalizacja” enzymów, która prowadzi do tego, ˝e katalizowane sà tylko reakcje po˝àdane.
Innà korzyÊcià jest wysoka sprawnoÊç takich wàskich specjalistów. W porównaniu z kataliza-
torami nieorganicznymi osiàgane pr´dkoÊci sà o kilka rz´dów wielkoÊci wi´ksze. Wymaga to
od enzymów zdolnoÊci do bardzo dok∏adnego rozpoznawania substratów, czyli specyficzno-
Êci substratowej.
Zasadniczo dany rodzaj enzymu przeprowadza tylko jeden rodzaj reakcji. Jest to pewne
rozwini´cie znanej Ci zasady: jeden enzym – jedna reakcja. Nie oznacza to wcale, ˝e enzym
przeprowadza jednà reakcj´ i ulega zniszczeniu. Czàsteczka biokatalizatora nie zu˝ywa si´
bowiem w pojedynczej przemianie – mo˝e przeprowadziç miliony takich operacji (mo˝na to
z grubsza porównaç do popularnego magnetowidu, który odtwarza bezawaryjnie wiele kaset).
Dlatego czàsteczka E widoczna po prawej stronie równania ogólnego (por. ryc.18) mo˝e po-
nownie przeprowadziç t´ reakcj´ z kolejnà czàsteczkà substratu. OczywiÊcie ˝ywotnoÊç ka˝-
dej struktury ma swoje granice – dlatego po jakimÊ czasie czàsteczki enzymu ulegajà
zestarzeniu (zu˝yciu) i ich liczba b´dzie musia∏a byç uzupe∏niona (z magnetowidem b´dzie
podobnie).
Enzymy ró˝nià si´ od siebie specyficznoÊcià, co jest zjawiskiem po˝àdanym, ale tak˝e nie-
bezpiecznym (por. ni˝ej inhibicja). Dla przyk∏adu
α-amylazy naszego przewodu pokarmowe-
go rozk∏adajà wiàzania typu
α-glikozydowego i nie ma tu wi´kszego znaczenia, czy
substratem jest skrobia, glikogen czy dekstryna.
Uwaga:
Nie oznacza to jeszcze, ˝e amylazy dzia∏ajà „na Êlepo” – por. CZ¢Âå: ANATOMIA I …,
ROZDZ. 5.
Ryc. 18. Model ilustrujàcy przestrzenne dopasowanie substratu i enzymu wed∏ug modelu zamka
i klucza
Podobnie lipaza trzustkowa rozk∏ada wiàzania estrowe w ró˝nych t∏uszczach. Inne enzy-
my wykazujà znacznie wi´kszà dok∏adnoÊç w rozpoznawaniu substratu, na przyk∏ad anhydra-
za w´glanowa katalizuje tylko reakcje pomi´dzy dwutlenkiem w´gla i wodà. Inny
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
34

biokatalizator – polimeraza DNA I (przeprowadzajàca reakcje kopiowania DNA) w∏àcza do
syntetyzowanej nici nowe nukleotydy z dok∏adnoÊcià jednego b∏´du na 10 000 000 przepro-
wadzonych operacji.
SpecyficznoÊç enzymów dobrze oddaje model zamka
i klucza, sformu∏owana przez pana Fishera ju˝ w 1890 r.
Mówi ona, ˝e substrat pasuje do centrum aktywnego
enzymu tak jak klucz do zamka (por. ryc. 18).
Po prostu – odpowiednie ukszta∏towanie przestrzenne substratu znajduje odbicie w kon-
formacji miejsca aktywnego enzymu. Pozwala to na wejÊcie grup katalitycznych enzymu
w nietrwa∏e po∏àczenia z substratem i przeprowadzenie reakcji. Model Fishera jest prosty
i przekonywajàcy, nie wyjaÊnia jednak wszystkich aspektów katalizy enzymatycznej w ˝ywych
uk∏adach. Modelowanie matematyczne udowodni∏o bowiem, ˝e samo dopasowanie S do
E daje tylko dwie pierwsze z wymienionych wczeÊniej korzyÊci (przypomnij je sobie!). Nie po-
zwoli∏yby one na tak znaczne obni˝enie energii aktywacji. WyjaÊnienie tego problemu przed-
stawi∏ pan Koshland w swojej teorii indukcyjnego dopasowania (wymuszonego
dopasowania). W tym uj´ciu substrat niezbyt dok∏adnie pasuje przestrzennie do centrum ak-
tywnego. Inaczej mówiàc, konformacja obu sk∏adowych kompleksu E-S nie jest identyczna.
Otó˝ wchodzàc w s∏abe oddzia∏ywania z enzymem, S jest przez niego „wciàgany” do zag∏´bie-
nia centrum. W czasie dopasowywania nast´puje pewne odkszta∏cenie centrum i substratu.
Mo˝na wi´c oczekiwaç niewielkiego napr´˝enia (mniej wi´cej naderwania) wiàzaƒ w obu
komponentach. W tej sytuacji ju˝ niewielka porcja energii aktywacji wystarcza do pokonania
progu energetycznego reakcji. To, ˝e dochodzi do zmiany wiàzaƒ tylko w substracie, t∏umaczy
si´ czasem wielkoÊcià czàsteczki enzymu – przewaga masy daje mu wi´kszà stabilnoÊç i mniej-
szà podatnoÊç na odkszta∏cenia. Mamy wi´c tutaj wszystkie trzy przedstawione korzyÊci.
Czasem dla obrazowego wyjaÊnienia teorii Koshlanda mówi si´, ˝e substrat pasuje do
miejsca aktywnego jak r´ka do r´kawiczki (przemyÊl to dok∏adnie).
KINETYK¢ REAKCJI ENZYMATYCZNEJ OBRAZUJE MODEL MICHAELIS-MENTEN
Ju˝ w 1913 roku panie Michaelis i Menten przedstawili bardzo prosty model charaktery-
zujàcy dynamik´ przebiegu katalizowanych reakcji. Za∏o˝eniem by∏o oczywiÊcie, ˝e zawsze
tworzy si´ kompleks E-S jako poÊredni etap ca∏ego procesu. W swojej podstawowej wersji
tzw. równanie Michaelis-Menten przyjmuje nast´pujàcà postaç (po uproszczeniu!):
gdzie V – pr´dkoÊç katalizowanej reakcji, V
max
– pr´dkoÊç maksymalna jakà mo˝na by∏o-
by teoretycznie osiàgnàç w warunkach optymalnych, [S] – st´˝enie substratu, K
M
– sta∏a Mi-
chaelis. Ta ostatnia jest równa takiej wartoÊci st´˝enia substratu, przy którym pr´dkoÊç reakcji
jest równa po∏owie pr´dkoÊci maksymalnej (por. ni˝ej).
Nie wdajàc si´ w zawi∏e wyjaÊnienia, spójrzmy na to równanie kraƒcowo:
1. JeÊli st´˝enie substratu jest bardzo du˝e, to wówczas mo˝emy w u∏amku pominàç K
M
(sta-
∏a ta ma niewielkà wartoÊç rz´du od 10
–1
do 10
–7
mola/litr). Równanie uproÊci si´ i przyjmie
praktycznà postaç:
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
35
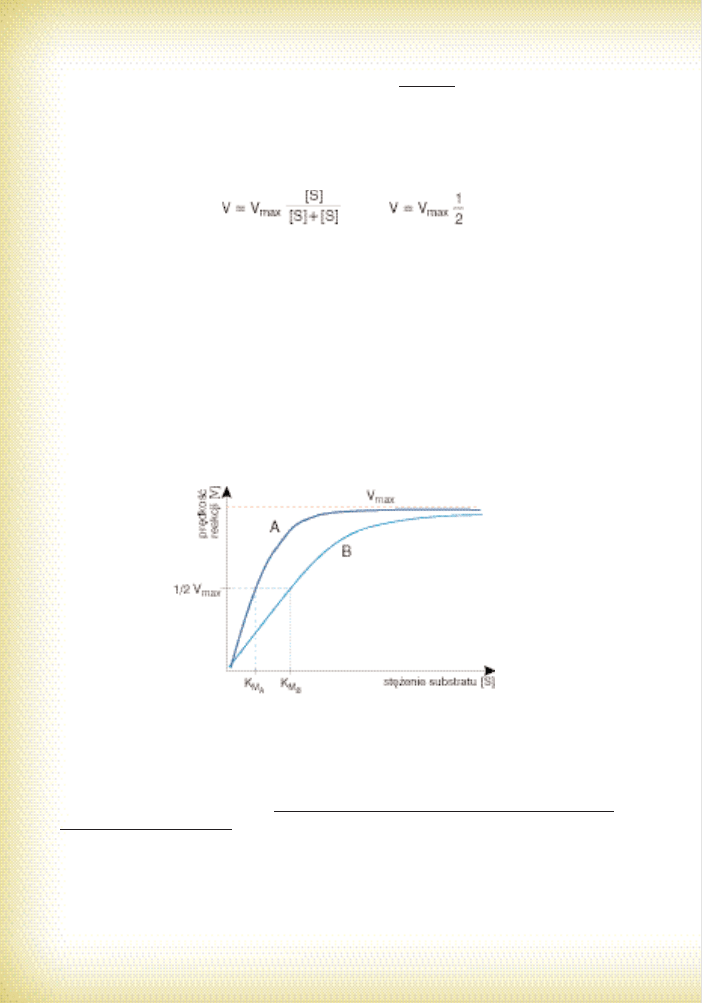
Logiczne wi´c jest, ˝e przy du˝ym st´˝eniu substratu wszystkie czàsteczki biokatalizatora
b´dà „pracowaç”, a wi´c pr´dkoÊç reakcji b´dzie maksymalna dla danego enzymu. ÂciÊle
mówiàc, b´dzie prawie maksymalna (por. ryc. 19; porozmawiaj te˝ ze swoim chemikiem,
szczególnie, jeÊli tego nie rozumiesz);
2. JeÊli niewielkie st´˝enie substratu (wynoszàce S) b´dzie takie jak wartoÊç sta∏ej K
M
, to
wówczas równanie przyjmie postaç:
Przy takim st´˝eniu substratu, które jest równe K
M
pr´dkoÊç reakcji osiàgnie wi´c po∏ow´
pr´dkoÊci maksymalnej. Zastanawiajàc si´ dalej, dojdziesz do wniosku, ˝e sta∏a Michaelis
dobrze odzwierciedla aktywnoÊç enzymu i (lub) jego powinowactwo z substratem. Jest to
bowiem wygodny sposób rozró˝niania enzymów o odmiennej aktywnoÊci (zale˝noÊç
V
max
od st´˝enia substratu jest ma∏o precyzyjna). Przyjrzyjmy si´ teraz hiperbolicznej krzy-
wej Michaelis przedstawionej na ryc. 19. Przy ma∏ych st´˝eniach substratu, gdy [S] jest
mniejsze od K
M
szybkoÊç reakcji jest wprost proporcjonalna do st´˝enia substratu. Na tym
odcinku krzywa jest stromotorowa – dodanie nawet niewielkiej iloÊci substratu wyraênie
przyspieszy reakcj´. Mo˝na powiedzieç, ˝e w tych warunkach enzym dysponuje du˝à nad-
wy˝kà „mocy przerobowej”. Przy du˝ych st´˝eniach substratu, gdy [S] jest wyraênie wi´k-
sze od K
M
, pr´dkoÊç jest zbli˝ona do V
max
i nie ulega zmianie. Na tym odcinku krzywa jest
p∏askotorowa – i nawet doÊç znaczne odchylenia [S] nie zmieniajà szybkoÊci (dlatego ta za-
le˝noÊç jest ma∏o precyzyjna).
Ryc. 19. Krzywa Michaelis ilustrujàca zale˝noÊç szybkoÊci reakcji enzymatycznej od st´˝enia sub-
stratu (A – w wypadku enzymu o du˝ej aktywnoÊci zwraca uwag´ ma∏a wartoÊç K
MA
,
B – dla enzymu ma∏o aktywnego wartoÊç K
MB
jest znacznie wi´ksza).
O AKTYWNOÂCI ENZYMU ÂWIADCZY TAK˚E LICZBA OBROTÓW
Poj´cie to oznacza praktycznie liczb´ czàsteczek substratu, która ulega przekszta∏ceniu
w produkt w jednostce czasu (w warunkach pe∏nego wysycenia enzymu). Liczba obrotów dla
jednego z najszybszych enzymów ustrojowych – anhydrazy w´glanowej wynosi ok. 600 000 s
–1
(600 000 czàsteczek „przerobionych” w ciàgu jednej sekundy). W wypadku innych biokatali-
zatorów wartoÊç ta jest najcz´Êciej znacznie mniejsza i wynosi na przyk∏ad dla dehydrogena-
zy mleczanowej 26 000 s
–1
, a dla syntetazy tryptofanowej ju˝ tylko 2 s
–1
. Jak widaç, sà one
bardzo ró˝ne, ale w zupe∏noÊci wystarczajà na nasze potrzeby (zale˝y to od czynników, któ-
rych tutaj poruszaç nie b´dziemy).
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
36

NA AKTYWNOÂå ENZYMÓW MA WP¸YW SZEREG CZYNNIKÓW USTROJOWYCH
JeÊli wszystkie znane enzymy ustrojowe sà bia∏kami, to nale˝y oczekiwaç, ˝e b´dà wykazy-
wa∏y wszystkie cechy tych makroczàsteczek (por. ROZDZ. 2). Oznacza to wi´c „delikatnoÊç”
i podatnoÊç ich aktywnoÊci na wp∏yw czynników zewn´trznych. WÊród najistotniejszych para-
metrów Êrodowiskowych nale˝y znaç:
1. Wp∏yw temperatury – dla wi´kszoÊci enzymów do ok. 37–40°C nast´puje wzrost szybkoÊci
reakcji. Jest to zgodne z regu∏à van Hoffa (Twój chemik!). Powy˝ej tej temperatury pr´d-
koÊç gwa∏townie spada – spowodowane to jest denaturacjà bia∏ka (sporzàdê sobie odpo-
wiedni wykres!). Wi´kszoÊç naszych enzymów ustrojowych ma optimum termiczne w∏aÊnie
w okolicach 38°C, co jest zgodne z hipotezà maksitermii (por. CZ¢Âå: ANATOMIA I …,
ROZDZ. 9). Dla organizmów zmiennocieplnych optimum termiczne przypada w ni˝szym
zakresie. Wyjàtkowo tylko u goràcolubnych bakterii ˝yjàcych w przegrzanych êród∏ach
stwierdzono obecnoÊç enzymów pracujàcych w temperaturze ok. 80°C.
2. Wp∏yw pH – w tym wypadku enzymy wykazujà powa˝ne ró˝nice. Zdecydowana wi´kszoÊç
komórkowych ma optimum w okolicach oboj´tnego – lekko kwaÊnego (pH = 6,4–6,6).
Enzymy trawienne dzia∏ajàce w przewodzie pokarmowym majà optimum pH zale˝ne od
ich budowy i miejsca dzia∏ania. I tak na przyk∏ad pepsyna najlepiej dzia∏a przy pH = 1,5–2,
amylaza Êlinowa przy pH = 7, a trypsyna przy pH = 8,7 (sporzàdê odpowiedni wykres).
Uwaga:
Omawiane zale˝noÊci sà cz´sto wykorzystywane przy tworzeniu pytaƒ egzaminacyjnych!
Posiadanie enzymów pozwala komórce na regulowanie w∏asnego metabolizmu (por.
ROZDZ. 3.1). Mo˝e si´ to odbywaç na kilku poziomach organizacji, a przejawem tego sà:
1. Sterowanie transkrypcjà (przepisywaniem informacji genetycznej z DNA na mRNA). Wy-
starczy teraz wyobraziç sobie, ˝e mo˝na biochemicznie zablokowaç lub odblokowaç odci-
nek DNA zawierajàcy informacje o budowie enzymu – w ten sposób albo zostanie on
zsyntetyzowany, albo nie. Wiesz zaÊ, co to oznacza dla konkretnej reakcji (por. te˝
CZ¢Âå: GENETYKA).
2. Obróbka posttranskrypcyjna i kontrolowanie translacji – powsta∏y transkrypt mo˝e byç
u˝yty lub „od∏o˝ony na potem” (por. tigroidy w neuronach).
3. Regulowanie aktywnoÊci ju˝ zsyntetyzowanego enzymu. Jest to najszybszy i zwykle najlep-
szy sposób sterowania procesami metabolicznymi. W zale˝noÊci od rodzaju enzymu i miej-
sca u˝ycia istnieje tu kilka mo˝liwoÊci:
A) Posiadanie enzymów rozk∏adajàcych bia∏ka jest dla komórki korzystne, ale i niebez-
pieczne. Dlatego syntetyzowane sà w formie nieczynnych proenzymów (czasem zymo-
genów). Formy te sà uaktywniane dopiero w miejscu gwarantujàcym zabezpieczenie
przed samostrawieniem (tzn. w przewodzie pokarmowym – albo w skali komórki euka-
riotycznej – w wakuoli trawiennej). Uczynnienie takiego biokatalizatora cz´sto polega
na odci´ciu fragmentu ∏aƒcucha polipeptydowego, który zas∏ania miejsce aktywne. Opi-
sany tutaj sposób aktywacji jest nieodwracalny.
B) W wypadku w pe∏ni sprawnego (gotowego) enzymu sytuacja mo˝e si´ przedstawiaç na-
st´pujàco:
a) Je˝eli pewien zwiàzek chemiczny jest na tyle podobny chemicznie i fizycznie do sub-
stratu, ˝e centrum aktywne enzymu ich nie odró˝nia to dochodzi do hamowania
kompetycyjnego (inhibicji kompetycyjnej, hamowania wspó∏zawodniczàcego; por.
ryc. 20A). Ten typ oddzia∏ywania polega, krótko mówiàc, na ubieganiu si´ o to samo
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
37
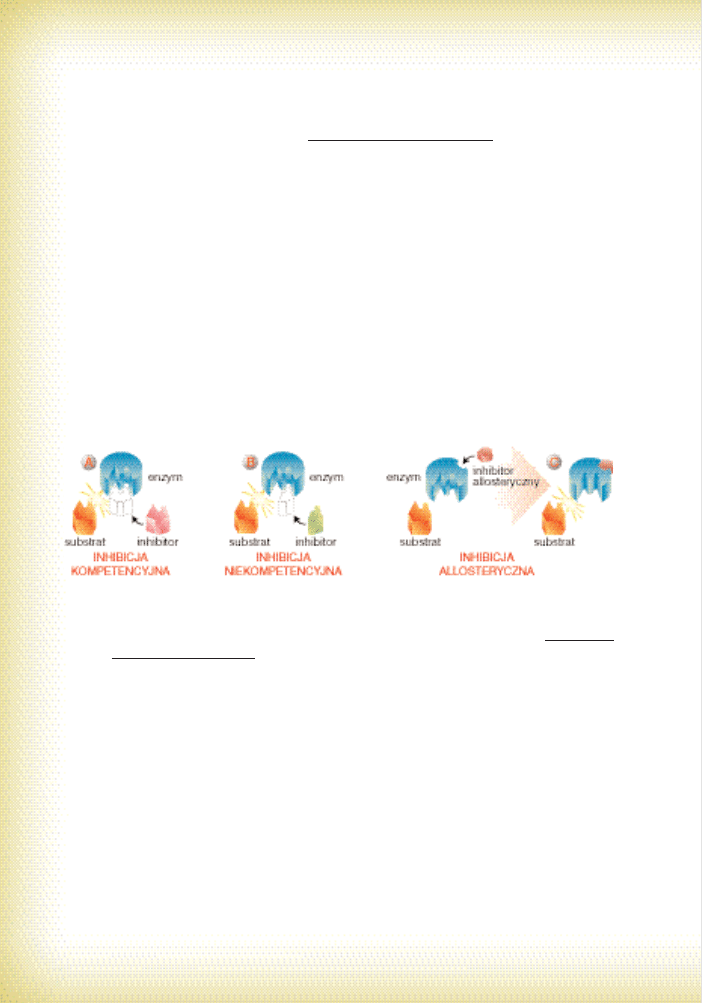
– dwa rodzaje czàsteczek (substrat i inhibitor) wspó∏zawodniczà o jedno centrum ak-
tywne. JeÊli st´˝enie takiego inhibitora zwi´kszy si´, to iloÊç czàsteczek enzymu, któ-
ra „obs∏uguje” substrat ulegnie zmniejszeniu – spadnie wi´c szybkoÊç katalizy. Takie
hamowanie mo˝na znosiç przez zwi´kszanie st´˝enia substratu (nastàpi z kolei wy-
pieranie inhibitora). Jak widaç, opisywany proces jest w pe∏ni odwracalny, wp∏ywa
tak˝e na wzrost K
M
(nie ma natomiast wp∏ywu na V
max
). Przyk∏adem klinicznym jest
leczenie ludzi zatrutych metanolem – alkohol ten jest w ustroju utleniany do niebez-
piecznego aldehydu mrówkowego przez dehydrogenaz´ alkoholowà. Enzym ten nie
odró˝nia metanolu od etanolu i dlatego ten ostatni mo˝e pe∏niç funkcj´ inhibitora
kompetycyjnego. Nie liczy∏bym jednak zbytnio na to, gdy˝ proces zatrucia post´puje
szybko. Lepiej wi´c nie piç napojów wysokoprocentowych niepewnego pochodzenia
(a najlepiej nie piç ich wcale).
b) JeÊli jakaÊ substancja niepodobna do substratu blokuje cz´Êciowo centrum aktywne,
to mamy do czynienia z hamowaniem niekompetycyjnym (por. ryc. 20B). Substrat
jest wi´c wiàzany, ale reakcja ulega zahamowaniu – skutkiem jest spadek liczby ob-
rotów i pr´dkoÊci maksymalnej reakcji (K
M
nie ulega zmianie). Sam proces hamowa-
nia jest odwracalny, ale nie mo˝na tutaj osiàgaç efektu jego znoszenia przez
zwi´kszanie st´˝enia substratu. Ten typ inhibicji nie ma wi´kszego znaczenia dla re-
gulacji metabolizmu w uk∏adach ˝ywych. Przyk∏adem sà zwiàzki miedzi i rt´ci bloku-
jàce grupy tiolowe aminokwasów katalitycznych.
Ryc. 20. Regulacja aktywnoÊci enzymów: hamowanie kompetencyjne (A), niekompetencyjne (B)
i inhibicja allosteryczna (C).
c) JeÊli pewna czàsteczka oddzia∏uje odwracalnie na aktywnoÊç enzymu, ale w innym miej-
scu ni˝ centrum aktywne, to mamy do czynienia z regulacjà allosterycznà (por. ryc. 20C).
Poj´cie to nie odnosi si´ wy∏àcznie od biokatalizatorów – oznacza bowiem zmian´ struk-
tury przestrzennej i aktywnoÊci danej makroczàsteczki pod wp∏ywem jakiejÊ substancji
(regulacji tego typu podlega np. hemoglobina – por. CZ¢Âå: ANATOMIA I …,
ROZDZ. 4.4). Enzymy, które podlegajà takiej modyfikacji wykazujà kilka cech:
– sà zbudowane z kilku podjednostek peptydowych;
– ich kinetyka odbiega od modelu Michaelis-Menten; przede wszystkim krzywa ich
aktywnoÊci ma kszta∏t sigmoidalny, a nie hiperboliczny; upraszczajàc – istniejàce
oddzia∏ywania pomi´dzy modu∏ami prowadzà do u∏atwiania wiàzania nast´pnych
czàsteczek substratu;
– majà oprócz centrum aktywnego jeszcze tak zwane centrum allosteryczne, które
mo˝e przy∏àczaç regulator allosteryczny.
Regulacja mo˝e polegaç na inhibicji albo indukcji – odpowiednio mówimy wów-
czas o dzia∏aniu inhibitora albo induktora allosterycznego. Tego typu oddzia∏ywa-
nia sà cz´sto wykorzystywane przez komórk´. Hamowanie allosteryczne wyst´puje
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
38
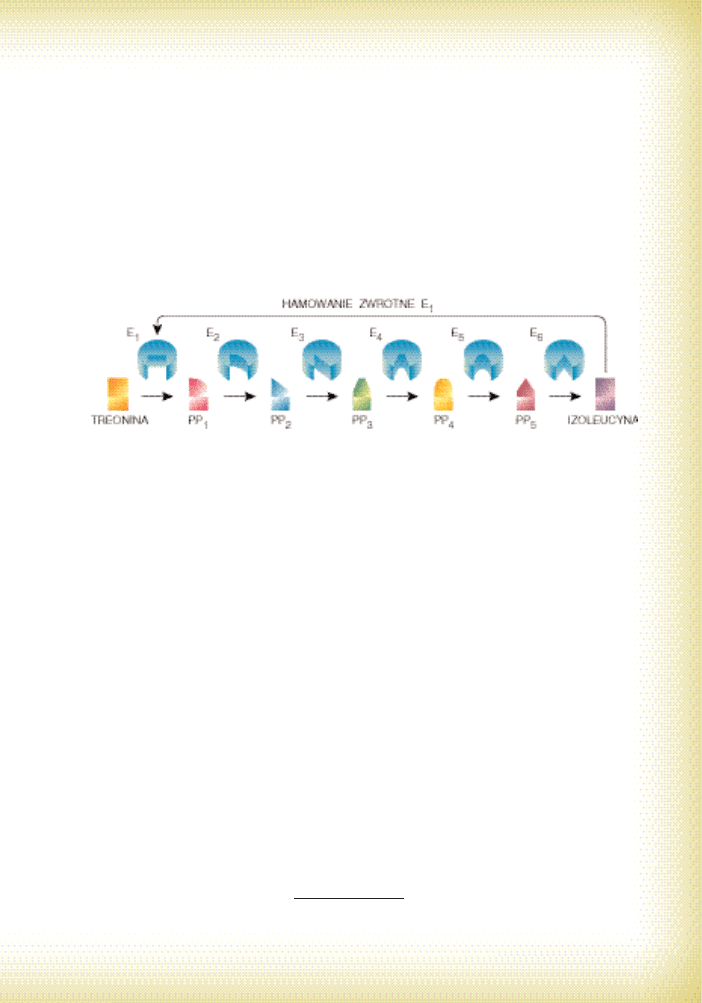
najcz´Êciej w d∏ugich szlakach metabolicznych. Jest to jednoczeÊnie wykorzystanie
mechanizmów sprz´˝eƒ zwrotnych ujemnych. Przyk∏adem niech b´dzie synteza izo-
leucyny (Ile) z treoniny, przebiegajàca w szeÊciu reakcjach (nie musisz ich znaç!).
Pierwsza reakcja jest katalizowana przez enzym dehydrataz´ treoninowà. Jednocze-
Ênie produkt koƒcowy szlaku – izoleucyna jest dla tego biokatalizatora inhibitorem
allosterycznym. W ten sposób komórka chroni si´ przed nadprodukcjà izoleucyny
– ona sama hamuje swojà syntez´. Ponadto blokowanie nast´puje ju˝ na etapie
pierwszej reakcji przez co oszcz´dza si´ koszty zwiàzane z niepotrzebnym wytwa-
rzaniem produktów poÊrednich (chytre?). JeÊli st´˝enie Ile spadnie (bo zu˝yto jà do
biosyntezy bia∏ka), szlak zostanie odblokowany (por. ryc. 21). Wynika to z tego, ˝e
kompleks enzym-inhibitor jest nietrwa∏y i rozpada si´ (jeÊli nie rozumiesz tego, do-
kszta∏ç si´ w zakresie równowag chemicznych).
Ryc. 21. Schemat sprz´˝enia zwrotnego ujemnego w kontroli szlaku syntezy izoleucyny (PP
1–5
– ko-
lejne produkty przejÊciowe, E
1–6
– kolejne enzymy szlaku). Widoczne jest hamujàce oddzia-
∏ywanie
Ile
na pierwszy enzym szlaku – dehydrataz´ treoninowà (E
1
)
ENZYMÓW JEST TAK DU˚O, ˚E MUSIANO JE PODZIELIå NA GRUPY
NiegdyÊ stosowano jedynie nazwy zwyczajowe enzymów. Od 1961 roku obowiàzuje nas
podzia∏ zaproponowany przez Komisj´ Enzymowà Mi´dzynarodowej Unii Biochemicznej.
Dzieli on biokatalizatory na szeÊç klas g∏ównych (i tylko ten poziom podzia∏u musisz znaç).
Kryterium jest tutaj rodzaj przeprowadzanej reakcji.
Uwaga: 1.
Dla wi´kszoÊci uczniów opanowywanie tego podzia∏u to niezas∏u˝ona katorga.
Tymczasem zaczàç trzeba od zastanowienia si´, co mo˝na zrobiç z dowolnym obiek-
tem chemicznym? Otó˝ mo˝na go: utleniç, przenieÊç jakiÊ jego fragment, roz∏o˝yç na
mniejsze cz´Êci, u˝ywajàc wody (lub bez jej udzia∏u), zmieniç wzajemne po∏o˝enie
elementów budulcowych (przebudowaç bez zmiany sk∏adników), wreszcie dwa
obiekty po∏àczyç ze sobà w jednà ca∏oÊç. Wszystko to znalaz∏o odbicie w przyj´tym
podziale.
2.
Nazwy enzymów zawierajà najcz´Êciej charakterystyczne koƒcówki. Przyj´∏o si´,
˝e aktywny enzym ma koƒcówk´ -aza, natomiast proenzym -gen.
Poni˝ej przedstawiono wszystkie klasy z przyk∏adami (jest ich te˝ mnóstwo w ca∏ej ksià˝ce!):
1. Oksydoreduktazy – przeprowadzajà reakcje typu redox. Mówiàc proÊciej, sà to enzymy ka-
talizujàce reakcje, w których dochodzi do zmiany stopnia utlenienia. Przyk∏adami sà: de-
hydrogenaza mleczanowa (uczestniczàca w wàtrobie w pozbywaniu si´ szkodliwego kwasu
mlekowego) i oksydaza L-aminokwasowa (bezpoÊrednio utleniajàca aminokwasy w mi-
krocia∏kach).
2. Transferazy – katalizujà reakcje przenoszenia grup funkcyjnych z jednej czàsteczki na dru-
gà. Trywializujàc – jednemu zabiera si´ jakàÊ istotnà cz´Êç, aby daç jà drugiemu. Przyk∏ada-
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
39

mi sà: transaminaza glutaminianowa (przenoszàca grup´ aminowà na ketoglutaran, przez
co powstaje mi´dzy innymi kwas glutaminowy; por. transaminacja) i syntaza laktozowa
(w gruczo∏ach mlecznych ssaków przenosi galaktoz´ na glukoz´, przez co powstaje laktoza).
3. Hydrolazy – zwykle sà to bia∏ka proste przeprowadzajàce reakcje rozpadu z udzia∏em wody. In-
nymi s∏owy: enzymy te rozk∏adajà wiàzania w czàsteczkach, u˝ywajàc do tego celu wody (mó-
wimy o hydrolizie). Przyk∏adami sà wszystkie enzymy trawienne przewodu pokarmowego.
Tutaj stwierdêmy tylko, ˝e zhydrolizowaç mo˝na wiàzania peptydowe, glikozydowe i estrowe.
4. Liazy – katalizujà reakcje rozpadu, tyle ˝e bez udzia∏u wody. Zwykle wówczas tworzà si´
wiàzania podwójne. Przyk∏adem jest dekarboksylaza pirogronianowa odpowiedzialna za
pozbawienie pirogronianu dwutlenku w´gla, w wyniku czego powstaje aldehyd octowy
(por. fermentacja alkoholowa).
5. Izomerazy – przeprowadzajà reakcje przegrupowaƒ wewnàtrzczàsteczkowych. Inaczej mó-
wiàc, przebudowujà one struktur´ czàsteczki bez zmiany jej sk∏adu atomowego. Przyk∏a-
dem jest izomeraza cytrynianowa, katalizujàca reakcj´ przekszta∏cania cytrynianu
w izocytrynian (por. cykl Krebsa).
6. Ligazy (syntetazy) – katalizujà tworzenie nowych wiàzaƒ. ÂciÊle mówiàc, odpowiedzialne
sà za wytwarzanie wiàzaƒ pomi´dzy dwoma substratami, a wi´c za reakcje syntezy. Pro-
blem dostarczenia niezb´dnej energii rozwiàzujà one przez sprz´˝enie syntezy z reakcjà
silnie egzoergicznà (tu: hydrolizy wiàzania wysokoenergetycznego, najcz´Êciej ATP).
Uwaga:
W zapami´taniu powy˝szych nazw mo˝e pomóc Ci skrót utworzony z ich pierwszych
liter – OTHLIL.
4.3. Kofaktory enzymatyczne
Jak zapewne pami´tasz, wi´kszoÊç enzymów zbudowanych jest z bia∏kowego apoenzymu
i niebia∏kowego koenzymu, zawsze po∏àczonych stechiometrycznie. Ten ostatni jest niezb´d-
nà cz´Êcià takiego biokatalizatora, zapewniajàcà mo˝liwoÊç przeprowadzenia danej reakcji.
Tak si´ sk∏ada, ˝e to koenzymy (inaczej: kofaktory) przyjmujà i (lub) oddajà elektrony, wodo-
ry albo grupy atomowe. Umo˝liwia to przekazywanie tych czàstek na inne substancje, tak˝e
na inne enzymy. W ten sposób mo˝liwe jest wymienianie na szerokà skal´ ró˝nych substancji
w warunkach ustrojowych. Na nasze nieszcz´Êcie kofaktory ulegajà zu˝yciu (najcz´Êciej przez
utlenianie). Dlatego ich zapas musi byç wcià˝ uzupe∏niany. Problem w tym, ˝e nasz organizm
nie potrafi sam zsyntetyzowaç wszystkich tych zwiàzków (por. ni˝ej).
WIELE KOENZYMÓW WYKAZUJE POKREWIE¡STWO Z WITAMINAMI
Ka˝dy wie, co to jest witamina – wa˝ny sk∏adnik po˝ywienia, niemo˝liwy do zastàpienia,
gdy˝ organizm nie jest zdolny do jego samodzielnej syntezy. W niektórych wypadkach ˝ywy
ustrój zsyntetyzuje sobie witamin´, jeÊli otrzyma bezpoÊredni prekursor – prowitamin´. Jest
w stanie przeprowadziç wi´c tylko ostatnià reakcj´ otrzymywania witaminy – tak jest w wy-
padku karotenu, który jest dla nas prowitaminà A.
Witaminy sà zwiàzkami oligodynamicznymi, to znaczy, ˝e ich iloÊç wyklucza zastosowanie
budulcowe lub „energodajne”. Jest ich na to zbyt ma∏o – dzienne zapotrzebowanie cz∏owieka
mierzone jest w miligramach (pomijam wit. C).
W tym miejscu dodajmy, ˝e nie wszystkie witaminy sà sk∏adnikami koenzymów. ˚eby si´
nie pogubiç w tym wszystkim, proponuj´ przeprowadzenie biochemicznej charakterystyki wi-
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
40
tamin (wa˝nych dla cz∏owieka) z zaznaczeniem, czy wchodzi w sk∏ad kofaktora i jaki on jest.
Zastosujmy tutaj klasyczny podzia∏ witamin:
1. Rozpuszczalne w t∏uszczach – grupa: A, D, E, K jak WALDEK, tyle ˝e nie ma witamin
W i L. Jest to niewyszukany sposób, ˝eby zapami´taç ca∏à klasyfikacj´ (reszta witamin b´-
dzie wi´c rozpuszczalna w wodzie).
A) Witamina A (retinol, akseroftol) powstaje z lipidowego prekur-
sora – karotenu (tego z marchewki). Jednà z funkcji biochemicz-
nych tego zwiàzku jest wspó∏tworzenie rodopsyny (purpury
wzrokowej). Niedobór witaminy A wywo∏uje tak zwanà Êlepot´
zmierzchowà. Wed∏ug niektórych êróde∏ retinol i karotenoidy
dzia∏ajà przeciwnowotworowo, poniewa˝ sà przeciwutleniacza-
mi, co w nab∏onkach jest bardzo istotne (pomyÊl dlaczego). Zbyt
du˝y deficyt witaminy A mo˝e doprowadziç do keratynizacji na-
b∏onków: oka, dróg oddechowych, pokarmowych, potem nawet do tak zwanej suchoÊci ro-
gówki (stàd ju˝ tylko krok do nieodwracalnej Êlepoty). Pami´taj te˝, ˝e nadmiar retinolu
mo˝e doprowadziç do zatrucia organizmu. Dzienne zapotrzebowanie na t´ witamin´ wyno-
si ok. 2 mg. èród∏a to m. in. wàtroba, tran, jaja, niektóre jarzyny. Nie jest ona koenzymem.
B) Witamina D (kalcyferol) wp∏ywa na gospodarkà wapniowà
organizmu. W∏aÊciwie jest ona steroidowym prohormonem,
który powstaje w skórze pod wp∏ywem Êwiat∏a (g∏ównie ul-
trafioletu). Substratem wyjÊciowym sà tu pochodne choleste-
rolu (ergosterol z roÊlin i dehydrosterol z tkanek
zwierz´cych). Z witaminy D w wàtrobie i potem w nerkach
powstaje hormon – kalcytriol. Dzia∏a on pobudzajàco na
transport wapnia ze Êwiat∏a przewodu pokarmowego. Niedo-
bór kalcytriolu powoduje, ˝e procesy koÊciotwórcze ulegajà zwolnieniu, a przebudowa
elementów szkieletowych zak∏óceniu. U dzieci skutkiem sà deformacje koÊci, czyli krzy-
wica (u doros∏ych jej odpowiednikiem jest osteomalacja), objawiajàca si´ mi´dzy inny-
mi nadmiernym rozmi´kczeniem koÊci. Witamina D nie jest koenzymem. Dzienne
zapotrzebowanie wynosi ok. 0,02 mg. èród∏em tej witaminy mi´dzy innymi sà: wàtroba,
mleko, mas∏o, jaja, dro˝d˝e.
C) Witamina E (
α-tokoferol) zwana jest tak˝e witaminà p∏odnoÊci.
Jednak jej podstawowa funkcja to zapobieganie nadmiernemu
utlenianiu substancji organicznych w ustroju – g∏ównie lipidów
b∏on komórkowych. Brak wit. E wywo∏uje mi´dzy innymi os∏a-
bienie mi´Êni i obni˝onà p∏odnoÊç. Dzienne zapotrzebowanie
ok. 20 mg. Nie jest najprawdopodobniej koenzymem. G∏ówne
êród∏a to olej s∏onecznikowy, sojowy, orzechy lub kie∏ki pszenicy.
D) Witamina K (fitochinon) spe∏nia wa˝nà funkcj´ w krzepni´ciu
krwi. Jej deficyt powoduje krwawienia i opóênienie krzepni´cia krwi (por. ANATO-
MIA I …, ROZDZ. 4.5). Najprawdopodobniej witamina K jest kofaktorem enzymu
odpowiedzialnego za modyfikacj´ bia∏kowych czynników krzepni´cia (m.in. czynniki
Rosenthala oraz Christmasa). Dzienne zapotrzebowanie wynosi ok. 1 mg i normalnie
jest ca∏kowicie pokrywane przez produkcj´ flory jelitowej.
E) Witamina Q (ubichinon) jest sk∏adnikiem mitochondrialnego ∏aƒcucha przenoÊników
elektronów. Zalicza si´ jà do witamin, ale poniewa˝ nie stwierdzono jej niedoborów
w organizmie mo˝esz jà pominàç.
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
41
2. Rozpuszczalne w wodzie:
A) Witamina B
1
(tiamina, aneuryna) jest sk∏adnikiem koenzymu
– pirofosforanu tiaminy. Umo˝liwia on przeprowadzanie mi´-
dzy innymi reakcji oksydacyjnych dekarboksylacji: ketoglutara-
nu (por. cykl Krebsa) i pirogronianu (por. reakcja pomostowa).
Dzienne zapotrzebowanie na t´ substancj´ wynosi Êrednio
1,5 mg. Deficyt wywo∏uje chorob´ beri-beri na skutek zaburzeƒ
w pracy neuronów i w∏ókien mi´Êniowych (bóle ràk i nóg, dr˝e-
nie, os∏abienie mi´Êni, niewydolnoÊç uk∏adu krà˝enia). èród∏a:
wàtroba, dro˝d˝e, nieoczyszczone ziarna zbó˝, mi´so.
B) Kompleks witaminy B
2
, na który sk∏adajà si´:
a) Kwas nikotynowy (witamina PP) i nikotynoamid (lepiej kojarzàce si´ nazwy to nia-
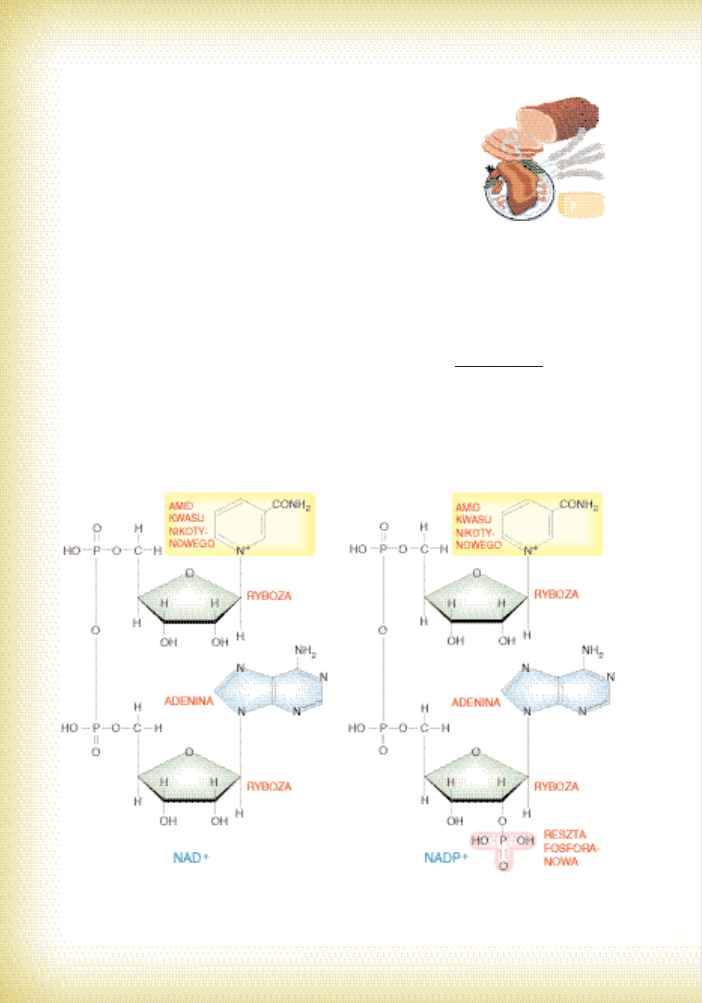
cyna i niacynamid). Zwiàzki te wchodzà w sk∏ad nukleotydów nikotynoamidowych.
Zalicza si´ do nich: NAD
+
(dinukleotyd nikotynoamidoadeninowy) i jego ufosforylo-
wanà pochodnà NADP
+
(fosforan dinukleotydu nikotynoamidoadeninowego; por.
ryc. 22). Oba te zwiàzki sà koenzymami enzymów z grupy dehydrogenaz, przeprowa-
dzajàcych wa˝ne reakcje odwodorowania (dok∏adniej przenoszenia elektronów
i H
+
). Ró˝nià si´ jednak wyst´powaniem i zastosowaniem. Ten pierwszy oddaje za-
brane substratowi elektrony i protony na ∏aƒcuch oddechowy. Drugi s∏u˝y jako re-
duktor w reakcjach syntezy, na przyk∏ad w fotosyntezie lub w budowie kwasów
t∏uszczowych. Poza tym NAD
+
jest koenzymem takich enzymów, jak dehydrogenazy:
alkoholowa, mleczanowa i glutaminianowa.
Ryc. 22. Budowa NAD
+
i NADP
+
z uwzgl´dnieniem elementów sk∏adowych (zwróç uwag´ na ich
prostot´). Fragmenty pochodzàce z witaminy PP zaznaczono ˝ó∏tym kolorem.
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
42

Nasz organizm potrafi syntetyzowaç nikotynoamid z tryptofanu (jeÊli ma go w dosta-
tecznej iloÊci). Rzadko wi´c stwierdza si´ objawy awitaminozy. JeÊli ju˝ do niej doj-
dzie, to jednostk´ chorobowà nazywa si´ pelagrà (rumieniem lombardzkim).
Charakteryzuje si´ ona przebarwieniami i zapaleniem skóry, biegunkà, goràczkà
i utratà przytomnoÊci.
Uwaga:
Zapis postaci NAD
+
i NADP
+
po zredukowaniu powinien przedstawiaç si´ nast´pu-
jàco: NADH + H
+
i NADPH + H
+
. Jednak w ksià˝ce czasem pojawi si´ odmiana
uproszczona: NADH i NADPH – nie jest to powa˝ny b∏àd.
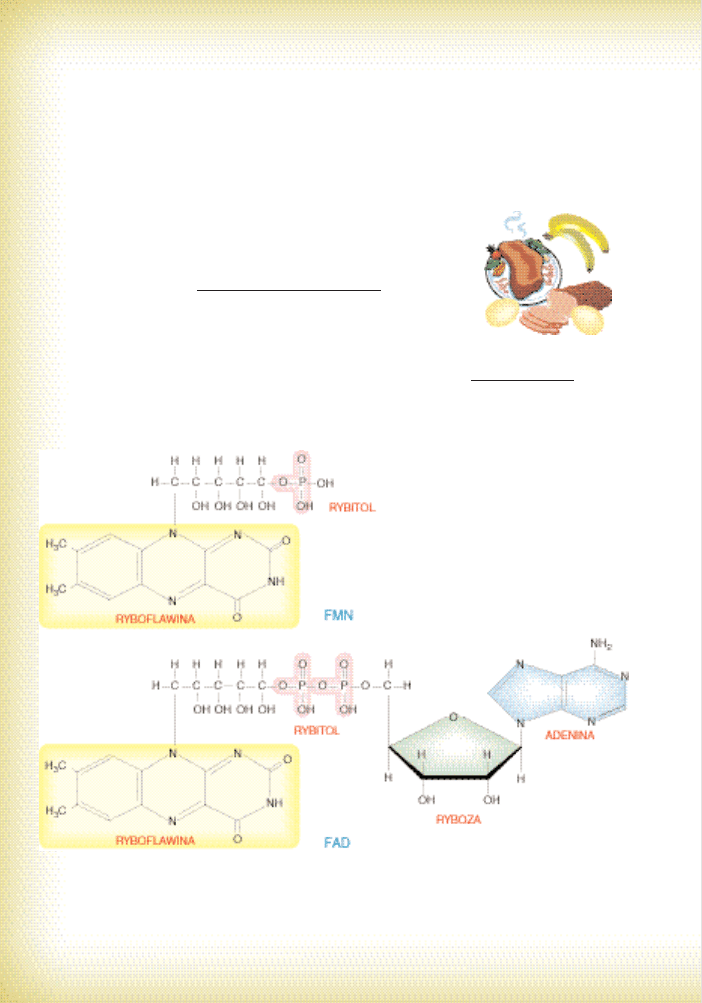
b) Ryboflawina (witamina B
2
sensu stricto) jest sk∏adnikiem grup
prostetycznych, tzw. flawoprotein. Enzymy te majà jako ko-
faktor FMN (mononukleotyd flawinowy) albo FAD (dinukle-
otyd flawinoadeninowy; por. ryc. 23). Charakterystyczne jest
dla nich silne, sta∏e zwiàzanie z cz´Êcià niebia∏kowà (stàd
okreÊlenie – grupy prostetyczne). Flawoproteiny przeprowa-
dzajà reakcje dehydrogenacji, uczestniczà wi´c w przenosze-
niu elektronów i jonów H
+
, ale o mniejszym potencjale ni˝ te,
którymi „interesuje si´” NAD
+
. Enzymy flawinowe sà licznà grupà (ponad 90 rodza-
jów), tutaj wymieƒmy wi´c tylko najwa˝niejsze. Sztandarowym przyk∏adem jest dehy-
drogenaza bursztynianowa, uczestniczàca w cyklu Krebsa oraz reduktazy ∏aƒcucha
oddechowego. W mikrocia∏kach wyst´pujà oksydazy flawinowe, przeprowadzajàce
bezpoÊrednie reakcje dehydrogenacji z wytworzeniem nadtlenku wodoru (por.
CZ¢Âå: CYTOLOGIA I …, ROZDZ. 2.7). Zapotrzebowanie dzienne na witamin´
B
2
wynosi oko∏o 1,7 mg. èród∏a: wàtroba, sery, mleko, jaja, niektóre jarzyny. Niedobór
powoduje zapalenia skóry oraz p´kanie kàcików ust.
c) Kwas foliowy (folian; wit. B
11
). Pochodna kwasu foliowego
– kwas tetrahydrofoliowy (koenzym F) jest kofaktorem
umo˝liwiajàcym przenoszenie reszt jednow´glowych nie-
zb´dnych do syntezy nukleotydu tymidynowego, który jest
sk∏adnikiem DNA (ÊciÊle mówiàc, umo˝liwia wykszta∏cenie
dTMP – por. ROZDZ. 8.1). Deficyt folianu powoduje mi´-
dzy innymi zak∏ócenia replikacji DNA, co upoÊledza proces
krwinkotwórczy (bardzo intensywne podzia∏y mitotyczne
w czerwonym szpiku kostnym wymagajà szybkiej syntezy kwasów nukleinowych w ery-
troblastach!). Skutkiem fizjologicznym jest anemia z∏oÊliwa, taka jak w wypadku nie-
doborów wit. B
12
. G∏ównymi êród∏ami kwasu foliowego sà: jarzyny liÊciaste, wàtroba
i dro˝d˝e. Dzienne zapotrzebowanie wynosi ok. 0,3 mg. Kwas foliowy stosuje si´ w pro-
filaktyce wad cewy nerwowej, chorób uk∏adu krwionoÊnego oraz nowotworów drà˝ni-
cy. Biochemicy sugerujà wi´c, ˝e korzystnie by∏oby dodawaç wit. B
11
do ˝ywnoÊci
(szczególnie w spo∏eczeƒstwach, gdzie problem niedo˝ywienia i braku witamin jest
bardzo ostry). Na koniec dodajmy jeszcze, ˝e folian sk∏ada si´ z trzech elementów. Jed-
nym z nich jest kwas p-aminobenzoesowy (PABA) – substancja egzogenna zarówno dla
zwierzàt, jak i bakterii. Ró˝nica polega na tym, ˝e organizmy zwierz´ce przyjmujà od
razu ca∏e czàsteczki folianu, natomiast drobnoustroje potrafià sk∏adaç je z cz´Êci prost-
szych (u˝ywajàc m. in. PABA). Wykorzystano to w leczeniu niektórych chorób bakte-
ryjnych. Otó˝ mo˝na zablokowaç przyswajanie kwasu p-aminobenzoesowego przez
podawanie zwiàzków do niego podobnych (tzw. analogów). W tym wypadku sà nimi
sulfonamidy (np. preparaty o nazwie biseptol albo sulfaguanidyna). Leki te sà wi´c in-
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
43
hibitorami wzrostu bakterii. Dla nas sà zasadniczo nieszkodliwe, chocia˝ nieoboj´tne!
Ponadto wszystkie leki bakteriobójcze przyjmowane doustnie niszczà, niejako po dro-
dze, naszà flor´ jelitowà. To zaÊ skutkuje du˝ym spadkiem poziomu witamin z grupy
B i K.
d) Kwas pantotenowy jest zmodyfikowanym dipeptydem szeroko rozpowszechnionym
w Êwiecie organizmów ˝ywych. Komórki u˝ywajà go do syntezy koenzymu A i ACP
(por. ROZDZ. 3.3). U ludzi nie stwierdza si´ objawów awitaminozy.
C) Witamina B
6
(pirydoksal, adermina) jest bliskà krewnà piry-
doksyny. Z kwasem fosforowym tworzy fosforan pirydoksalu
– kofaktor umo˝liwiajàcy przemiany enzymatyczne amino-
kwasów. Konkretnie fosforan pirydoksalu jest koenzymem
aminotransferaz przenoszàcych grupy aminowe w reakcjach
transaminacji i dezaminacji (por. ROZDZ. 7.4.2). Braki samej
wit. B
6
stwierdza si´ bardzo rzadko, np. u dzieci karmionych
przez matki, które d∏ugo przyjmowa∏y doustne Êrodki anty-
koncepcyjne, oraz wÊród potomstwa alkoholików. JeÊli jednak wystàpià, powodujà zaha-
mowanie syntezy bia∏ek (stàd pirydoksyn´ okreÊla si´ mianem czynnika wzrostu). Inne
skutki to m.in.: stany zapalne skóry, drgawki, nadmierna pobudliwoÊç. G∏´boki deficyt
wp∏ywa te˝ na zmniejszenie odpornoÊci typu komórkowego. Dzienne zapotrzebowanie
wynosi ok. 2 mg. G∏ównymi êród∏ami wit. B
6
sà: wàtroba, jaja, jarzyny, mi´so i banany.
D) Witamina B
12
(kobalamina, poniewa˝ zawiera kobalt) jest bardzo skomplikowanym zwiàz-
kiem chemicznym tworzàcym koenzym, który uczestniczy w przemianach aminokwasów,
kwasów t∏uszczowych i poÊrednio zasad azotowych. Brak wit. B
12
uniemo˝liwia normalnà
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
44
Ryc. 23.
Budowa FMN i FAD. ˚ó∏tà
barwà zaznaczono „witami-
nowe” fragmenty tych kofak-
torów.

erytropoez´, skutkiem czego powstaje anemia z∏oÊliwa. Poda-
nie nawet niewielkich dawek wit. B
12
dzia∏a wi´c przeciwane-
micznie. Zapotrzebowanie wynosi ok. 10–15
µg. Kobalamina
wytwarzana jest wy∏àcznie przez drobnoustroje, a wi´c nie wy-
st´puje w roÊlinach (powinni na to zwróciç uwag´ jarosze!).
U zwierzàt magazynowana jest mi´dzy innymi w wàtrobie
– warto wi´c jà jeÊç, podobnie jak dro˝d˝e.
E) Witamina H (biotyna) wyst´puje jako grupa prostetyczna en-
zymu transferazy karboksylowej, który umo˝liwia w∏àczanie dwutlenku w´gla w zwiàz-
ki organiczne (por. reakcje anaplerotyczne karboksylacji pirogronianu). Dzienne
zapotrzebowanie wynosi do 200
µg, jednak objawy niedoboru wyst´pujà bardzo rzadko.
Mo˝na je mimo to wywo∏aç przez spo˝ywanie du˝ych iloÊci surowych jaj (zawierajà one
termolabilne bia∏ko – awidyn´, która doÊç ∏atwo wià˝e biotyn´). Pojawiajà si´ wówczas
halucynacje, depresje, choroby skóry i bóle mi´Êni.
F) Witamina C (kwas askorbinowy) jest pochodnà w´glowodanów.
Dla wi´kszoÊci kr´gowców zwiàzek ten nie jest witaminà (syn-
tetyzujà jà z glukozy!). Jedynie cz∏owiek, ma∏py, Êwinka mor-
ska, nietoperze i ptaki muszà jà otrzymywaç w po˝ywieniu, i to
w sporych iloÊciach (75–100 mg w naszym wypadku). Kwas
askorbinowy uczestniczy w reakcjach redox i dzia∏a w nich ja-
ko donor równowa˝ników redukujàcych (oczywiÊcie, sama wi-
tamina C ulega przy tym utlenieniu). Fizjologiczne dzia∏anie
kwasu askorbinowego jest szerokie i nie do koƒca poznane. Wiadomo jednak, ˝e mi´-
dzy innymi umo˝liwia hydroksylowanie proliny do hydroksyproliny (jest to wst´pny wa-
runek sprawnej syntezy kolagenu – por. ni˝ej). Ponadto uczestniczy w syntezie kwasów
˝ó∏ciowych, wch∏anianiu ˝elaza, syntezie hormonów kory nadnerczy i mechanizmach
odpornoÊciowych. Niedobór witaminy C by∏ kiedyÊ zmorà dalekich wypraw morskich
i polarnych. Brak Êwie˝ych jarzyn i owoców prowadzi∏ do szkorbutu (gnilca), objawia-
jàcego si´ krwawieniem dziàse∏, wypadaniem z´bów, nieprawid∏owym zrastaniem koÊci
(zastanów si´, jaki ma to zwiàzek z kolagenem). Obecnie nie ma wi´kszych problemów
z utrzymaniem odpowiedniego poziomu witaminy C w organizmie, chocia˝ nale˝y pa-
mi´taç, ˝e jest ona termolabilna i cieplna obróbka pokarmów niszczy jà skutecznie. Dla-
tego nieodpowiednie nawyki ˝ywieniowe mogà zachwiaç bilansem kwasu
askorbinowego w organizmie.
Uwaga:
Jak widaç, koenzymami sà w zasadzie tylko witaminy rozpuszczalne w wodzie (i to
nie wszystkie).
4.4. Akumulatory i przenoÊniki energii
Wiesz ju˝, ˝e reakcje rozpadu sà egzoergiczne, a syntezy endoergiczne. O ile przeprowadza-
nie tych pierwszych wydaje si´ doÊç proste i logiczne z fizycznego punktu widzenia, o tyle te dru-
gie nastr´czajà sporo k∏opotów. Problemy zasadnicze sà dwa:
1. Przede wszystkim, skàd wziàç energi´ do przeprowadzenia syntez? Najprostszym rozwià-
zaniem jest skorzystanie z energii wyzwalanej w czasie reakcji rozpadu.
2. Nawet jeÊli mo˝emy przekazaç energi´ z jednej reakcji do drugiej, to k∏opotliwe stanie si´
zgrywanie (fachowo: sprz´ganie) tych przemian w czasie i przestrzeni. Mo˝emy akurat
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
45
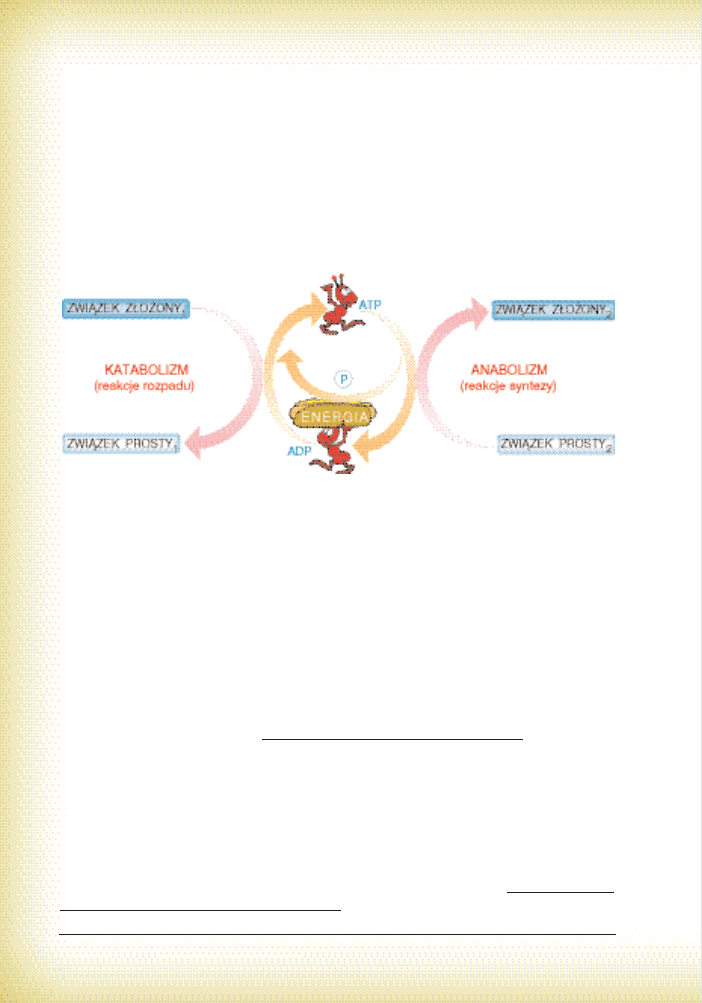
gwa∏townie potrzebowaç energii w pewnym miejscu. Tymczasem brak tam jest surowca
energetycznego albo jego obróbka zajmie sporo czasu. Pos∏u˝my si´ prostym przyk∏adem –
˝eby uruchomiç samochód (proces endoergiczny), nale˝y w∏àczyç rozrusznik. Urzàdzenie
to do pracy wymaga energii elektrycznej. W tej sytuacji mo˝na by∏oby udaç si´ do elektrow-
ni i poprosiç, ˝eby ktoÊ uruchomi∏ turbin´ pràdotwórczà. O wadach tego rozwiàzania nie
b´d´ pisa∏. Mo˝na te˝ problem rozwiàzaç inaczej – skorzystaç z energii zgromadzonej
wczeÊniej w akumulatorze. Co zyskujemy? Mo˝emy skorzystaç z niej natychmiast, a ponad-
to ten sposób nie wymaga w danym momencie przeprowadzania procesu egzoergicznego.
Wszystkie organizmy ˝ywe wykorzystujà ró˝norodne przemiany w celu wyzwolenia ener-
gii. JednoczeÊnie magazynujà jà w taki sposób, który umo˝liwia jej ca∏kowite wykorzystanie.
Ryc. 24. Ogólna zasada sprz´gania reakcji egzo- i endoergicznych (widoczna jest rola ATP)*
ENERGI¢ ZMAGAZYNOWANÑ W ODPOWIEDNI SPOSÓB NAZYWA SI¢
U˚YTECZNÑ BIOLOGICZNIE
Gromadzenie energii u˝ytecznej biologicznie odbywa si´ na zasadzie chemicznej. Otó˝
w czasie reakcji egzoergicznych syntetyzowane sà równolegle specjalne substancje posiadajà-
ce wiàzania makroergiczne. Do ich wytworzenia potrzeba du˝ych porcji energii, jednak gdy
zachodzi koniecznoÊç, ich roz∏o˝enie daje du˝e korzyÊci. Mo˝na wi´c mówiç o sprz´˝eniu
energetycznym przemian endoergicznych z egzoergicznymi. Rol´ poÊredników i „usprawnia-
czy” odgrywajà jednoczeÊnie zwiàzki nazywane akumulatorami i przenoÊnikami energii (por.
ryc. 24). Muszà one posiadaç kilka cech:
1. PowszechnoÊç, czyli du˝à dost´pnoÊç sk∏adników, z których sà zbudowane.
2. ¸atwoÊç w syntetyzowaniu i rozpadzie – oznacza to ograniczonà stabilnoÊç, ale nie za bar-
dzo. Wadà tej cechy jest to, ˝e takich czàsteczek nie mo˝na transportowaç. Dlatego zu˝y-
wane sà tylko w miejscu syntezy, na lokalnym rynku metabolicznym. Przyk∏adowo
przenoszenie ATP jest nierealne, mo˝liwy jest natomiast ruch mitochondrium (por.
CZ¢Âå: CYTOLOGIA I …, ROZDZ. 2.9).
3. RozpuszczalnoÊç w wodzie, co gwarantuje mo˝liwoÊç przeprowadzania przemian w kolo-
idach wodnych.
4. Stosunkowo ma∏à mas´ czàsteczkowà.
W 1941 roku panowie Lipmann i Kalckar udowodnili, ˝e takim zwiàzkiem jest adenozy-
notrifosforan („s∏ynny” ATP; por. ryc. 25). Okaza∏o si´ póêniej, ˝e jest to powszechny prze-
noÊnik energii we wszystkich uk∏adach ˝ywych. Najciekawsza jest w tym budowa chemiczna
tej substancji. ATP jest zwyk∏ym nukleotydem, tyle ˝e nieco zmodyfikowanym. W jego sk∏ad
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
46
*Przedstawiona na rycinie mrówka to oczywiÊcie ˝art.
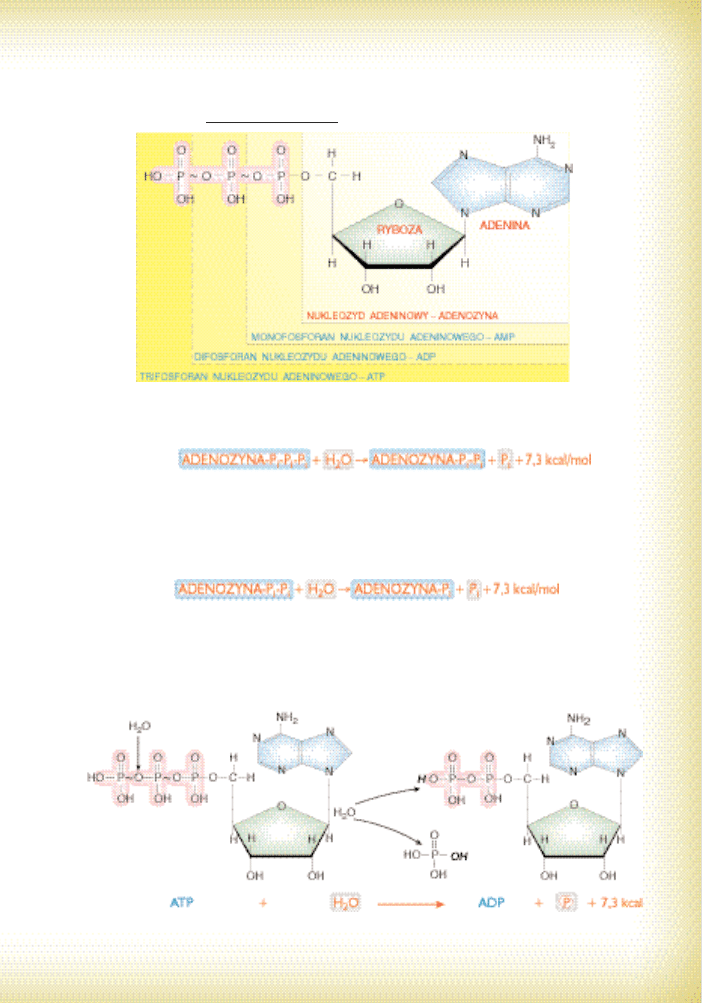
wchodzi bowiem: adenozyna (po∏àczenie cukru – rybozy z zasadà azotowà – adeninà), do któ-
rej przy∏àczone sà reszty fosforanowe. W wypadku ATP sà trzy takie reszty, stàd inna nazwa
tej substancji brzmi trifosforan adenozyny.
Ryc. 25. Budowa ATP, ADP i AMP; (~) – wiàzanie wysokoenergetyczne
Zanalizujmy teraz reakcj´ rozpadu ATP w drodze hydrolizy. Przemiana ta przedstawia si´
nast´pujàco:
Jak widaç, ATP rozpad∏ sià do ADP (adenozynodifosforanu) i reszty fosforanowej. Jedno-
czeÊnie wydzieli∏a si´ spora porcja energii 7,3 kcal z mola wiàzaƒ (30,5 kJ/mol), którà mo˝na
wykorzystaç (por. reakcja 3). ATP ma dwa wysokoenergetyczne wiàzania bezwodnikowe, dla-
tego dalsza hydroliza ADP tak˝e jest êród∏em energii. Reakcja przebiega nast´pujàco (pa-
mi´taj jednak, ˝e nie jest nigdy wykorzystywana w ustroju!):
Tym razem rozpad ADP doprowadzi∏ do powstania AMP (adenozynomonofosforanu)
i kolejnej reszty fosforanowej oraz energii w iloÊci 7,3 kcal/mol (jest to wartoÊç Êrednia obu
hydroliz!). AMP nie posiada wiàzaƒ wysokoenergetycznych (chocia˝ mo˝na go roz∏o˝yç).
Uwaga:
Wiàzanie wysokoenergetyczne to takie, które wyzwala w wyniku hydrolizy wi´cej ni˝
20 kJ/mol.
Reakcja 3. Hydroliza wiàzania bezwodnikowego w ATP
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
47

Ka˝dy akumulator trzeba na∏adowaç – w wypadku opisywanego przenoÊnika proces pole-
ga na tworzeniu ATP z ADP i reszty fosforanowej. Energia czerpana jest g∏ównie z procesów
utleniania wewnàtrzkomórkowego (por. odpowiednie rozdzia∏y). Mamy tu wi´c swoisty cykl
ATP
↔ ADP + P
i
, który jest podstawowym sposobem wymiany energii w uk∏adach ˝ywych.
Zapas adenozynotrifosforanu nie jest przeznaczony do pracy ciàg∏ej. W mi´Êniach szkieleto-
wych starcza tylko na u∏amek sekundy (zu˝ycie energii w uk∏adzie ruchu jest bardzo du˝e,
gdy˝ chodzi o zamian´ energii chemicznej w mechanicznà na znacznà skal´). Wystarcza to
jednak w zupe∏noÊci, poniewa˝ w∏ókna zdà˝à w tym czasie uruchomiç inne êród∏a energii.
Ryc. 26. Energia w organizmie nie mo˝e byç uwalniana na raz (A), przenoÊniki i akumulatory two-
rzà mo˝liwoÊç magazynowania i uwalniania energii ma∏ymi porcjami (B). PrzemyÊl dobrze
powy˝szà rycin´.
AKUMULATORAMI MOGÑ BYå TAK˚E INNE ZWIÑZKI ORGANICZNE
Istnieje kilka rodzajów nukleotydów, które mogà odgrywaç rol´ analogicznà do ATP. Sà
to: guanozynotrifosforan (GTP) wykorzystywany w cyklu Krebsa, urydynotrifosforan (UTP)
i cytydynotrifosforan (CTP). Sà to, jak widaç, tak˝e trifosforany nukleozydów i reakcje ich
hydrolizy sà takie jak ATP (odpowiednio do GDP, UDP i CDP). Wykorzystanie tych zwiàz-
ków ma jednak ograniczony charakter i dlatego nie musisz ich znaç dok∏adnie.
Uwaga:
Za inny specyficzny rodzaj przenoÊników energii mo˝na uznaç NAD
+
i FAD. Zwiàzki
te poÊredniczà bowiem w przekazywaniu zasobnych w energi´ elektronów na tlen – po-
zwala to na wytwarzanie wysokoenergetycznych wiàzaƒ w ATP (por. ROZDZ. 5.5.2).
TWORZENIE WIÑZA¡ WYSOKOENERGETYCZNYCH ODBYWA SI¢ NAJCZ¢ÂCIEJ PRZEZ
PRZY¸ÑCZANIE RESZTY FOSFORANOWEJ
Proces, w którym powstaje wiàzanie zawierajàce du˝à iloÊç tak zwanej energii swobodnej
(ogólnie: zgromadzonej do u˝ytku), zwykle polega na kowalencyjnym zwiàzaniu reszty fosfo-
ranowej. Dlatego nazywa go si´ fosforylacjà, pami´taj jednak, ˝e nie ka˝de przy∏àczenie P
i
jest fosforylacjà. Poj´cia tego mo˝esz u˝ywaç tylko wówczas, gdy chodzi o tworzenie zwiàzku
makroergicznego (np. sama reakcja F w glikolizie nie jest fosforylacjà).
Istniejà trzy mo˝liwoÊci fosforylowania ADP do ATP:
1. Fosforylacja substratowa – zachodzi, gdy reszta fosforanowa zostanie przeniesiona bezpo-
Êrednio na ADP z wykorzystaniem energii organicznego substratu (cz´sto to on sam jest
dawcà reszty fosforanowej). Ten sposób ∏adowania ATP jest ewolucyjnie najstarszy, jednak
M O L E K U L A R N E P O D ¸ O ˚ E B I O L O G I I
48

niezbyt korzystny energetycznie. Nie wymaga udzia∏u tlenu i zachodzi w glikolizie oraz cy-
klu Krebsa.
2. Fosforylacja fotosyntetyczna (fotofosforylacja) – zachodzi wy∏àcznie u fotoautotrofów. W pro-
cesie tym nast´puje konwersja energii Êwietlnej na chemicznà ATP. Wyst´pujà dwa typy foto-
fosforylacji – ich przebieg jest doÊç skomplikowany i zosta∏ omówiony w ROZDZ. 5.2.1.
3. Fosforylacja oksydacyjna – zachodzi u wszystkich organizmów tlenowych. Ogólnie rzecz
bioràc, jest to wydajny sposób magazynowania energii u˝ytecznej biologicznie. Do syntezy
ATP wykorzystywana jest energia elektronów i protonów przekazywanych na tlen. Proces
jest skomplikowany i zosta∏ dok∏adniej omówiony w ROZDZ. 5.5.2.
4 . W p r o w a d z e n i e d o e n z y m o l o g i i
49
Wyszukiwarka
Podobne podstrony:
Enzymologia 4
AKTYWNOŚĆ ENZYMÓW PRZECIWUTLENIAJĄCYCH!
07 Modyfikacje struktury enzymówid 7062 ppt
Enzymologia materiały do ćwiczeń
Enzymologia wyniki egzaminu (I termin)
pytania-enzymy, Technologia żywności UWM, enzymologia
KLASYFIKACJA ENZYMÓW
Zastosowanie enzymow w syntezie- wyniki, PWR, III semestr
enzy 2011-11-23, enzymologia, notatki
Inhibitory enzymów jako leki, materiały medycyna SUM, biochemia, Kolokwium II
Budowa enzymów ściąga
Enzymologia wykłady ściąga
EnzymologiaTZ wyklad 4
ściąga z enzymologii
enzymologia
enzymologia cw 1
enzymologia ćwiczenie 3
9) Oznaczanie aktywności enzymów amylolitycznych
Zastosowanie enzymów
więcej podobnych podstron