Choroby układowe tkanki
łącznej
KOLAGENOZY

Etiopatogeneza
Wciąż nie jest w pełni poznana; uważa się, że autoimmunizacja,
czyli zaburzenie tolerancji autoimmunologicznej, związane jest
z wpływem czynników środowiskowych (wirusy, leki, toksyny,
promieniowanie UV), genetycznych (HLA-DR2, HLA-DR3,
wrodzone niedobory składowych dopełniacza- delecja genu dla
C4a), hormonalnych (ciąża, estrogeny) i metabolicznych
(endogenna hiperkortyzolemia)
Sugeruje się, że czynniki te mogą zmieniać specyficzne
mechanizmy zapewniające w organizmie autotolerancję, takie
jak: delecja klonalna- odpowiedzialna za apoptozę
autoreaktywnych limfocytów; anergia klonalna- czynnościowa
inaktywacja autoreaktywnych limfocytów; aktywna supresja
pod kontrolą limfocytów T supresorowych i przeciwciał
antyidiotypowych (Ab2)
Wytwarzane nadmiernie autoprzeciwciała to wyraz
poliklonalnej aktywacji limf.B i zaburzenia czynności limf.T!

Rozpoznanie
Powinno opierać się na ocenie całości danych z wywiadu,
badania przedmiotowego oraz badań laboratoryjnych
Należy wykluczyć choroby powodujące występowanie
podobnych objawów, np. przewlekłe zakażenia (gruźlica),
choroby nowotworowe narządów wewnętrznych, choroby
rozrostowe (białaczki, ziarnica, chłoniaki, szpiczak)
UWAGA: trudności diagnostyczne towarzyszą zwykle
początkowemu obrazowi choroby, w którym dominować
mogą nieswoiste objawy ogólne

Badania laboratoryjne
Wyniki mogą wskazywać na obecność procesu zapalnego!
W układowych chorobach tkanki łącznej stwierdza się:
wzrost OB, niedokrwistość (normo- lub niedobarwliwa, a
nieraz autoimmunohemolityczna), leukocytozę (eozynofilia
w guzkowym zapaleniu tętnic) lub leukopenię (toczeń
układowy), podwyższoną frakcję α1 i α2-globulin, a w
stanach przewlekłych również podwyższoną frakcję gamma-
globulin
Dla zapalenia charakterystyczna jest także obecność w
surowicy białek ostrej fazy (haptoglobina, ceruloplazmina,
fibrynogen, białko C-reaktywne,składowa C3 komplementu);
synteza tych białek przez kom.wątrobowe mediowana jest
przez cytokiny zapalne

Diagnostyka immunologiczna
Z praktycznego punktu widzenia opiera się na badaniu
obecności przeciwciał przeciwjądrowych (antinuclear
antibodies- ANA).
ANA są skierowane przeciwko różnym składowym jądra
komórkowego:
a)
antygenom związanym z chromatyną
b)
antygenom związanym z cząsteczkami RNA
c)
antygenom związanym z gr. heterogenną
! Przeciwciała mają znaczenie diagnostyczne tylko w
wysokim mianie, a nie przypisuje im się roli w samej
patogenezie choroby.
Należy pamiętać, że przeciwciała takie pojawiają się również u
ludzi zdrowych, szczególnie w wieku starczym i dlatego nie są
jedynym elementem rozpoznania.

Niektóre z ANA uważane są za markery zespołów chorobowych, w
których szczególnie często występują.
Na przykład, przeciwciała przeciw DNA obecne są u 75% chorych
na toczeń układowy, przeciwciała przeciwko dezoksyrybonukleidowi
(DNP) występują u 70% chorych na toczeń, przeciwciała przeciw
histonom u 80% chorych na toczeń indukowany lekami.
Oprócz ANA obserwuje się występowanie:
a) Przeciwciał przeciwko cytoplazmie granulocytów (antineutrophil cytoplasm
antibodies- ANCA)/przeciwciała C-ANCA związane z proteinazą 3 występują u
ok..100% chorych z ziarniniakiem Wegenera; przeciwciała antyleukocytarne
P-ANCA związane z mieloperoksydazą u chorych z r.z.s. i równoczesnym
zap.naczyń oraz w n.postaciach zap.nerek/
b) Przeciwciał przeciwko fosfolipidom (antiphospholipid antibodies- APA)- są to
IgG,M,A skierowane przeciw białkom związanym z ujemnie naładowanymi
fosfolipidami; należą do nich p. antykardiolipinowe (anticardiolipin
antibodies-ACA) oraz antykoagulant toczniowy (lupus anticoagulant-LA)/ APA
występują w z-le antyfosfolipidowym, charakteryzującym się obecnością obj.
Naczyniowych związanych z zakrzepicą żylną i tętniczą, trombocytopenią,
nawracającymi poronieniami i skórnymi zmianami martwiczymi; wtórnie taki
z-ł może wystąpić u chorych na toczeń/

Tu była grafika – Napisz do nas
by otrzymać pełną wersję

Toczeń rumieniowaty układowy (lupus
erythematosus systemicus, ang. systemic
lupus erythematosus- SLE)
Przewlekła zapalna choroba tkanki łącznej
dotycząca wielu narządów.
Rzadka u dzieci poniżej 5 roku życia i nieczęsta
poniżej 10 roku życia, występuje sporadycznie w
wieku dojrzewania, głównie u dziewcząt.
Częstość występowania: 15-50 przypadków na
100 tys. ludzi; dotyczy głównie młodych kobiet
(początek choroby przypada najczęściej na okres
między 20 a 40 rokiem życia), które stanowią od
60-80 % chorych, częściej u rasy czarnej;
częstość zachorowań większa u członków rodziny
chorego.
Obserwuje się objawy zaostrzeń i remisji!

Etiopatogeneza
Czynnik indukujący dotąd nieznany. Możliwa współzależność
między czynnikami środowiskowymi, genetycznymi i
hormonalnymi; zmiamy we wzajemnej iterakcji limfocytów mogą
być indukowane przez wirusy.
Predyspozycje genetyczne- rodziny z wrodzonym niedoborem
składowych dopełniacza, częstsza obecność antygenów zgodności
tkankowej HLA-DR2 i HLA-DR3 u chorych.
!Utrata tolerancji wobec własnych antygenów- autoimmunizacji
może towarzyszyć genetyczna predyspozycja do produkcji
autoprzeciwciał oraz zwiększona reaktywność limfocytów B i
zaburzenia czynności limfocytów T.
U chorych stwierdzamy: spadek ilości limfocytów T, głównie CD8+,
spadek wytwarzania IL-2, wzrost ilości limfocytów B.
Estrogeny mogą modulować pojawienie się tocznia- ułatwiają
aktywację limfocytów B.
Promieniowanie UV- indukcja uwolnienia keratynocytów skórnych
IL-1, wpływającej na zaburzenia aktywacji limfocytów B.

Czynnikami uszkadzającymi w SLE są przeciwciała
i kompleksy antygen-przeciwciało.
Krążące kompleksy odkładane są w ścianie naczyń
i kłębuszkach nerkowych, prowadząc do
uszkodzenia tych tkanek (w procesie odkładania w
tkankach mają znaczenie: wielkość,
rozpuszczalność, stężenie, zdolność wiązania
składowych dopełniacza ze strony kompleksów, a
ze strony układu siateczkowo- śródbłonkowego-
zdolność usuwania kompleksów z krążenia.
Można stwierdzić obecność przeciwciał przeciw
erytrocytom, leukocytom, płytkom, co powoduje
spadek liczby tych komórek.

Zmiany histopatologiczne.
Martwica włóknikowa- kompleksy
składające się z DNA, przeciwciał
przeciwko DNA i dopełniacza w ścianie
naczyń i tkance łącznej.
Ciałka hematoksylinowe: w miejscu
uszkodzenia tkanki łącznej występuje
zawartość jądra komórkowego, komórki LE
są to granulocyty, które pochłaniają in vitro
ciałka hematoksylinowe.
Koncentrycznie odkładający się kolagen.
Wokół tętnic śledziony na kształt cebuli.

Tu była grafika – Napisz do nas
by otrzymać pełną wersję

Objawy kliniczne- RÓŻNORODNOŚĆ,
ZMIENNE NASILENIE, OKRESY
REMISJI I NAWROTÓW.
Skóra: typowa wysypka na twarzy z zajęciem
policzków i nosa, o kształcie motyla u 50%
chorych, nasilająca się pod wpływem prom.
słonecznego; obecność rumienia
obrączkowatego; możliwe zmiany o typie
pęcherzy i wysypka grudkowo-plamista
(przypomina wysypkę uczuleniową); łysienie
plackowate- dotyczy 70% chorych; możliwy
rumień dłoni oraz zapalenie naczyń skórnych
prowadzące do owrzodzeń.

Tu była grafika – Napisz do nas
by otrzymać pełną wersję

Jama ustna: niebolesne owrzodzenia błony śluzowej
gł.podniebienia, mogą ponadto pojawić się w przegrodzie
nosowej, powodując czasem jej przebicie z krwawieniem.
Układ ruchu: zajęty u 80% chorych; symetryczne zapalenie
stawów międzypaliczkowych bliższych z towarzyszącą
bolesnością! i obrzękiem; zapalenie stawów odróżnieniu od
RZS nie prowadzi do nadżerek na ich powierzchni i rzadko
powoduje zniekształcenia; sztywność poranna, bóle mięśniowe.
Serce: zmiany u 20% chorych; zapalenie osierdzia, mięśnia
sercowego, wsierdzia oraz naczyń wieńcowych. Możliwe
wystąpienie tarcia osierdzia, tamponady serca, zgrubienia
osierdzia, tachykardii, zaburzeń przewodnictwa, objawów
niedokrwiennych i zmian zastawkowych; !charakterystyczne
dla tocznia jest niebakteryjne zapalenie wsierdzia typu
Libmana- Sachsa- najczęściej u chorych z obecnością
przeciwciał antyfosfolipidowych.

Tu była grafika – Napisz do nas
by otrzymać pełną wersję

Płuca: u 30%- zapalenie wysiękowe opłucnej; „zespół
kurczącego się płuca”, jako wynik zmian zapalnych w
mięśniach międzyżebrowych i przeponie; toczniowe
zapalenie płuc- nacieki w tkance śródmiąższowej mogące
powodować duszność i krwioplucie.
Nerki: !zmiany u większości pacjentów: mezangialne,
ogniskowe, rozplemowe oraz błoniaste zapalenie kłębuszków.
Układ nerwowy: zaburzenia u 50% chorych; obwodowe
porażenia nerwów, o typie czuciowo-ruchowym; zapalenie
nerwów czaszkowych z porażeniem n. twarzowego;
zapalenie n. wzrokowego/ są wynikiem zapalenia naczyń
nerwów/; depresje, psychozy, zaburzenia ukrwienia, napady
padaczkowe, porażenia połowicze.
Przewód pokarmowy: o jego zajęciu świadczą bóle brzucha,
nudności czy biegunki, przyczyną dolegliwości może być
zapalenie naczyń prowadzące do owrzodzeń martwiczych
jelit, zawału, perforacji i krwawień.

Badania laboratoryjne
Hematologiczne: obraz niedokrwistości hemolitycznej, normocytowej
normochromicznej (upośledzenie erytropoezy), leukopenia<4tys./mm3,
limfocytopenia i granulocytopenia, trombocytopenia, wzrost OB.
Układ krzepnięcia: wydłużenie czasu kaolinowo-kefalinowego w wyniku
obecności przeciwciał antyfosfolipidowych (antykoagulant toczniowy); !
pomimo wydłużenia t krzepnięcia paradoksalnie stwierdza się zwiększoną
tendencję do zakrzepicy tętniczej i żylnej.
Serologiczne: dodatnie odczyny kiłowe (bo obecne przeciwciała
antyfosfolipidowe)
Immunologiczne: niskie stęż. C3 i C4 dopełniacza, hipergammaglobulinemia,
obecność autoprzeciwciał- najbardziej charakterystyczne są:
a) przeciwciała przeciwko podwójnemu łańcuchowi DNA (dsDNA) wykrywane w
aktywnym procesie chorobowym, dają obwodowy typ świecenia.
b) przeciwciała przeciw histonom- homogenny lub rozlany typ świecenia
c) przeciwciała przeciwko małym, jądrowym rybonukleoproteinom:
- przeciw antygenowi Sm (Smith): !typowe dla tocznia
- przeciwciało reagujące z U1RNP: w toczniu i mieszanej chorobie tkanki
łącznej
Oba dają plamisty typ świecenia.
- przeciwko antygenowi Ro składowej snRNP: !charakterystyczne dla tocznia
ze zmianami skórnymi i nadwrażliwością na światło, też w z-le Siogrena
Leczenie
Uwzględnić przewlekły charakter schorzenia i informować
chorych o czynnikach nasilających jego przebieg (prom.
słoneczne, stres, n. leki, np. sulfonamidy) oraz przeciwdziałanie
zaostrzeniom.
Stosuje się NLPZ, leki przeciwmalaryczne, glikokortykosteroidy
(podstawowe leki w leczeniu tocznia o ciężkim przebiegu i
utrzymujących się objawach, wskazaniem do ich włączenia są:
hemoliza, tombocytopenia, zap .śródmiąższowe płuc, zab. OUN
i ObwUN, zmiany w sercu i mm. szkieletowych, zmieny w
nerkach i zapalenie naczyń), cytostatyki- w opornych na
leczenie objawach tocznia, szczególnie ze zmianami w
nerkach, cyklosporyna- gdy przebieg tocznia ciężki i oporność
na sterydy, plazmafereza- w celu usunięcia przeciwciał i
kompleksów gł. w zaburzeniach krzepnięcia , niedokrwistości i
zmianach w OUM.
!!!Najczęstsze przyczyny zgonu w toczniu to powikłania w
przebiegu zmian w NERKACH oraz w przebiegu zakażeń
(długotrwałe stosowanie sterydów).

Tu była grafika – Napisz do nas
by otrzymać pełną wersję

Toczeń polekowy (Lupus
postmedicamentosus)
Niektóre leki przyjmowane przez dłuższy czas
mogą prowadzić do wystąpienia objawów
typowych dla tocznia rumieniowatego (najczęściej
hydralazyna, ale również Li, fenytoina,
sulfonamidy, monocyklina, karbamazepina).
Przebieg tej postaci tocznia jest zwykle
łagodniejszy od tocznia rumieniowatego.
Rzadko wymaga włączenia do leczenia
glikokortykoidów.

Neonatal LE
Newborn babies born to mothers with
subacute LE may develop a temporary
ring-like or annular rash, known as
neonatal LE. Although the rash clears
within a few months, the baby is at risk of
congenital heart block. A paediatrician
should assess all babies born to mothers
with subacute LE (or carrying the antibody
for this condition) at birth.

Twardzina układowa (Sclerodermia
systemica)
Charakteryzuje się zmianami zapalnymi, naczyniowymi,
zwłóknieniem gł. okolicy koniuszków palców oraz zajęciem
wielu narządów.
Istnieje również postać ograniczona twardziny, w której
zmiany dotyczą skóry i tk. podskórnej z tym, że bez zajęcia
skóry palców i bez objawów Raynauda i zmian w narządach
(sclerodermia circumscripta, morphea).
Występuje rzadko, rocznie rozpoznaje się 5-10 nowych
zachorowań na 1 mln mieszkańców.
3-4 razy częściej chorują kobiety.
Pierwsze objawy pojawiają się między 30. a 50. r.ż.

Etiopatogeneza
Nieznana
Zwraca się uwagę na udział czynników środowiskowych
(toksyny, wirusy), które miałyby przez uszkodzenie śródbłonka
prowadzić do odpowiedzi immunolog., zapalenia i włóknienia.
Zespoły zwłóknienia twardzinopodobne opisano u chorych
narażonych na działanie PVC, żywic epoksydowych, silikonu,
zanieczyszczonego oleju rzepakowego oraz po spożyciu L-
tryptofanu w z-le eozynofilia-mialgia (EMS).
Dochodzi do uszkodzenia śródbłonka (czynnik środowiskowy,
immunolog.) z następowym postępującym włóknieniem skóry
i narządów; w małych naczyniach skóry i narządów
wewnętrznych stwierdza się rozrost bł.podstawnej powodujący
zwężenie światła naczyń,a w konsekwencji niedokrwienie
tkanek.

Zwłóknienie tkanek jest następstwem niedokrwienia i
toczącego się procesu immunolog. wywołującego wzrost
aktywności fibroblastów.
Obecne komórki zapalne uwalniają cytokiny i czynniki
wzrostu, prowadząc z kolei do nasilenia uwalniania
kolagenu, substancji podstawowej przez fibroblasty i
rozwoju włóknienia

Obraz kliniczny
Początkowe objawy twardziny układowej mogą wystąpić pod
postacią: *obj. Raynauda,*nieznacznego obrzęku dystalnych
odc. kończyn,*stopniowego pogrubienia skóry palców.
Wcześnie pojawiają się objawy ze strony małych stawów
(symetryczne bóle i okresowa sztywność).
Czasami pierwszymi objawami są dolegliwości gastryczne
(dysfagia, zgaga) bądź oddechowe (duszność).
Skóra: zajęta u 90% pacjentów; stwardnienie jest symetryczne
i może być ograniczone do skóry palców (SKLERODAKTYLIA) i
części dystalnych kończyn górnych albo może obejmowac
prawie całe ciało; ! Najczęściej zmiany pojawiają się
dystalnie,stopniowo przechodzą na ramiona, tułów i szyję;
skóra jest napięta, błyszcząca i nadmiernie pigmentowana,
trwale połączona z tkanką podskórną; twarz przybiera wygląd
maskowaty; naskórek ulega ścieńczeniu, zanikają bruzdy
skórne, gruczoły i włosy. Po kilku latach- zaniki skóry,
teleangiektazje na klp, twarzy, wargach i języku oraz
owrzodzenia atroficzne.

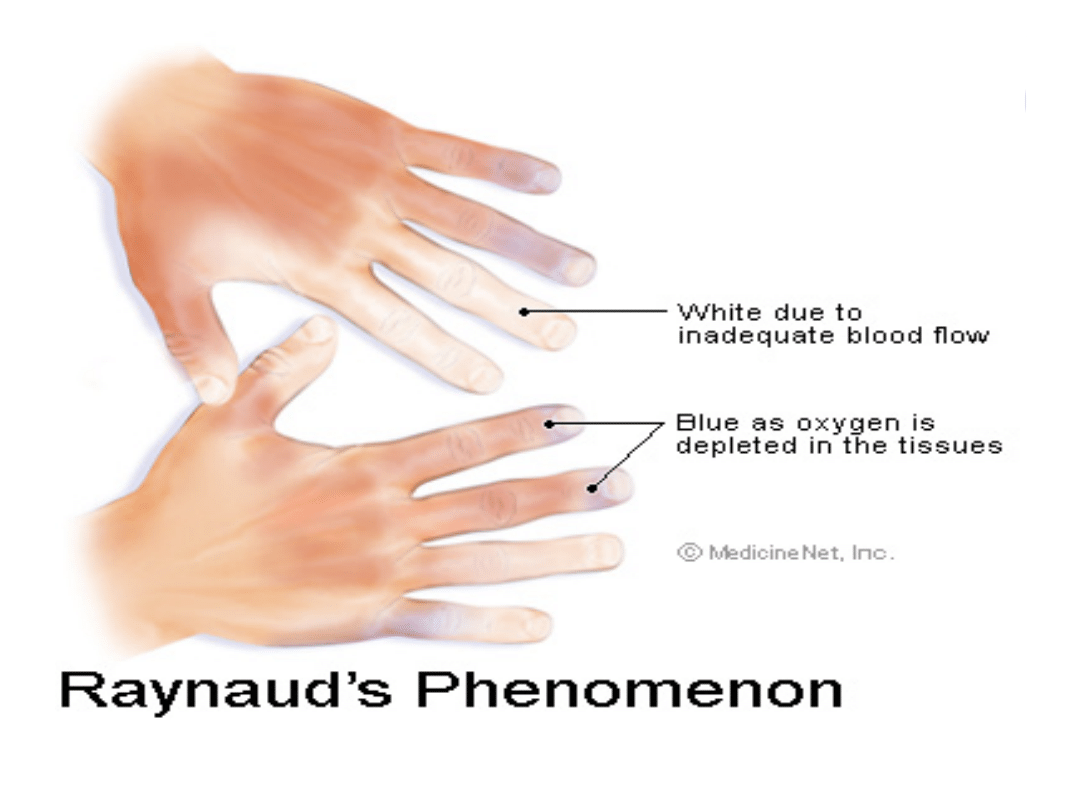
Objaw Raynauda: u 95% chorych; jest to napadowy skurcz naczyń pod
wpływem zimna lub stresu, spadek przepływu krwi wywołuje
zblednięcie palców lub sinicę z towarzyszącym zdrętwieniem i
bolesnością, powrót ukrwienia powoduje zaczerwienienie.
Układ mięśniowo-kostny: włóknienie struktur okołostawowych powoduje
zapalenie pochewek ścięgnistych, zgięciowy przykurcz palców,
nadgarstków i łokci oraz osłabienie siły mięśniowej z ich zanikiem.
!!! U większości chorych najczęstszym obj. zajęcia narządów jest
upośledzenie funkcji przełyku: często występuje dysfagia, refluks
spowodowany niewydolnością dolnego zwieracza przełyku oraz
zapalenie przełyku, prowadzące zwykle do owrzodzeń błony śluzowej i
zwężeń./w badaniu stwierdza się usztywnienie ścian przełyku i brak
czynności motorycznej oraz zmniejszoną kurczliwość jego dolnego
zwieracza, obecność uchyłków i owrzodzeń; u 30% chorych stwierdza
się metaplazję Barretta, która może dpprowadzić do zwężenia i
gruczolakoraka. Błona śluzowa żołądka zanika i stwierdza się
upośledzenie opróżniania żołądka, zmniejszenie perystaltyki jelit
ułatwia rozwój bakterii beztlenowych,…

Tu była grafika – Napisz do nas
by otrzymać pełną wersję

upośledzający trawienie i wchłanianie pokarmu/ w badaniu
jelita grubego- poszerzenia i uchyłkowatość/
Jama ustna: !zanik warg, ograniczający otwieranie ust;
zanik brodawek językowych; ubytki w uzębieniu;
rozszerzenie drobnych naczyń na śluzówkach
Układ krążeniowo-oddechowy: włóknienie śródmiąższowe
płuc, zapalenie opłucnej i osierdzia z obecnością wysięku;
nadciśnienie płucne jest wynikiem włóknienia płuc albo
przerostu błony wewnętrznej małych tętnic płucnych
(CREST); zmiany w sercu- włóknienie mięśnia, zaburzenia
rytmu- prowadzić mogą do niewydolności krążenia opornej
na działanie naparstnicy.
Nerki: przerost błony wewn. małych tętnic nerkowych z
następowym rozwojem złośliwego nadciśnienia- nieleczone
szybko prowadzi do niewydolności nerek i śmierci.
Postacie kliniczne twardziny
Rozróżnienie postaci układowej i ograniczonej opiera się na
stwierdzeniu zmian narządowych.
TWARDZINA UKŁADOWA POSTĘPUJĄCA: ze zmianami
skórnymi dotyczącymi odcinków bliższych kończyn górnych
(do stawów śródręczno-paliczkowych) oraz szybkim
rozwojem zmian w narządach; w tej postaci częściej
dochodzi do zajęcia nerek i włóknienia płuc, co wpływa na
gorsze rokowanie.
TWARDZINA OGRANICZONA: dotycząca jedynie skóry
odcinków dystalnych kończyn oraz twarzy; wyróżnia się
dwie zasadnicze odmiany tej postaci: PLACKOWATĄ
(morphea) i LINIJNĄ (sclerodermia linearis); przebieg jest
powolny, u części chorych po wielu latach może rozwinąć
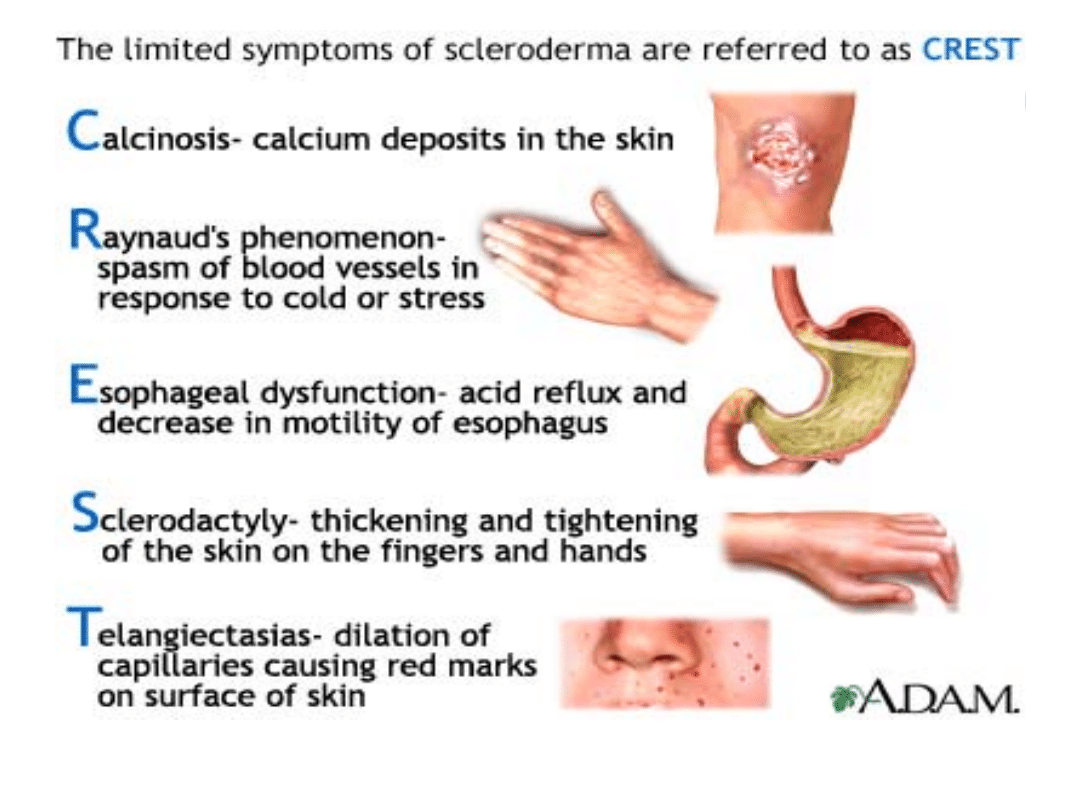
się pełnia charakterystycznych objawów- zespół
CREST

Tu była grafika – Napisz do nas
by otrzymać pełną wersję


Tu była grafika – Napisz do nas
by otrzymać pełną wersję

Mieszana choroba tkanki łącznej
(MCTD= Mixed Connective Tissue
Disease)= zespół nakładania=
zespół Sharpa
Współistnienie zmian typowych dla:
1)
tocznia rumieniowatego układowego
2)
postępującej twardziny układowej
3)
zapalenia wielomięśniowego
Pojawić się mogą także objawy RZS i zespołu Sjogrena.
!u większości pacjentów z czasem dochodzi do rozwoju
pełnoobjawowej twardziny lub tocznia
W zespole tym może wystąpić objaw Raynaud, bóle i obrzęki
dłoni.
Stwierdza się obecność przeciwciał U1RNP w wysokim mianie.
W leczeniu stosuje się przewlekle glokokortykosteroidy.

Rozpoznanie
Pojawienie się nieswoistego objawu Raynaud (występuje też w
innych chorobach tkanki łącznej) może na kilka lat poprzedzać
wystąpienie innych objawów twardziny.
W badaniach laboratoryjnych: częsta obecność przeciwciał
przeciwjądrowych anty-Scl-70 w postaci układowej i przeciwciał
przeciwko centromerom w postaci ograniczonej (CREST).
Należy różnicować ze stanami rzekomotwardzinowymi,
eozynofilowym zapaleniem powięzi, zespołem eozynofilia-mialgia
(EMS).
Leczenie: !brak skutecznej metody
Uważa się, że penicylaminą można hamować włóknienie skóry i płuc;
próbuje się stosować kolchicynę, która zapobiega ostrym zapalnym
zespołom bólowym z odkładaniem soli Ca w tkance podskórnej;
wystąpieniu objawu Raynaud zapobiega się unikając kontaktu z
zimnem, zaprzestanie palenia i stosowanie antagonistów kanału
wapniowego; refluks żołądkowo-przełykowy wymaga stosowania
leków osłaniających błonę śluzową, blokerów receptorów H2 i pompy
protonowej; glukokortykoidy; NLPZ-w bólach stawów

Zespół Sjögrena (dry eye
syndrome)
Charakteryzuje się objawami suchości w jamie ustnej (xerostomia)
i brakiem wydzielania łez (keratoconiunctivitis sicca).
Znacznie częściej choroba dotyczy kobiet, głónie w wieku 45-55
lat.
PIERWOTNY ZESPÓŁ SJOGRENA: obecność zmian narządowych i
zaburzenia czynności gruczołów wydzielania zewnętrznego;
wykazuje związek z antygenem HLA-DR3; występuje suchość
skóry, pochwy, objaw Raynauda, plamica, nawracające zakażenia i
śródmiąższowe włóknienie płuc; w jamie ustnej: zapalenie
kącików, kandydiaza, ciemnoczerwony ze szczelinowatymi
pęknięciami język, gwałtownie postępujące zmiany próchnicze,
częste wypadanie wypełnień; chorzy skarżą się na trudności w
żuciu i połykaniu, zaburzenia odczuwania smaku i zapachu,
nawracające zapalenia przyusznic; częściej stwierdza się u nich
zanikowe zapalenie żołądka, przewlekłe aktywne zapalenie
wątroby, marskość żółciową, zapalenie trzustki;…

Niedobór wydzielania łez daje uczucie obecnośći piasku pod
powiekami, pieczenia i swędzenia oczu; wystąpić mogą także
zapalenia mięśni, neuropatie obwodowe, przewlekłe zapalenie
tarczycy, splenomegalia z neutropenią, niedokrwistość
normochromiczna oraz częstsze występowanie chłoniaków( non-
Hodgkin 40 razy częściej).
WTÓRNY ZESPÓŁ SJOGRENA: !pojawia się w przebiegu innej układowej
choroby autoimmunologicznej (np. RZS, tocznia, twardziny); częściej
występuje u osób z obecnym antygenem HLA-DR4; objawy dotyczą
głównie zaburzeń czynności gruczołów łzowych i ślinowych.
ROZPOZNANIE: oparte na objawach suchości w obrębie spojówek,
błony śluzowej jamy ustnej i powiększenia ślinianek; pomocne jest
badanie okulistyczne w lampie szczelinowej i wykonanie testu
Schirmera (pasek bibuły umieszczony w worku spojówkowym po 5
minutach powinien zawierać wilgotną plamę o średnicy 5 mm); w
surowicy często obecne są przeciwciała przeciwjądrowe.
LECZENIE: higiena jamy ustnej!; unikanie leków powodujących
suchość jamy ustnej, częste picie wody, stosowanie sztucznej śliny;
częste zakraplanie sztucznych łez (metyloceluloza) oraz stosowanie
wilgotnej komory szczególnie podczas snu ma chronić rogówkę przed
wysychaniem; unikać stosowania sterydów do oczu;zmiany
narządowe wymagają stosowania glikokortykosteroidów i leków
immunosupresynych.

Obraz kliniczny
Początek choroby jest zwykle ostry u dzieci, a u dorosłych bardziej podstępny.
Wystąpienie pierwszych objawów może być poprzedzone ostrą infekcją.
Osłabienie mięśni: rozwija się zwykle powoli; występują trudności z wchodzeniem
po schodach, wstawaniem, podnoszeniem przedmiotów czy czesaniem włosów; !
osłabienie mięśni obręczy barkowej lub miednicy w zaawansowanych postaciach
choroby zmusza do korzystania z wózka inwalidzkiego; częste zajęcie mięśni
krtani i przełyku powoduje zaburzenia artykulacji i połykania; trudność z
podnoszeniem głowy z poduszki jest skutkiem zmian zapalnych mięśni szyi; może
dojść do zajęcia mięśni oddechowych, co nieraz prowadzi do niewydolności
oddechowej; postępujące procesy zapalne prowadzą do zaników mięśniowych,
charakteryzujących się zachowaniem czucia i prawidłowych lub upośledzonych
odruchów ścięgnistych.
Zmiany skórne: w zapaleniu skórno-mięśniowym mogą czasami wyprzedzać
objawy zapalenia mięśni; wykwity skórne są śniade i rumieniowate, mają
tendencję do tworzenia na twarzy zmian przypominających kształtem motyla (!
jak w toczniu); typowe są obrzęki o odcieniu fioletowym wokół oczu oraz
wyniesiona ponad powierzchnię skóry i złuszczająca się wysypka na czole (objaw
Gottrona), szyi, klatce, przedramionach, podudziach, łokciach i kolanach, okolicy
stawów śródręczno-paliczkowych i kostek przyśrodkowych; objaw
zaczerwienionej, chropowatej skóry i hipertroficzne zmiany na powierzchni
dłoniowej rąk i palców to tzw. ręce mechanika; objaw szala to obecność zmian
rómieniowatych na szyi; typowe jest pogrubienie płytek paznokciowych z
obecnością ciemnych poprzecznych linii.

Tu była grafika – Napisz do nas
by otrzymać pełną wersję

Zapalenie wielomięśniowe
(polymyositis) i skórno-mięśniowe
(dermatomyositis)
Zapalenie wielomięśniowe charakteryzuje się zmianami zapalnymi i
zwyrodnieniowymi w mięśniach oraz często również w skórze
(zapalenie skórno-mięśniowe) które prowadzą do symetrycznego
osłabienia mięśni i nieraz ich zaniku, szczególnie w zakresie obręczy
barkowej i miednicy; objawy czasem mogą przypominać twardzinę,
toczeń czy zapalenie naczyń.
Etiologia jest nieznana; w naciekach obecnych w zapaleniu mięśni
dominują limfocyty B, wśród limfocytów T przeważają pomocnicze
nad supresorowymi; w śródbłonku naczyń odkładane złogi składnika
C3 dopełniacza i przeciwciał klasy IgM i IgG uważa się za przyczynę
rozwoju zapalenia naczyń towarzyszącego zapaleniu mięśni; rolę
odgrywają niewątpliwie autoprzeciwciała skierowane przeciwko
różnym składnikom mięśni.
Zapalenie wielomięśniowe występuje rzadziej niż toczeń i twardzina (1
na 200 tysięcy osób) ale częściej niż guzkowe zapalenie naczyń;
dwukrotnie częściej choroba dotyczy kobiet i pojawia się najczęściej
między 40. a 60. r. życia, a w przypadku dzieci między 5. a 15. r.
życia.

Zmiany w narządach: oprócz krtani i przełyku dotyczą głównie płuc i
serca (śródmiąższowe zapalenie płuc, niewydolność oddechowa,
kardiomiopatia z zastoinową niewydolnością krążenia, zaburzenia
rytmu i przewodnictwa).
Z dermatomyositis często współistnieją nowotwory złośliwe głównie
piersi, żołądka, jajnika i płuc.
MŁODZIEŃCZE ZAPALENIE SKÓRNO-MIĘŚNIOWE: u dzieci ta postać
zapalenia mięśni występuje częściej niż zapalenie wielomięśniowe; w
późnej fazie choroby obserwujemy przykurcze spowodowane złogami
wapnia w uszkodzonych mięśniach; obecność zmian zapalnych w
naczyniach prowadzić może do niedokrwienia w zakresie mięśni, skóry i
jelit.
Swoistymi przeciwciałami dla zapalenia wielomięśniowego jest
przeciwciało przeciwko syntetazie oraz przeciwciało Jo-1 (przeciw
syntetazie histydylo-tRNA), przeciw cząsteczce rozpoznającej sygnał
(SRP) i przeciw Mi-2 (białko jądra komórkowego)
Leczenie: na początku glikokorykosteroidy, następnie dawkę
zmniejszamy kierując się poprawą siły mięśniowej i spadkiem
aktywności enzymów mięśniowych, przy braku poprawy po 3
miesiącach leczenia sterydami rozważyć trzeba podanie dodatkowo
leków immunosupresyjnych; inhibitory konwertazy angiotensyny w
leczeniu nadciśnienia.

Zapalenie naczyń (Vasculitis)
Zmiany zapalne ściany naczyń, którym często towarzyszy
martwica tkanek, występować mogą w różnych
kolagenozach. Zapalenie naczyń dotyczy głównie tętnic.
Dla wielu postaci zapalenia naczyń wspólne są zmiany
histopatologiczne; w poszczególnych zespołach można
wykazać zaburzenia odpowiedzi humoralnej i komórkowej;
w ścianie naczyń odkładać się mogą rozpuszczalne
kompleksy immunologiczne, które, wiążącskładowe
dopełniacza, wykazują aktywność chemotaktyczną dla
komórek zapalnych niszczących ścianę naczynia; w
zespołach chorobowych przebiegających z tworzeniem
ziarniniaków decydującą rolę odgrywają mechanizmy
komórkowe z udziałem limfocytów T klasy CD4+.

Zespoły kliniczne
Alergiczne zapalenie naczyń:
a)
Plamica Schönleina-Henocha: dotyczy małych naczyń,
występuje u dzieci po przebytym paciorkowcowym zapaleniu
gardła; typowe zmiany to plamica skóry, obrzęki i bóle stawów,
bóle brzucha lub krwawienia z przewodu pokarmowego oraz
kłębuszkowe zapalenie nerek; w biopsji skóry stwierdzamy
złogi IgA w zmienionych naczyniach; leczenie
glikokortykosteroidami.
b)
Choroba posurowicza: zapalenie naczyń poprzedzone
ekspozycją na leki (penicylina, sulfonamidy) lub
obcogatunkowe białko.
c)
Samoistna, mieszana krioglobulinemia: wytrącanie się
kompleksów pod wpływem zimna; poza zmianami skórnymi,
stawowymi i kłębuszkowym zapaleniem nerek możliwy objaw
Raynauda.

Guzkowe zapalenie tętnic: najczęściej między 40. a 50.
rokiem życia.
Inne: choroba Churga-Straussa, ziarniniak Wegenera,
choroba Takayasu, olbrzymiokomórkowe zapalenie tętnic.

Tu była grafika – Napisz do nas
by otrzymać pełną wersję
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
Wyszukiwarka
Podobne podstrony:
Choroby układowe tkanki łącznej
CHOROBY UKŁADOWE TKANKI ŁĄCZNEJ, INTERNA, reumatologia
Choroby układowe tk łącznej pediatria
Choroby kolagenowe tkanki łacznej
Układowe choroby tkanki łącznej, Pediatria
Zajęcie układu oddechowego w przebiegu układowych chorób tkanki łącznej, INTERNA, reumatologia
Zmiany w ukł odd w przebiegu układowych chorób tkanki łącznej, INTERNA, reumatologia
Układowe choroby tkanki łącznej
choroby tkanki lacznej
Diagnostyka chorob tkanki lacznej studenci ppt
Przewlekłe choroby tkanki łącznej, MEDYCYNA i RATOWNICTWO, Pediatria
Diagnostyka chorób tkanki łącznej, dermatologia
Kolagenozy - choroby tkanki łącznej, Dermatologia
wrodzone choroby tkanki łącznej(w tym z ł Ehlersa Danlosa)
Immunopatogeneza chorób tkanki łącznej
więcej podobnych podstron