
Journal of Basic Microbiology 2012, 52, 27 – 34
27
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Research Paper
Simultaneous analysis of foodborne pathogenic bacteria
by an oligonucleotide microarray assay
Yushan Hu, Junhua
Liu, Dan Xia and Shouyi Chen
The Center for Disease Control and Prevention of Guangzhou, Guangzhou, China
A rapid and accurate method for simultaneous identification of foodborne infectious patho-
gens was developed based on oligonucleotide microarray technology. The proposed identifi-
cation method is based on PCR amplification of the target region of the groEL genes with de-
generate primers, followed by the PCR products hybridization with oligonucleotide probes
specific for species. The groEL gene amplification products of seventeen species of pathogenic
bacteria were hybridized to the oligonucleotide array. Hybridization results were analyzed
with digoxigenin-linked enzyme reaction. Results indicated that fifteen species of pathogenic
bacteria showed high sensitivity and specificity for the oligonucleotide array, while two other
species gave cross-reaction with the E. coli. Our results
suggested that microarray analysis of
foodborne infectious pathogens
might be very useful for simultaneous identification of bacte-
rial
pathogens. The oligonucleotide array can also be applied to samples collected in clinical
settings of foodborne infections. The superiority of oligonucleotide array over other tests lies
on its rapidity, accuracy and efficiency in the diagnosis, treatment and control of foodborne
infections.
Keywords: groEL / Foodborne infection / Oligonucleotide microarray
Received: November 16, 2010; accepted: March 07, 2011
DOI 10.1002/jobm.201000458
Introduction
*
Foodborne diseases, caused by consuming contaminat-
ed foods or beverages, are worldwide serious public
health problem. Annually, thousands cases of out-
breaks of foodborne diseases occur around the world [1,
4]. Although natural toxins, parasites and poisonous
chemicals also cause foodborne diseases, the patho-
genic bacteria are the major cause of foodborne dis-
eases [2, 18, 19]. In Guangzhou, China, the species of
bacteria causing foodborne infections have become
more and more diversified and the Salmonella is the
most common pathogen.
Foodborne infections often involve quite a lot of in-
dividual and can spread rapidly in schools, factories
and other institutions [10, 30]. Microbial pathogens are
conventionally identified
by surrogate biochemical and
Correspondence: Yushan Hu, The Center for Disease Control and Pre-
vention of Guangzhou, Guangzhou, Qide Road, Jiahe Guangshou,
510440, China
E-mail: huyushan1976@gmail.com
Phone: +862083828291
Fax: +862083822400
immunological markers. These
conventional appro-
aches are well established but often time-consuming,
culture-based, and have room for improvement in
terms of sensitivity and precision [2, 5]. Besides, these
traditional methods are impractical for the detection
and identification of a large group of related bacteria
with significant antigenic similarities. In addition,
many foodborne infectious bacteria can alter their bio-
logical characters, such as colony formation and anti-
gencity [3]. These changes often make conventional
detection methods inefficient. In order to control infec-
tious diseases effectively, it is important to identify and
detect pathogens rapidly. Therefore, there is still a need
for a rapid and specific method for simultaneous detec-
tion and identification of foodborne infection bacteria
for diagnostic and epidemiological purposes. Microar-
ray technology has great potential in diagnostic micro-
biology [7–9, 14, 16]. To select a common gene frag-
ment for microarray-based identification of foodborne
infection pathogens, such gene must contain conserved
regions common to these pathogens, and on the other
hand sufficient sequence diversity for species identifi-

28 Y.
Hu
et al.
Journal of Basic Microbiology 2012, 52, 27 – 34
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
cation. The groEL gene, which encodes a 60 kDa sub-
unit, is highly conserved and mainly exists in bacteria
and eukaryotic cells [6, 11–13, 20]. Despite the conserv-
ed nature of the groEL gene, the level of interspecies
variation in groEL sequence is quite high, and thus pro-
vides suitable resolution for bacteria identification [15,
17, 21].
In this study, we described a rapid and reliable mi-
croarray-based assay for simultaneous detection and
identification of foodborne infectious pathogens. The
method includes PCR amplification of part of the groEL
gene with universal primers, followed by analysis of
amplicons by hybridization with specific -oligonucleo-
tide probes immobilized on the microarray.
Materials and methods
Bacterial strains
In total, seventeen common bacteria species causing
foodborne infections were selected in our studies which
were shown in Table 1. These included Escherichia coli,
Campylobacter jejuni, Vibrio cholerae, Vibrio parahaemolyti-
cus, Vibrio alginolyticus, Staphylococcus aureus, Streptococcus
hemolyticus, Yersinia enterocolitica, Proteus vulgaris, Bacillus
cereus, Salmonella enterica, Salmonella typhimurium, Listeria
monocytogenes, Shigella dysenteriae, Shigella flexneri, Clostrid-
ium perfringens and Clostridium botulinum. Streptococcus
pneumonia, Klebsiella pneumoniae and Neisseria gonorrhoeae
were chosen as control species which are unrelated to
foodborne infections. These standard bacterial species
were stored and revived in culture for 18–24 h in the
laboratory, then transferred to suitable media and cul-
tivated for 18–24 h. They were identified by conven-
tional methods and by the API test system (bioMerieux,
France). Pure cultures were diluted as foodborne infec-
tion mock samples. Each species of bacterium was di-
luted from 10
6
to 1 cfu/ml. Mock sample no. 1 contains
bacteria species of E. coli, mock sample no. 2 contains
bacteria species of S. enterica, mock sample no. 3 con-
tains bacteria species of L. monocytogenes, mock sample
no. 4 contains bacteria species of C. jejuni, mock sample
no. 5 contains bacteria species of V. parahaemolyticus,
mock sample no. 6 contains bacteria species of P. vul-
garis. Fifty foodborne infectious samples were collected
from Guangzhou center for disease control and preven-
tion between June 2000 and February 2009.
Total DNA preparation
DNA was extracted from freshly grown bacterial cells
by phenol-chloroform extraction. The presence, con-
centration, and purity of genomic DNA in the prepared
samples were detected by measuring the absorbances at
260 and 280 nm with an Ultraspec 3000 spectropho-
tometer (Pharmacia, Peapack, N.J.).
PCR primers and oligonucleotide probes
groEL gene sequences of seventeen foodborne pathogens
together with other twenty-six species (genera) of bac-
teria were retrieved from the GenBank sequence li-
brary. Homology was analyzed using CLUSTAL W soft-
ware (Version 1.5). A 600 bp mutation fragment was
selected as detection target and a PCR primer pair was
designed from the conservation region of the both ends
of the fragment with Primer Premier Software (Version
5.0). The designed primer pair was verified to amplify
the groEL gene fragment of all target bacterial species.
The primer pairs were as follows: P1:-5′AGTTACCCT
XGG YCCZ AAAG-3′, X is C or T, Y is T or C, Z is A or G;
P2: -5′CAGCAACCACGCCTTCTTC-3′. The expected length
of the PCR product was about 600 bp. Digoxigenin was
incorporated to 5′-end of P2 primer for color develop-
ment. Twenty-three oligonucleotide probes were se-
lected from the target region of different species, re-
spectively (Table 1). All primer pair and probes were
synthesized in Takara Bio.
PCR amplification of bacteria groEL gene fragment
The PCR amplification was performed in 50 μl reaction
volume of mixture containing 5 μl of 10 × PCR buffer,
4 μl of 20 mM dNTP, 3 μl of 2 mM MgCl
2
, 1 μl of
25 pmol forward and reverse primers, 5 μl of temple
DNA, and 1 μl of 5U Taq DNA polymerase (Takara Bio).
In order to amplify groEL gene fragment of all target
bacteria, we used the following thermal cycling condi-
tions for screening: the PCR mixture was held at 95 °C
for 4 min prior to the 35 cycles of PCR amplification in
a thermal cycler (Eppendorf, Hamburg, Germany), the
first 5 cycles are consisted of denaturation at 94 °C for
40 s, annealing at 46 °C for 50 s and extension at 72 °C
for 1.5 min, and followed the other 30 cycles: denatura-
tion at 94 °C for 30 s, annealing at 53 °C for 45 s, and
extension at 72 °C for 1.5 min.
Oligonucleotide array fabrication
Positive charge nylon membrane was used as array
base. A 4 × 4 mm grid was formed with blunt pencil.
The nylon membrane was immersed in distilled water
for 10 min and then dried in WhatsmanR paper. Each
of the 23 probes was suspended to make a 50 pmol/μl
solution. The solution was heated at 95 °C for 3 min
and 1 μl of the solution was spotted on a corresponding
position of the nylon membrane. The membrane spot-
ted with probes was air-dried at room temperature and
Journal of Basic Microbiology 2012, 52, 27 – 34
Simultaneous analysis of foodborne pathogenic bacteria
29
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
heated at 120 °C for 30 min to allow binding of probes
onto the nylon membrane. The unbounded probes were
removed by two washes in 0.5 × SSC 0.1% sodium dode-
cyl sulfate (SDS) for 2 min at 37 °C. Then the membrane
was air-dried and stored at room temperature, ready for
use.
Hybridization, washing, and detection
Membranes with 23 probes were immersed in a Petri
dish containing 0.5 ml DIG Easy Hyb solution (Roche,
Indianapolis, USA) pre-warmed at hybridization tem-
perature and pre-hybridization was performed at 50 °C
for 30 min with gentle shaking. Five-microliter PCR
products were heated at 95 °C for 5 min, immediately
cooled on ice, then added to newly pre-warmed hy-
bridization solution. The membranes were hybridized
in the solution at 50 °C for 4 h with gentle shaking.
After hybridization, the membranes were washed four
times in 0.25 × SSC –0.1% SDS for 2 min at 37 °C. Be-
fore blocking, the membranes were washed in washing
buffer (Roche) for 1 min, then immersed in 10 ml
blocking buffer (Roche) for 30 min, and put to react
with anti-digoxigenin antibody for another 30 min with
gentle shaking. Colour development was made with
NBT-BCIP for 30–60 min in the dark without shaking.
The reaction was stopped in tap water. The resulting
images were visible and photographed.
Results
Amplification of groEL gene fragment
The groEL gene in foodborne infection pathogens, such
as Escherichia coli, Salmonella enterical, Shigella flexneri,
Vibrio parahaemolyticus, Campylobacter jejuni was success-
fully amplified by PCR, and a DNA fragment of 600 bp
could be obtained from all the pathogenic bacteria,
indicating that the universal degenerate primer can
amplify the groEL gene effectively.
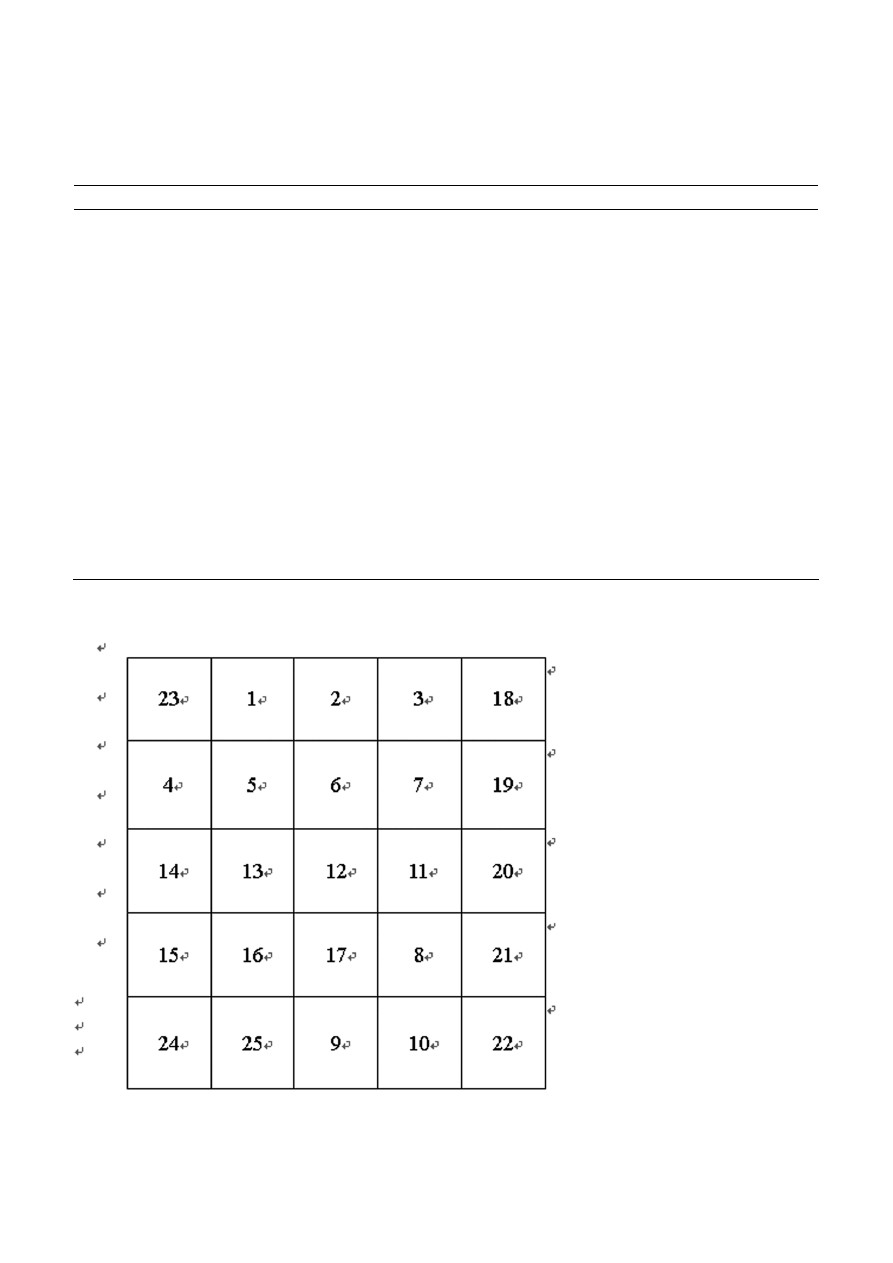
Array of oligonucleotide probes
In our study, 23 oligonucleotide probes were designed
and synthesized (Table 1) to match bacteria of different
species (genera). Probe no. 23 was universal to all bacte-
ria species (conservative sequence of bacteria groEL
gene). Probe nos. 18–22 was family- or genus-specific.
Probes nos. 1–17 were species-specific. To facilitate the
hybridization result analysis of the different bacterial
species, the twenty-three oligonucleotide probes were
arrayed in suitable grids on the nylon membrane
(Fig. 2). Positive, negative probes were spotted on the
last row to verify validity of the detection results. Dif-
ferent family- or genera-specific probes were spotted on
the grids in the last line of the array. Genus- or species-
specific probes were spotted on the grids of the first to
the fourth line of the array.
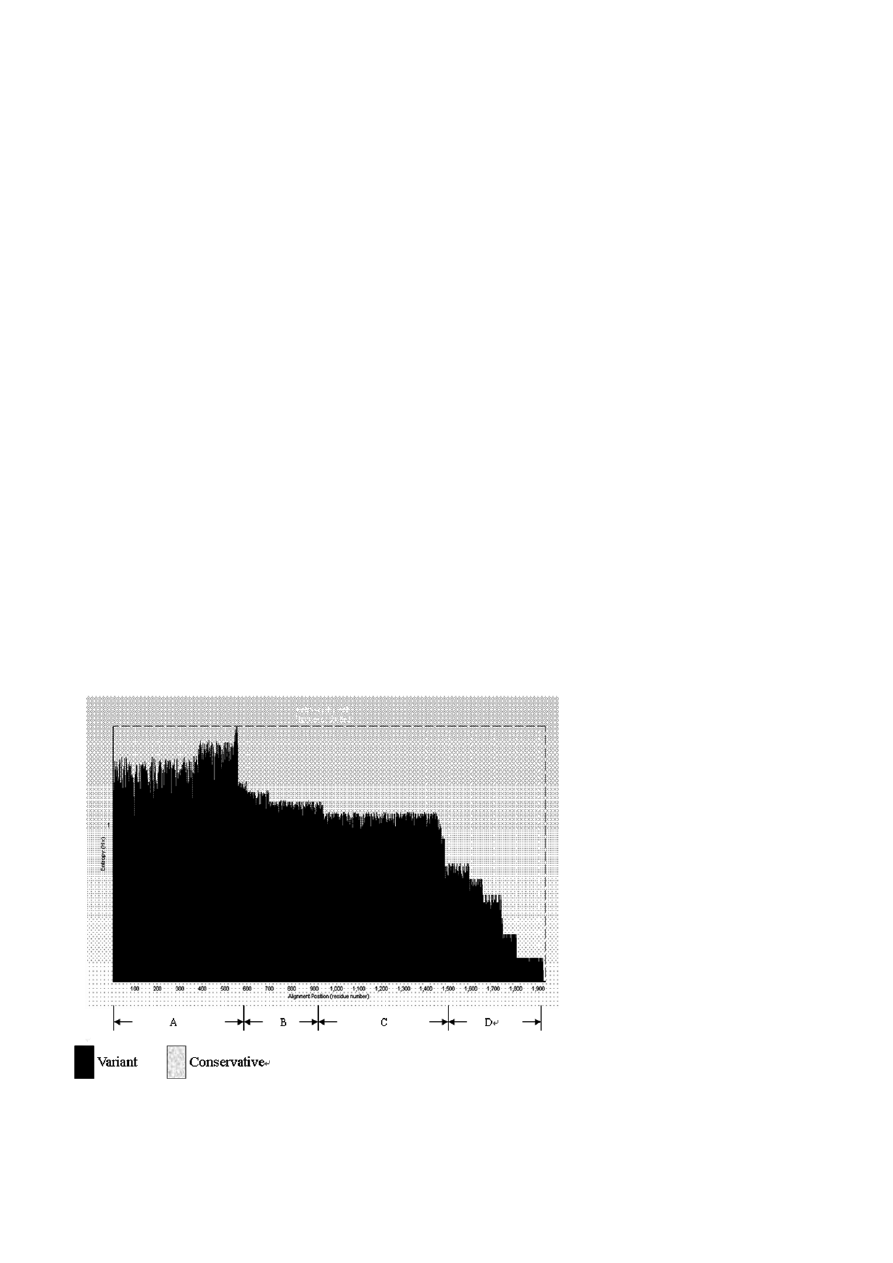
Figure 1. Analysis of groEL gene divergence. There are four variant regions in groEL gene. A region is the most mutable one; D region is
the least mutable one.
30 Y.
Hu
et al.
Journal of Basic Microbiology 2012, 52, 27 – 34
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Table 1. Probes and bacteria used in the study.
Probe no.
Probe sequence (5′ to 3′)
Genbank accession number
Detection range
1
TATTGAACTGCGGCGAAGAACC M11294
Escherichia coli
2
TGAACCCGATGGACCTGAAACG AY044102
Salmonella enterica
3
CTCCGCTAACTCCGACGAAACC AY044105
Salmonella typhimurium
4
TCTCCGCTAACTCCGACGAAA AY044103
Shigella flexneri
5
ACCATCTCCGCTAACTCCGACG NC007606
Shigella dysenteriae
6
AACCCGATGGATCTGAAAC
AY123739 Proteus
vulgaris
7
TTGCGGAAGATGTTGAAGG AY922346
Listeria monocytogenes
8
AAGCGGGCAGCGTTGAGC AF230952
Vibrio parahaemolyticus
9
GAAGATGTTGAAGGCGAAGCG
AF230930 Vibrio
alginolyticus
10
GCGGTTATCGCTGCGGTAGA AF230940
Vibrio cholerae
11
GGAAAGCCCATTCATCCTGC X59367
Yersinia enterocolitica
12
AAGTGGGCAAAGATGGTGT AY628401
Campylobacter jejuni
13
GAGGATGCTCTAAATGCCACA X89236
Streptococcus hemolyticus
14
ATCGTGCTAAACCGTATGCGTG
AF053568
Staphylococcus aureus
15
AAGACTAATGATGTGGCAGG X62914
Clostridium perfringens
16
GCTACTGAAGCAGGCGTT EU372231
Clostridium botulinum
17 GGCAAATCTTCTATCGCACA
EF685191 Bacillus
cereus
18
GAACCCGATGGACCTGAAACG
Salmonella spp.
19
ACCATCTCCGCTAACTCCGACG
Shigella spp.
20
CAAGTAGGTGCGATTTCTGC AF053568
Staphylococcus spp. and Streptococcus spp.
21
CTAATGATGTGGCAGGAGAT X62914
Clostridium spp.
22
GCAGAAGATGTGAAGGCGAAGC AF230940
Vibrio spp.
23
CTAAAGCGATTGCTCAGGTTG
X62914
Universal probes
24
Positive control
25
Negative control
Postive control is digoxigenin labeling plasmid pBR328/BamHI. Negative control is plasmid pBR328/BamHI.
Figure 2. The probe position of the oligonucleotide array. The numbers in the grids are probe nos. as indicated in Table 1.

Journal of Basic Microbiology 2012, 52, 27 – 34
Simultaneous analysis of foodborne pathogenic bacteria
31
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Figure 3. Oligonucleotide hybridization assay results of 20 species (genera) pathogenic bacteria. 1 – 17 are hybridization results of different
bacterial species: Escherichia coli, Campylobacter jejuni, Vibrio cholerae, Vibrio parahaemolyticus, Vibrio alginolyticus, Staphylococcus
aureus, Streptococcus hemolyticus, Yersinia enterocolitica, Proteus vulgaris, Bacillus cereus, Salmonella enterica, Salmonella typhimurium,
Listeria monocytogenes, Shigella dysenteriae, Shigella flexneri, Clostridium perfringens, Clostridium botulinum Lane 18 – 20 are hybridiza-
tion results of three
control species Lane: Streptococcus pneumoniae, Klebsiella pneumoniae and Neisseria gonorrhoeae. The data shown
are representative of five independent tests per species.
Hybridization results
Seventeen bacterial species were tested with the oligo-
nucleotide array method (Fig. 3). For each species, five
unrelated isolates were tested and all of the five tests
could show a consistent result. The results showed that
high sensitivity and specificity of hybridization results
were obtained with fifteen species of bacteria, includ-
ing Escherichia coli, Campylobacter jejuni, Vibrio cholerae,
Vibrio parahaemolyticus, Vibrio alginolyticus, Staphylococcus
aureus, Streptococcus hemolyticus, Yersinia enterocolitica, Pro-
teus vulgaris, Bacillus cereus, Salmonella enterica, Salmonella
typhimurium, Listeria monocytogenes, Clostridium perfringens
and Clostridium botulinum. As to Shigella dysenteriae and
Shigella flexneri, we found cross-reaction with the E. coli
species-specific probe. No hybridization signal was de-
tected with Streptococcus pneumoniae, Klebsiella pneumo-
niae, Neisseria gonorrhoeae.
Sensitivity
Each species of bacterium of mock samples was diluted
from 10
6
to 1 cfu/ml. Different titers of dilutions were
mixed and tested with the oligonucleotide array
method. Positive signal could be obtained from dilu-
tions between 10
6
and 10
2
cfu/ml. If the titer of dilu-
tions was below 10 cfu/ml, the results would be nega-
tive (Table 2). Fifty foodborne true samples were tested
with the oligonucleotide array method. At the same
time, they were identified by conventional culture
methods. Results manifested those samples except 2, 7,
10, 32, 39, 40, 41 show consistent results with culture
method as the probes used are highly specific (Table 3).
In samples 2, 7, 10, 32, 39, 40 and 41, oligonucleotide
array shows a slightly different result with culture
method, as Shigella spp. have cross-reaction with E. coli.
With oligonucleotide array method, we only concluded

32 Y.
Hu
et al.
Journal of Basic Microbiology 2012, 52, 27 – 34
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Table 2. Sensitivity test of foodborne infection mock samples with oligonucleotide array test.
Mock sample
Test 1
Test 2
Test 3
Test 4
Test 5
Test 6
Test 7
1 E. coli
10
6
/+ 10
5
/+ 10
4
/+ 10
3
/+ 10
2
/+ 10/± 1/–
2 S. enterica 10
6/
+ 10
5
/+ 10
4
/+ 10
3
/+ 10
2
/+ 10/+ 1/–
3 L. monocytogenes 10
6
/+ 10
5
/+ 10
4
/+ 10
3
/+ 10
2
/+ 10
/± 1/–
4 C. jejuni 10
6
/+ 10
5
/+ 10
4
/+ 10
3
/+ 10
2
/+ 10/+ 1/–
5 V. parahaemolyticus 10
6
/+ 10
5
/+ 10
4
/+ 10
3
/+ 10
2
/+ 10/± 1/–
6 P. vulgaris 10
6
/+ 10
5
/+ 10
4
/+ 10
3
/+ 10
2
/+ 10/+ 1/–
+, – and ±
represents positive signal, negative signal and weak signal respectively in oligonucleotide array test. 10
6
– 1 represents
bacterium titers in mock samples.
that these samples contain E. coli or Shigella spp. With
culture test, samples 2 and 40 contain Shigella dysente-
riae, samples 7 and 32 contain Shigella flexneri, samples
39 and 41 contain ETEC, while sample 10 contains ETEC
and S. dysenteria. Oligonucleotide array method gave
genus results and culture can discriminate to species
level.
Discussion
Although the pathogens causing foodborne infections
have become diversified in recent years, the epideomi-
ological study showed that pathogenic bacteria are
mainly involved. The conventional methods of identify-
ing foodborne infection pathogens are cultivation, bio-
chemistry reaction, and serologic or immunological
test, which are trivial, time-consuming and labor-inten-
sive. Furthermore, these routine methods are less sensi-
tive than DNA-based methods. In recent years, DNA and
oligonucleotide microarray technology has played an
increasingly important role in genomic
studies, drug
discovery, and toxicological research [14, 23, 25, 26, 28,
29, 33, 35, 37]. A rapid and precise DNA-based diagnos-
tic test would be of great value for the foodborne in-
fection pathogens. Although several molecular genetic
methods, such as single-stranded conformational poly-
morphism (SSCP) analysis, mismatch amplification mu-
tation assay (MAMA), and restriction fragment length
polymorphism (RFLP) analysis, have been used to inves-
tigate bacteria, all of them have limitations in different
aspects and are not yet established in clinical routine
diagnostics of microbial pathogens. As an example,
SSCP can detect only the region of the missense muta-
tion and not the exact position of the missense muta-
tion, MAMA can either detect one genotype or requires
the use of multiplex PCR, and RFLP can detect missense
mutations inside the recognition sequence of the re-
striction enzyme but not the exact position and the
substitution. In contrast, DNA microarray technology
provides a promising alternative for high-throughput
genotype-based diagnostics. The potential of miniaturi-
zation and multiplexing offers a considerable advan-
tage over other molecular genetic methods for clinical
application, which could be demonstrated, for example,
in the case of DNA microarray-based assays developed
for the detection of rifampin-resistant Mycobacterium.
Real-time PCR has the potential to provide a quicker
and more sensitive method for the detection of a di-
verse range of microorganisms. In real-time PCR ampli-
fication, the products can be detected through the use
of TaqMan probes. Although modern multiplex real-
time PCR has in many diagnostic applications replaced
array design, it needs expensive equipments and re-
agents. Microarrays are not in common use in average
laboratories today. However, like any new technology,
as more applications are developed for the microarray
technology, it will become more practical and may well
become widely used. More recently, oligonucleotide
microarray method has been applied to rapidly analyze
pathogens. For example, microarray analysis of micro-
bial virulence factors has been used for discrimination
among species of E. coli and S. aureus.
Heat shock protein is a highly conserved protein,
whose encoding gene groEL constitutes to be the most
conserved component in evolution [24, 31, 34, 36]. The
groEL gene has been used as the target gene in the typ-
ing and identification of Salmonella, Campylobacter jejuni
and Staphylococcus on account of its complete database
[22, 27, 32]. Compared with the 16S rRNA genes and 23S
rRNA genes, groEL sequences have higher divergence for
strains of bacteria. In the present study, sequence ho-
mology analysis was conducted with the full-length
fragments of the groEL genes obtained from 40 species
(genera) of bacteria. From the result of variance analy-
sis on groEL gene, it is evident that its variation is rather
high, especially among interspecies. There are four
variant regions in groEL gene (A–D), in which the A
region appears to be the most mutable one, while the D
region is the least mutable one (Fig. 1). According to the
distribution of groEL gene, a long fragment with greater
degree of mutation was selected as discriminative diag-

Journal of Basic Microbiology 2012, 52, 27 – 34
Simultaneous analysis of foodborne pathogenic bacteria
33
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Table 3. Oligonucleotide array test of foodborne infection true
samples.
Sample
no.
Oligonucleotide
array test
Culture test
1
Staphylococcus aureus
Staphylococcus aureus
2
E. coli and Shigella spp. Shigella dysenteriae
3
Vibrio parahaemolyticus Vibrio parahaemolyticus
4
Vibrio cholerae
Vibrio cholerae
5
Salmonella enterica
Salmonella enterica
6
Listeria monocytogenes
Listeria monocytogenes
7
E. coli and Shigella spp.
Shigella flexneri
8
Campylobacter jejuni
Campylobacter jejuni
9
Proteus vulgaris
Proteus vulgaris
10
E. coli and Shigella spp. ETEC and Shigella dysenteriae
11
Clostridium perfringens
Clostridium perfringens
12
Salmonella typhimurium Salmonella typhimurium
13
Clostridium botulinum
Clostridium botulinum
14
Bacillus cereus
Bacillus cereus
15
Yersinia enterocolitica
Yersinia enterocolitica
16
Vibrio alginolyticus
Vibrio alginolyticus
17
Streptococcus hemolyticus Streptococcus hemolyticus
18
Salmonella enterica
Salmonella enterica
19
Salmonella enterica
Salmonella enterica
20
Vibrio cholerae
Vibrio cholerae
21
Staphylococcus aureus
Staphylococcus aureus
22
Proteus vulgaris
Proteus vulgaris
23
Bacillus cereus
Bacillus cereus
24
Salmonella typhimurium Salmonella typhimurium
25
Vibrio parahaemolyticus Vibrio parahaemolyticus
26
Vibrio parahaemolyticus Vibrio parahaemolyticus
27
Vibrio cholerae
Vibrio cholerae
28
Vibrio alginolyticus
Vibrio alginolyticus
29
Salmonella typhimurium Salmonella typhimurium
30
Listeria monocytogenes
Listeria monocytogenes
31
Campylobacter jejuni
Campylobacter jejuni
32
E. coli and Shigella spp. Shigella flexneri
33
Salmonella typhimurium Salmonella typhimurium
34
Salmonella enterica
Salmonella enterica
35
Staphylococcus aureus
Staphylococcus aureus
36
Clostridium perfringens
Clostridium perfringens
37
Streptococcus hemolyticus Streptococcus hemolyticus
38
Bacillus cereus
Bacillus cereus
39
E. coli and Shigella spp. ETEC
40
E. coli and Shigella spp. Shigella dysenteriae
41
E. coli and Shigella spp. ETEC
42
Proteus vulgaris
Proteus vulgaris
43
Staphylococcus aureus
Staphylococcus aureus
44
Salmonella enterica
Salmonella enterica
45
Salmonella enterica
Salmonella enterica
46
Vibrio cholerae
Vibrio cholerae
47
Yersinia enterocolitica
Yersinia enterocolitica
48
Campylobacter jejuni
Campylobacter jejuni
49
Staphylococcus aureus
Staphylococcus aureus
50
Salmonella enterica
Salmonella enterica
nostic target gene sequence. We then have designed the
PCR universal primers from both ends of the fragment,
and by the use of a pair of primers thus synthesized all
the corresponding gene fragments in pathogenic bacteria
could be amplified by PCR under the proper amplifica-
tion conditions. Meanwhile, universal probe for different
genera can be designed from the conservative region and
the detection probe for genus and species of bacteria can
be also designed based on the variant region.
Our study showed that satisfactory hybridization
results with good sensitivity and specificity were ob-
tained with fifteen species of bacteria after considerable
design and redesign of the probes. Two other species
(Shigella dysenteriae and Shigella flexneri) gave weak cross-
reaction with E. coli. We used this method to test clini-
cal isolates analyzed previously and found that the
method is rapid and precise. Proper design of probes is
the key factor for successful oligonucleotide array hy-
bridization. In our microarray system, we used rela-
tively short oligonucleotides (18 to 25 nucleotides) for
that the shorter oligonucleotide probe sequences
(<25 bp) are often capable of detecting a single-nucleo-
tide mismatch and allow independent testing of several
species-specific regions. Hybridization conditions and
bacteria gene mutations often affect the sensitivity and
specificity of the detection. Oligonucleotide array tech-
nique has the merit of rapidity and of high reliability.
By combining it with PCR, the detection level of oli-
gonucleotide array in our study could reach 10 cfu/ml.
In addition to primers and probes, different labeling
methods also influence the result of detection. Digoxi-
genin-linked enzyme colour development method was
used in our study and the results could be evaluated by
naked eyes. For this reason, we believe that even small
laboratories can perform this rapid and accurate test.
The quality of the foodborne infection samples might
also affect the result of examination.
In conclusion, we have developed a microarray-based
assay for the simultaneous identification of foodborne
infection bacteria. Our method has great potential for
application in high-throughput screening and accurate
identification of genes, which are especially important
in epidemiological studies.
Acknowledgements
This study was supported by Guangzhou Scientific and
Technological Project (No. 2007J1-C0201).
References
[1]
Ana, M., Angela, D., Nuria, T., Laura, R., 2008. Epidemiol-
ogy of foodborne Norovirus outbreaks in Catalonia, Spain.
BMC Infect. Dis., 47, 102–109.
[2] Arvind, A.B., Medha, B., 2008. Methods and tools for com-
parative genomics of food-borne pathogens Foodborne
Pathog. Dis., 5, 487–497.
[3] Barry, R.B., 2009. Global phenotypic characterization of
bacteria. FEMS Microbiol. Rev., 33, 191–205.

34 Y.
Hu
et al.
Journal of Basic Microbiology 2012, 52, 27 – 34
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
[4] Bresee, J.S., Widdowson, M.A., Monroe, S.S., Glass, R.I.,
2002. Foodborne viral gastroenteritis: challenges and op-
portunities. Clin. Infect. Dis., 35, 748–753.
[5] Brian, K.C., Mark, E. W., Mariola, M., Samantha, G., Brett,
F., Mohamed, A.K., 2008. Molecular analysis as an aid to
assess the public health risk of non-O157 shiga toxin-
producing Escherichia coli strains. Appl. Environ. Micro-
biol., 74, 2153–2160.
[6] Brousseau, R., Hill, J.E., Prefontaine, G. et al., 2001. Strepto-
coccus suis serotype characterization by analysis of chaper-
onin 60 gene sequences. Appl. Environ. Microbiol., 67,
4828–4833.
[7] Carl, R. H., Sacha, L., Karyn, P. R., Udo, W., Tracy, J. E.,
2008. A short-oligonucleotide microarray that allows im-
proved detection of gastrointestinal tract microbial com-
munities. BMC Microbiol., 8, 195–214.
[8] Carole, L.Y., Lynn, B.M., 2007. Review of the literature
examining the correlation among DNA microarray tech-
nologies. Environ. Mol. Mutag., 48, 380–394.
[9] Christopher, W., Charlie, L., Wah, H. et al., 2007. Optimi-
zation and clinical validation of a pathogen detection mi-
croarray. Gen. Biol., 8, 93–105.
[10] Daniels, N.A., MacKinnon, L., Rowe, S.M. et al., 2002.
Foodborne disease outbreaks in United States schools. Pe-
diatr. Infect. Dis., 21, 623–628.
[11] Fares, M.A., Barrio, E., Sabater, B., Moya, A., 2002. The
evolution of the heat-shock protein GroEL from Buchne-
ra, the primary endosymbiont of aphids, is governed by
positive selection. Mol. Biol. Evol., 19, 1162–1170.
[12] Goh, S.H., Potter, S.J., Wood, O.S. et al., 1996. HSP60 gene
sequences as universal targets for microbialspecies identi-
fication: studies with coagulase-negative staphylococci. J.
Clin. Microbiol., 34, 818–823.
[13] Goh, S.H., Driedger, D., Gillett, S. et al., 1998. Streptococcus-
iniae, a human and animal pathogen: specific identifica-
tion by the chaperonin 60 gene identification method. J.
Clin. Microbiol., 36, 2164–2166.
[14] Hinanit, K., Carmiya, W., 2008. Specificity of DNA micro-
array hybridization: characterization, effectors and ap-
proaches for data correction. Nucl. Acid Res., 36, 2395–
2405.
[15] Hiroo, K., Blythe, E.J., Marek, B. et al., 2008. GroEL as a
molecular scaffold for structural analysis of the anthrax
toxin pore. Nat. Struct. Mol. Biol.,
15, 754–760.
[16] Il-Jin, K., Hio, C.K.,
Sang, G.J. et al., 2007. Development and
applications of a BRAF Oligonucleotide Microarray. J. Mol.
Diagn., 9, 55–63.
[17] Jian, W., Zhu, L., Dong, X., 2001. New approach to phy-
logenetic analysisof the genus Bifidobacterium based on
partial HSP60 gene sequences. Int. J. Syst. Evol. Micro-
biol., 51, 1633–1638.
[18] Jong-Yil, C., Eun-Hee, S., Soon-Hyung, L. et al., 2009. Food-
borne intestinal flukes in Southeast Asia. Korean J. Parasi-
tol., 47, 69–102.
[19] Joyce, V.D., Valerie, B., Thomas, B. et al., 2009. Occurrence
of foodborne bacteria in Alberta feedlots. Can. Vet. J., 50,
166–172.
[20] Jung-Hee, L., Hyo-Soon, P., Won-Jong, J., 2003. Differen-
tiation of Rickettsiae by groEL gene analysis. J. Clin. Mic-
robiol., 41, 2952–2960.
[21] Karenlampi, R.I, Tolvanen, T.P., Hanninen, M.L., 2004.
Phylogenetic analysis and PCR-restriction fragment
length polymorphism identification of Campylobacter spe-
cies based on partial groEL gene sequence. J. Clin. Micro-
biol., 42, 5731–5738.
[22] Kwok, A.Y., Su, S.C., Reynolds, R.P. et al., 1999. Species
identification and phylogenetic relationships based on
partial groEL gene sequences within the genus Staphylococ-
cus. Int. J. Sys. Bact., 49, 1181–1192.
[23] Ling-Xiang, Z., Zhi-Wei, Z., Can,W. et al., 2007. Use of a
DNA microarray for simultaneous detection of antibiotic
resistance genes among staphylococcal clinical isolates. J.
Clin. Microbiol., 45, 3514–3521.
[24] Marco, V., Carlos, C., Ralf, Z. et al., 2004. Characterization
of the groEL and groES loci in Bifidobacterium breve UCC
2003: genetic, transcriptional, and phylogenetic analyses.
Appl. Environ. Microbiol., 70, 6197–6209.
[25] Mathieu, M., Owen, Z.W., Alexandre, M.
et al., 2006. A meth-
odology for global validation of microarray experiments.
BMC Bioinform., 7, 333–349.
[26] Nikolay, S., Dmitriy, V., Vladimir, C. et al., 2004. Simulta-
neous analysis of multiple staphylococcal enterotoxin ge-
nes by an oligonucleotide microarray assay. J. Clin. Mi-
crobiol., 42, 2134–2143.
[27] Rebecca, S., Wong, Y., Chow, A.W., 2002. Identification of
enteric pathogenes by heat shock protein 60 kDa (GrOEL)
gene sequences. FEMS Microbiol. Lett., 206, 107–111.
[28] Richard, A.S., Lisa, F.D., Petra, C.F.
et al., 2008. Development
and application of the active surveillance of pathogens
microarray to monitor bacterial gene flux. BMC Micro-
biol., 8, 177–190.
[29] Roberto, V., Maricel, V., Rossana, L. et al., 2004. Multiplex
PCR for diagnosis of enteric infections associated with di-
arrheagenic Escherichia coli. J. Clin. Microbiol., 42, 1787–
1789.
[30] Scott, E., 2003. Food safety and foodborne disease in 21st
century homes. Can. J. Infect. Dis., 14, 277–280.
[31] Sensu, L., Hyo-Soon P., Jung-Hee L. et al., 2004. Evaluation
of groEL gene analysis for identification of Borrelia burgdor-
feri. J. Clin. Microbiol., 42, 1270–1273.
[32] Teng, L.J., Hsueh, P.R., Tsai, J.C. et al., 2002. groESL se-
quence determination, phylogenetic analysis, and species
differentiation for viridans group streptococci. J. Clin. Mi-
crobiol., 40, 3172–3178.
[33] Tim, L., Eric, G.,
2006. The emergence and diffusion of DNA
microarray technology. J. Biom. Disc Colla., 1, 11–49.
[34] Tobias, W., Laurence, D.H., 2010. GroEL dependency
affects codon usage-support for a critical role of misfold-
ing in gene evolution. Mol. Syst. Biol., 6, 340–351.
[35] Xiaolei, Y., Milorad, S., Cornelius, K. et al., 2004. Develop-
ment and validation of a diagnostic DNA microarray to
detect quinolone-resistant Escherichia coli among clinical
isolates. J. Clin. Microbiol., 42, 4083–4091.
[36] Yu-Hsiu, C., Yung-Hui, S., Hung-Chi, L. et al., 2003. PCR
assay of the groEL gene for detection and differentiation
of Bacillus cereus group cells. Appl. Environ. Microbiol., 69,
4502–4510.
[37] Zhengshan, Z., Gerardo, A., Francois, J.P. et al., 2008.
Plastic polymers for efficient DNA microarray hybridiza-
tion: application to microbiological diagnostics. J. Clin.
Microbiol., 46, 3752–3758.
((Funded by
• Guangzhou Scientific and Technologi-
cal Project; grant number: 2007J1-C0201))
Wyszukiwarka
Podobne podstrony:
jobm 201000013
jobm 201000298
jobm 201000191
jobm 201000321
jobm 201000018
jobm 201000214
jobm 201000067
jobm 201000037
jobm 201000074
jobm 201000280
jobm 201000385
jobm 201000198
jobm 201000147
jobm 201000520
jobm 201000327
jobm 201000342
jobm 201000420
jobm 201000364
jobm 201000317
więcej podobnych podstron