
VII Patogeneza wybranych
chorób układu nerwowego

STWARDNIENIE ROZSIANE
(ŁAC. SCLEROSIS
MULTIPLEX)

Stwardnienie rozsiane (łac. sclerosis
multiplex, SM, ang. multiple sclerosis,
MS) – przewlekła, zapalna,
demielinizacyjna choroba centralnego
układu nerwowego,
w której dochodzi do wieloogniskowego
uszkodzenia (demielinizacji i rozpadu
aksonów) tkanki nerwowej.

Choroba ma najczęściej przebieg
wielofazowy z okresami zaostrzeń i
poprawy.
Stwardnienie rozsiane zostało po raz
pierwszy opisane w 1868 przez Jeana-
Martina Charcota.
Dotyczy najczęściej osób młodych, ze
szczytem występowania między 20. a
40. rokiem życia, i nieznaczną przewagą
zachorowań u kobiet

SM jest chorobą dotyczącą komórek nerwowych
(neuronów), glejowych (oligodendrocyty), komórek
odpornościowych mózgu (mikroglej) ,
- w której dochodzi do uszkodzenia otoczki mielinowej
wokół wypustek komórek nerwowych, co powoduje
niemożność prawidłowego przekazywania impulsów
wzdłuż dróg nerwowych w mózgowiu i rdzeniu
kręgowym,
- Nazwa "stwardnienie rozsiane" odzwierciedla rozsianie
procesu patologicznego w różnych miejscach układu
nerwowego, jak również rozsiew zmian w czasie

Limfocyty T
Chociaż dużo wiadomo na temat mechanizmów
uszkodzeń zachodzących w stwardnieniu
rozsianym, to przyczyny występowania
stwardnienia rozsianego nie są znane.
W chorobie tej dochodzi do degeneracji mieliny
(substancji tłuszczowej pokrywającej aksony
komórek nerwowych).
Według teorii autoimmunologicznej patogenezy SM w
inicjacji procesu zapalnego główną rolę odgrywają
komórki T.
U chorych z SM regulatorowe limfocyty T posiadają
kilkukrotnie mniejszą zdolność hamowania
podziałów innych populacji limfocytów

Limfocyty T rozpoznają mielinę jako obcą
substancję i kierują przeciwko niej swoją
odpowiedź immunologiczną
Zainicjowany proces zapalny angażuje także inne
komórki układu odpornościowego jak również
cytokiny i przeciwciała.
Zostaje naruszona bariera krew-mózg, co z kolei
sprzyja takim uszkadzającym procesom jak
obrzęk, aktywacja makrofagów i silniejsza
aktywacja cytokin i enzymów niszczących
tkanki jak metaloproteinazy.

Infekcje wirusowe w
rozwoju SM
Najpopularniejsze hipotezy to również ta o udziale
infekcji wirusowej lub reaktywacji utajonego
zakażenia retrowirusowego, które miałyby
zainicjować późniejszą odpowiedź układu
odpornościowego.
Na poziomie molekularnym istniałoby podobieństwo
(mimikra) między antygenem wirusa a własnym
antygenem komórek OUN, co wyjaśniałoby
skierowanie odpowiedzi układu odpornościowego
przeciwko własnym autoantygenom.

Witamina D
Ponieważ SM występuje częściej u ludzi żyjących dalej od
strefy równikowej, postulowano udział obniżonej
ekspozycji na światło słoneczne w patogenezie SM, np.
za sprawą obniżonej syntezy witaminy D3.
Teorię tę wspierają niedawne odkrycia, wskazujące, że
witamina D jest istotnym regulatorem układu
odpornościowego. Duże badanie przeprowadzone w
2006r. wykazało większe ryzyko zachorowania przy
niskim poziomie witaminy D tylko wśród osób białych.
Badanie z 2007r. wykazało, że przy braku predyspozycji
genetycznych ekspozycja na światło w dzieciństwie
zmniejsza ryzyko zachorowania na SM.

Chlamydophila pneumoniae
Nadal spotyka się teorie, że SM może się rozwinąć pod
wpływem przewlekłej infekcji krętkami, którą to
hipotezę wspiera badania na małych grupach
pacjentów, u których z PMR izolowano postaci
przetrwalnikowe tych bakterii.
Inną bakterią, której udział w patogenezie SM wysuwano,
jest Chlamydophila pneumoniae; DNA tej bakterii
izolowano wielokrotnie z PMR pacjentów z SM, a w
jednym badaniu udowodniono, że prążki oligoklonalne u
14 z 17 przebadanych pacjentów składały się z
przeciwciał skierowanych przeciwko antygenom
Chlamydophila

Geny
SM nie jest chorobą dziedziczną. Gromadzone są jednak
dowody na udział czynników genetycznych w predyspozycji
do zachorowania na tę chorobę.
Szczególnym zainteresowaniem badaczy cieszy się region
chromosomu 6, w którym znajdują się geny głównego
układu zgodności tkankowej (HLA). Kodowane tam białka są
istotnymi dla funkcjonowania układu odpornościowego.
Uważa się, że być może pewien zestaw HLA może mieć
działanie selekcjonujące do 15 roku życia – tzn. może
uwrażliwiać lub uodparniać organizm na określone
zakażenia wirusowe, układ HLA może odpowiadać za
obecność receptorów i przeciwciał przeciw antygenom
wirusowym.

Badania nad rodzinnymi przypadkami SM i badania porównujące
ekspresję genów u ludzi z SM i u myszy z EAE sugerują, że inne
locus związane z predyspozycją do SM znajduje się na
chromosomie 5.
Inne loci na chromosomach 2, 3, 7, 11, 17, 19 i X zostały wskazane
jako potencjalne loci genów biorących udział w patogenezie SM.
Badania te umacniają teorię o wieloczynnikowej etiologii SM.
Rozwój SM wydaje się zależeć od interakcji szeregu genów, z
których każdy osobno ma niewielki wpływ na rozwój choroby.
Dalsze badania są konieczne, by określić jakie geny znajdują się we
wskazanych loci, jaka jest ich funkcja, i w jaki sposób zależności
tych genów i czynników środowiskowych prowadzą do rozwoju
choroby.

OSTRE ROZSIANE
ZAPALENIE MÓZGU I
RDZENIA (ADEM)

Ostre rozsiane zapalenie mózgu i rdzenia (
Acute Disseminated Encephalomyelitis -
ADEM) –
Autoimmunologiczna, demielinizacyjna
choroba mózgu bardzo podobna
patologicznie do SM, które zazwyczaj jest
choroba chroniczną nawracającą chorobą
młodych dorosłych,
podczas gdy ADEM jest zazwyczaj
jednofazową chorobą dzieci.

Nieprawidłowy poziomu immunoglobulin w PMR jest
znacznie rzadszy w ADEM niż w SM.
ADEM zwykle występuje zwykle po infekcji z gorączką
lub po szczepieniu.
U części chorych z początkowym rozpoznaniem ADEM
diagnozuje się później SM.
Śmiertelność w ADEM wynosi ok. 5% i większość chorych
przeżywających ma częściową niepełnosprawność.

Mglistość rozdziału między ADEM i SM sugeruje kontinuum,
w skład którego miałyby wchodzić także zapalenie nerwu
wzrokowego i poprzeczne zapalenie rdzenia (zespół
Devica)
Z innej strony ADEM zlewa się z różnymi zapaleniami
mózgu, chorobami zapalnymi naczyń, innymi
jednofazowymi chorobami poinfekcyjnymi jak ostra afazja
móżdżkowa.
Kolejna płynna granica jest z zespołem Guillaina-Barrégo w
postaci zespołu Millera-Fishera i encefalomielo-
radikuloneuropatią.

MIASTENIA

Pierwszy opis przypadku miastenii podany
został w 1672 roku przez angielskiego
lekarza Tomasza Willisa.
Jednak dopiero na przełomie XIX i XX
wieku pojawiły się znakomite opisy serii
przypadków miastenii podane przez
profesora z Heidelbergu, Wilhelma H.
Erba i neurologa z Warszawy, Samuela
Goldflama.

Choroba Erba-Goldflama
(MG)
Myasthenia Gravis jest przewlekłą
autoimmunologiczną chorobą objawiającą się
nadmierną męczliwością mięśni prążkowanych
(szkieletowych).
Choroba nie dotyczy mięśni gładkich: serca, ścian
naczyń krwionośnych, jelit, pęcherza moczowego.
Nadmierna męczliwość mięśni spowodowana jest
zaburzeniami w przekazywaniu impulsu
nerwowego na połączeniu nerwowo-mięśniowym.

Choroba występuje u osób w każdym wieku,
niezależnie od płci. Występuje we wszystkich
szerokościach geograficznych i dotyczy wszystkich
ras.
Najczęściej ujawnia się u ludzi między 18 a 30 rokiem
życia - w tym okresie kobiety chorują od dwóch do
czterech razy częściej.
Kolejny szczyt zachorowań występuje w wieku 45-55
lat i wtedy proporcje między kobietami i
mężczyznami wyrównują się, a w grupie powyżej
50-60 roku życia częściej chorują mężczyźni.

Miastenia jest chorobą rzadko spotykaną,
a co za tym idzie - mało znaną w
społeczeństwie. Choruje na nią ok. 8 do
12 osób na 100 000. Nie jest chorobą
dziedziczną.
W ok. 15% przypadków miastenia
występuje łącznie z innymi chorobami
autoimmunoligicznymi, takimi jak:
reumatoidalne zapalenie stawów, toczeń
rumieniowaty, nadczynność tarczycy,
pęcherzyca, łuszczyca, zapalenie
wielomięśniowe lub stwardnienie
rozsiane.
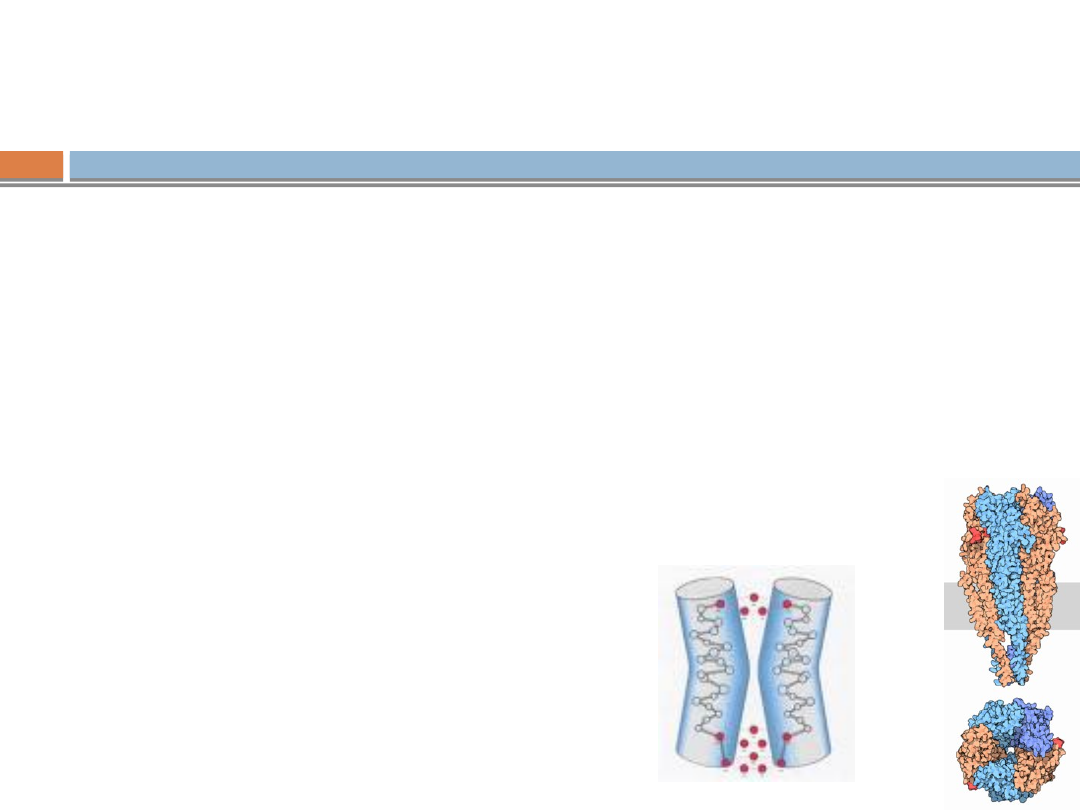
Patogeneza miastenii
W normalnych warunkach impuls
docierający do zakończenia nerwu (do
płytki nerwowo-mięśniowej) uwalnia z
zakończenia nerwu mediator
acetylocholinę, która łączy się ze swoim
receptorem na części mięśniowej płytki
nerwowo-mięśniowej co powoduje
transmisję
impulsu z nerwu do mięśnia.

W miastenii wytwarzanie acetylocholiny
jest prawidłowe, natomiast zmniejszona
jest znacznie (nawet do 80%) ilość
receptorów po stronie mięśniowej
mogących przekazać impuls do mięśnia.
Redukcja ilości receptorów acetylocholiny
spowodowana jest zablokowaniem lub
zniszczeniem ich przez autoprzeciwciała.
Obecność autoprzeciwciał w miastenii
potwierdza autoimmunologiczny
(autoagresywny) charakter tej choroby.

Kryteria przemawiające za zaliczeniem
miastenii do grupy chorób
autoimmunologicznych:
Obecność w surowicy przeciwciał przeciw AChR
Wiązanie się przeciwciał z antygenem, tj. AChR
Uzyskanie modelu miastenii po immunizacji
antygenem
Obniżenie się miana przeciwciał w miarę poprawy
klinicznej

Prawdopodobne działanie autoprzeciwciał
na synapsę nerwowo-mięśniową odbywa
się za pomocą kilku mechanizmów, do
których należą:
blokowanie receptorów - co uniemożliwia
wiązanie się w tych miejscach
acetylocholiny,
uszkadzanie strukturalne po stronie
mięśniowej synapsy (płytki nerwowo-
mięśniowej),

przyspieszanie naturalnego procesu
degradacji receptorów acetylocholiny - w
wyniku tego zostaje zaburzona
równowaga pomiędzy ich stałym
tworzeniem się i rozpadaniem.
Badania wykazały, że w płytce nerwowo-
mięśniowej u chorych na miastenię
znajduje się zaledwie 1/3 liczby
receptorów acetylocholiny w porównaniu
z mięśniem zdrowym.

Męczliwość mięśni objawia się wtedy, gdy
acetylocholina nie może aktywować
wystarczającej ilości receptorów na płytce
nerwowo-mięśniowej.
Na ogół zmniejszenie liczby AChR powoduje
nasilenie objawów klinicznych miastenii,
Ilość auto
przeciwciał
przeciw AChR we krwi
często, ale nie zawsze bezpośrednio koreluje
z objawami i przebiegiem choroby - np. w
miastenii ocznej miano przeciwciał jest
niskie lub nie stwierdza się ich w ogóle.

Typy miastenii wg
Ossermana
Miastenia oczna
W tej postaci miastenii zajęte są tylko
mięśnie oczu. Występuje: podwójne i
niewyraźne widzenie, zez, opadanie
powiek.
Miastenia uogólniona
W miastenii uogólnionej choroba
zaczynają przenosić się na mięśnie
innych części ciała, przechodząc przez
poszczególne podtypy.

Miastenia uogólniona - postać
łagodna
Objawy choroby rozwijają się powoli, poza
obawami ocznymi (w których występują
okresowe nasilenia i zmniejszenia
objawów) choroba zaczyna zajmować
jamę ustną i kończyny. Nie występują
problemy z oddychaniem. W tym okresie
leki przeciecholinoesterazowe przynoszą
choremu pełną poprawę.

Miastenia uogólniona - postać
podostra
Coraz ostrzejsze objawy choroby dotyczą
mięśni oczu, jamy ustnej i kończyn. W
dalszym ciągu nie występują zaburzenia
oddychania. Leki
przeciecholinoesterazowe nie przynoszą
choremu pełnej poprawy - często po
paru miesiącach leczenia, mimo
zwiększania dawki leku, nie zmniejszają
objawów w sposób wystarczający.
Miastenia uogólniona - postać ostra
Do tej grupy zaliczamy chorych z podtypu
II A i II B, u których nastąpiło nagłe
zaostrzenie objawów - dołączają się
zaburzenia oddechu. Chorych z tej grupy
najczęściej występują przełomy
miasteniczne, bardzo często stwierdza
się też u nich występowanie grasiczaków.

ZESPOŁY
MIASTENICZNE

Zespoły miasteniczne
wrodzone
Zdarzają się rzadko
Niektórzy twierdzą, iż pozostają one
nierozpoznane i w rzeczywistości są
o wiele częstsze
Patogeneza jest taka j.w opisywanej
Miastenii
Pierwsze objawy choroby występują
później, dotyczą tylko niektórych
grup mięśniowych, a ich nasilenie
jest zmienne i nietypowe

Zespoły presynaptyczne
Istotą ich jest defekt w resyntezie lub
rozmieszczeniu pęcherzyków synaptycznych w
części presynaptycznej synapsy
Choroba zaczyna się we wczesnym dzieciństwie
Znany jest również wrodzony zespół miasteniczny
z napadowym bezdechem
Innym defektem presynaptycznym jest
zmniejszenie wielkości pęcherzyków
synaptycznych połączone z upośledzeniem
uwalniania transmitera

Zmniejszenie liczby kwantów Ach
uwalnianych pod wpływem bodźca
Obniżenie to wynika z tzw. frakcji Ach
gotowej do natychmiastowego działania i
ze zmniejszonej gęstości pęcherzyków
synaptycznych
Jest do defekt w syntezie i aksonalnym
transporcie Ach od komórki rogu
przedniego rdzenia na obwód

Zespoły synaptyczne
Są to zespoły polegające na braku
cholinoesterazy w szczelinie
synaptycznej, co prowadzi do wtórnego
zmniejszania zakończeń nerwowych i
redukcji liczby uwalnianych kwantów Ach
Jednocześnie powstaje depolaryzacyjny
blok z niewrażliwości na poziomie
receptorów acetylocholiny
Zespół występuje u noworodków

Zespoły postsynaptyczne
Powstają one z powodu mutacji genów dla
poszczególnych podjednostek AChR
Najważniejszy jest
ZESPÓŁ POWOLNEGO KANAŁU AChR
Jego istota jest przedłużenie i występująca
częściej niż w normie aktywacja kanałów
jonowych, co prowadzi do wydłużenia MEPP i
EPP
Pojawia się podwójna odpowiedź elektryczna
na pojedynczy bodziec

ZESPÓŁU KANAŁU SZYBKIEGO
•
Przyczyną jest rzadkie i krótko trwające
otwieranie się kanałów jonowych AChR,
co powoduje obniżenie amplitudy i
skrócenie czasu MEPP i EPP
•
Zespół ten spowodowany jest przez
różne mutacje w podjednostkach AChR
•
objawy mogą pojawic się w okresie
noworodkowym

Zespół Lamberta-Eatona
Występuje u osób z choroba nowotworową
Jest to choroba autoimmunologiczna, mechanizm
bloku nerwowo-mięśniowego jest presynaptyczny
W zespole tym utrudnione jest uwalnianie Ach z
części presynaptycznej synapsy do szceliny
synaptycznej
Wapniowe kanały jonowe zlokalizowane w części
presynaptycznej, konieczne do wyrzucania
transmittera, blokowane są przez krążące w
surowicy przeciwciała skierowane przeciw
kanałom wapniowym

Zespoły miasteniczne
polekowe
Klasycznym przykładem jest wystąpienie
miastenii w wyniku leczenia penicylaminą
reumatoidalnego zapalenia stawów
Stwierdza się wówczas w surowicy obecność
przeciwciał przeciw AChR
mechanizm bloku nerwowo- mięśniowego
jest postsynaptyczny
poprawa nastepuje po kilku miesiącach od
odstawienia lekui towarzyszy jej obniżenie
miana przeciwciał

WIELOOGNISKOWA
NEUROPATIA RUCHOWA

opisana w latach 80. cechuje się
przewlekle postępującym,
asymetrycznym niedowładem i zanikiem
mięśni oraz częściowym lub całkowitym
blokiem przewodzenia we włóknach
ruchowych nerwów
przewodzenie we włóknach czuciowych
jest prawidłowe

Etiologia MMN jest nieznana, choć
przynajmniej w części przypadków
zakłada się mechanizm
autoimmunologiczny choroby z udziałem
przeciwciał IgM przeciwko gangliozydom,
zwłaszcza GM 1.
Za mechanizmem immunologicznym
może także przemawiać korzystna w
większości przypadków odpowiedź na
leczenie immunolgobulinam
podawanymi dożylnie

Badanie płynu mózgowo-rdzeniowego
bywa prawidłowe, choć czasami stężenie
białka jest podwyższone
W płynie mózgowo-rdzeniowym jak i w
surowicy, niekiedy nawet u 80% chorych,
podwyższone jest stężenie przeciwciała
przeciwko GM 1
należy podkreślić, że te przeciwciała
mogą występować w niskim mianie u
ludzi zdrowych oraz w innych chorobach
neurologicznych

ZESPÓL GUILLAINA-
BARREGO

Kryteria kliniczne to:
1.
Postępujący niedowład więcej niż jednej
kończyny
2.
Zniesienie lub osłabienie odruchów
3.
Narastanie objawów przez kilka dni do
czterech tygodni
4.
Niewydolności elektrofizjologiczne
5.
Rozszczepienie białkowo-komórkowe w
płynie mózgowo- rdzeniowym

Zespól ten jest ostrą zapalną demielinizacją
powstającą z udziałem mechanizmów
immunologicznych
Początkowo zakładano, że charakterystyczną
cechą patologiczna nerwów są nacieki
limfocytarne i demielinizacja z udziałem
makrofagów, a wzrost ekspresji cząsteczek
adhezyjnych (ICAM-1, ICAM-3, selektyna E) na
komórkach śródbłonka naczyń nerwu
przyczynia się do migracji przez ścianę naczyń
komórek zapalnych (limfocyty, makrofagi)

Stwierdzone w surowicy chorych
przeciwciała skierowane są przeciwko
składnikom mieliny i błony komórkowej
Najczęściej są to przeciwciała przeciw:
P2, P0, MAG, glikolipidom, sulfatydom,
gangliozydom (LM, 1, GM 1, GQ, SGPD,
SGLPG)

Dokładny patomechanizm daleki jest od
wyjaśnienia, gdyż np. podwyższone
miano przeciwciał przeciwko GM 1
występuje w wielu innych przypadkach
Udowodniono homologię między LPS
bakterii Campylobacter jejuni a GM 1,
GD 1a, GD 3 nerwu
Zespół ten może towarzyszyć
nowotworom, zwłaszcza płuc,
chłoniakom, infekcji HIV, boleriozie i
sarkoidozie

PRZEWLEKŁA ZAPALNA
POLIRADIKULONEUROPATI
A DEMIELINIZACYJNA

PZPD
Jest procesem autoimmunologicznym, w
wyniku którego dochodzi do uszkodzenia
korzeni nerwowych i nerwów
obwodowych.
Cechuje się postępujacym lub
nawracajacym wiotkim niedowładem
kończyn z różnego rodzaju zaburzeniami
czucia i niekiedy zajęciem nerwów
czaszkowych

Zakłada się, że mechanizm uszkodzenia
nerwów w PZPD jest podobny do
mechanizmu sugerowanego z Guillaina-
Barrego
Przeciwciałami zlokalizowanymi w
surowicy chorych przeciw białkom
mieliny są: Po, P2, PMP 22, przeciw
tubulinie i gangliozydom GM 1, LM 1 oraz
przeciw SGPG
W ogromniej większości antygeny
będące celem ataku pozostają nieznane

Dowodem na patogenną rolę komórek T
w PZPD jest zwiększona liczba krążących
aktywowanych komórek T i podwyższone
stężenie interleukiny 2 (IL-2)
zwiększone stężenie TNF-α koreluje z
aktywnościa procesu
TNF może być związany z demielinizacją
i przerwaniem bariery krew-nerw

Reakcja immunologiczna może rozwinąć
się pierwotnie także wewnątrz nerwu,
gdyż komórki Schwanna posiadają
zdolność prezentowania antygenu
W większości przypadków PZPD
obserwuje się kliniczne i
elektrofizjologiczne, oprócz
demielinizacji, cechy procesu
aksonalnego

Prawdopodobnie zwyrodnienie aksonalne
może być związane z efektem widza w
obrębie nacieków, atakiem
immunologicznym zwróconym przeciwko
epitopom aksonu i zwiększonym
ciśnieniem wewnątrz endoneurium

Częste są fluktuacje objawów, rzadko
dochodzi do zajęcia nerwów
czaszkowych, zaburzeń autonomicznych
i niewydolności oddechowej
Wg niektórych autorów prawidłowe
stężenie białka w płynie mózgowo-
rdzeniowym można obserwować nawet u
90% chorych
Choroba może wystąpić w każdym
wieku, ale rzadziej u dzieci i starszych

W wieloogniskowej zapalnej neuropatii
demielinizacyjnej (MIND)
Rozkład niedowładów jest niesymetryczny z przewagą
zajęcia kończyn górnych, z towarzyszącymi
zaburzeniami czucia
Odsiebna nabyta symetryczna neuropatia
demielinizacyjna (DADS)
Często u chorych klinicznie występują wyłącznie objawy
czuciowe, ale badania elektrofizjologiczne wykazują
nieprawidłowości także we włóknach ruchowych

SCHIZOFRENIA

Schizofrenia– zaburzenie psychiczne
zaliczane do grupy psychoz
endogennych.
Objawia się upośledzeniem postrzegania
lub wyrażania rzeczywistości, najczęściej
pod postacią omamów słuchowych,
paranoidalnych lub dziwacznych urojeń
albo zaburzeniami
mowy i myślenia, co powoduje
znaczącą dysfunkcję społeczną
lub zawodową.

W chorobie zwiększona jest aktywność
dopaminergiczna w szlaku
mezolimbicznym mózgu, co
konsekwentnie wykazują różne badania.
Szczególną rolę odgrywa tu receptor D2
(układ dopaminergiczny).
Podejrzewa się, że jego niewłaściwe
funkcjonowanie jest odpowiedzialne za
część objawów występujących w tej
chorobie.

Przesłanki epidemiologiczne
Schizofrenia występuje na całym świecie
Płeć żeńska opóźnia przeciętny wiek
zachorowania, jednakże najczęstszy
początek zachorowań ma miejsce w 3.
dekadzie życia (wiek 21-30 lat).
Płodność chorych jest mniejsza, a
śmiertelność większa niż w populacji, co
utrudnia wyjaśnienie nie zmieniającej się
zapadalności

Przesłanki genetyczne
Przekazywana jest nie tyle podatność na schizofrenię,
ile skłonność do wystąpienia spektrum zaburzeń:
trudności przystosowania, cech osobowości typu
schizotymii, zaburzeń osobowości należących do
osobowości schizoidalnej, paranoicznej, zaburzeń z
pogranicza schizofrenii, czy innych psychoz.
Wielkie nadzieje na znalezienie "genu schizofrenii",
pokładane w genetyce molekularnej, na razie nie
przyniosły wyników.
Poszukiwanie sprzężeń genetycznych (ang. linkage) w
miejscach genomu podejrzewanych o podatność na
schizofrenię nie dały przekonujących ani
powtarzalnych wyników.

Poszukiwano rzadkich nawet nieprawidłowości
chromosomalnych związanych z częstszym
występowaniem zaburzeń psychicznych o
obrazie schizofrenii.
Wiele ostatnich badań dotyczy delecji
chromosomu 22, przejawiającej się fenotypem
specyficznego wyglądu osoby, wad podniebienia
(najczęściej rozszczepem) i gardła, pnia
tętniczego, upośledzeniem umysłowym –
określonym jako zespół DiGeorge'a – z czego w
około 25% przypadków występowały psychozy
urojeniowe przypominające schizofrenię.

Przesłanki infekcyjno-immunologiczne
Podejrzenie wiąże się z częstym neurotropizmem
wirusów, możliwością ich wieloletniej,
bezobjawowej obecności w tkankach gospodarza,
inkorporacji jego genomu do komórek i
przekazywania następnym pokoleniom.
Ponadto choroby wirusowe mogą przebiegać w
postaci zaostrzeń i remisji, jak np. opryszczka –
gdzie wirus pozostaje w tkance osoby
zainfekowanej do końca życia i okresowo się
ujawnia, zaś pomiędzy epizodami opryszczki stan
zdrowia osoby prezentuje się jako prawidłowy:
choroba jest w remisji.

Dowodów na istnienie "schizowirusa"
obecnie nie przedstawiono, istnieją
jednakże nie odrzucone ostatecznie
podejrzenia dotyczące wirusów
neurotropowych (z grupy grypy czy
cytomegalii).

Przesłanki
neuromorfologiczne
Dotyczą odmienności budowy mózgu.
Przesłanki te badane są post mortem
oraz w przyżyciowych badaniach TK i
MR.
W badaniach post mortem stwierdzono, że
zmiany u osób chorych na schizofrenię
mają charakter raczej ogniskowy niż
rozlany i nie można mówić o jakimś
swoistym dla schizofrenii umiejscowieniu
zmian.

W przyżyciowych badaniach
obrazujących stwierdzono poszerzenie
komór bocznych w 75% badań, komory
III w 83% badań, poszerzenie przestrzeni
pomiędzy czaszką i mózgiem (tzw.
przestrzeń płynowa) w 63%. Większość
badań nad dynamiką zmian obrazu
mózgu w przebiegu choroby sugeruje, że
zmniejszenie objętości tkanki jest nie
postępujące i najprawdopodobniej
występuje już przed ujawnieniem się
choroby. Wyniki metaanaliz wskazują
przeciętne zmniejszenie objętości tkanki
mózgowej, choć u wielu stwierdza się
normę.

Przesłanki
neurofunkcjonalne
Najnowsza wersja hipotezy dopaminowej
głosi, że aktywność DA jest relatywnie
niska w okolicach czołowych, co pociąga
za sobą dysregulację DA w innych
częściach mózgu (układ limbiczny,
struktury podkorowe)
Aktualnie przypuszcza się, że układy DA i
5-HT wchodzą w niejasną obecnie
interakcję.
Zwrócono też uwagę na neuroprzekaźnik
GABA, oraz GLU.
Document Outline
- VII Patogeneza wybranych chorób układu nerwowego
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Limfocyty T
- Slide 7
- Infekcje wirusowe w rozwoju SM
- Witamina D
- Chlamydophila pneumoniae
- Geny
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Choroba Erba-Goldflama (MG)
- Slide 20
- Slide 21
- Patogeneza miastenii
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Typy miastenii wg Ossermana
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Zespoły miasteniczne wrodzone
- Zespoły presynaptyczne
- Slide 35
- Zespoły synaptyczne
- Zespoły postsynaptyczne
- Slide 38
- Zespół Lamberta-Eatona
- Zespoły miasteniczne polekowe
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Kryteria kliniczne to:
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- PZPD
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Przesłanki epidemiologiczne
- Przesłanki genetyczne
- Slide 63
- Przesłanki infekcyjno-immunologiczne
- Slide 65
- Przesłanki neuromorfologiczne
- Slide 67
- Przesłanki neurofunkcjonalne
Wyszukiwarka
Podobne podstrony:
11. INFEKCYJNE CHOROBY UKŁADU NERWOWEGO, FIZJOTERAPIA, Jednostki chorobowe
Choroby układu nerwowego ppt
SPECYFIKA CHORÓB UKŁADU NERWOWEGO OKRESU ROZWOJOWEGO
Choroby układu nerwowego
Ocena sprawności funkcjonalnej w chorobach układu nerwowego, Neurologia
Choroby układu nerwowego
CHOROBY UKLADU NERWOWEGO U DZIE Nieznany
Choroby układu nerwowego, Patologia i choroby
Choroby układu nerwowego, ZDROWIE-Medycyna naturalna, 01-Do uporządkowania, ZIOŁA
Choroby układu nerwowego, przydatne teksty (np. do szkoły)
CHOROBY UKŁADU NERWOWEGO KONI
Choroby układu nerwowego w ciąży
Choroby ukladu nerwowego 2
Stany nagłe w chorobach układu nerwowego, Medycyna Ratunkowa - Ratownictwo Medyczne
Choroby układu nerwowego 3
W1 Choroby układu nerwowego
Choroby układu nerwowego, Fizjoterapia, . fizjoterapia
27 Zastosowanie metod fizykoterapeutycznych w chorobach ukladu nerwowego (Osrodkowego i obwodowego
Choroby ukladu nerwowego w ciaz Nieznany
więcej podobnych podstron