
372
Journal of Basic Microbiology 2011, 51, 372 – 384
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Research Paper
Culture-dependent and -independent molecular analysis
of the bacterial community within uranium ore
Ekramul Islam and Pinaki Sar
Department of Biotechnology, Indian Institute of Technology, Kharagpur, India
The bacterial community structure within a uranium ore was investigated using culture-
dependent and -independent clone library analysis and denaturing gradient gel electrophoresis
of 16S rRNA genes. The major aerobic heterotrophic bacteria were isolated and identified, and
their resistance to uranium and other heavy metals was characterized. Together with near
neutral pH, moderate organic carbon content, elevated U and other heavy metals (V, Ni, Mn,
Cu, etc.), the ore showed high microbial counts and phylotype richness. The bacterial
community mainly consisted of uncultured Proteobacteria, with the predominance of
γ
- over
β
-
and
α
-subdivisions, along with Actinobacteria and Firmicutes. A phylogenetic study revealed
that nearly one-third of the community was affiliated to as yet uncultured and unidentified
bacteria having a closer relationship to Pseudomonas. Lineages of Burkholderiaceae and Mor-
axellaceae were relatively more abundant in the total community, while genera affiliated to
Xanthomonadaceae and Microbacteriaceae and Exiguobacterium were detected in the culturable
fraction. More than 50% of the bacterial isolates affiliated to Stenotrophomonas, Microbacterium,
Acinetobacter, Pseudomonas and Enterobacter showed resistance to uranium and other heavy
metals. The study showed for the first time that uranium ore harbors major bacterial groups
related to organisms having a wide range of environmentally significant functional attributes,
and the most abundant members are possibly new groups/taxa. These findings provide new
insights into U-ore geomicrobiology that could be useful in biohydrometallurgy and bio-
remediation applications.
Keywords: Uranium ore / Bacterial community / 16S rRNA gene / Heavy metals / Uranium resistance
Received: August 17, 2010; accepted: November 13, 2010
DOI 10.1002/jobm.201000327
Introduction
*
The vast wealth of microbial diversity that may exist in
extreme deep-subsurface environments such as those in
mineral ore deposits has so far remained largely unex-
plored [1–5]. The increasing interest in intra-terrestrial
life within subsurface mines may lead us to expand our
knowledge on diverse microbial communities with
unique metabolic properties [3, 5]. In culture experi-
ments of the recent years, independent approaches
have been introduced to unravel the metabolic poten-
tial of microbial communities inhabiting environments
Correspondence: Pinaki Sar, Department of Biotechnology, Indian
Institute of Technology, Kharagpur 721 302, India
E-mail: psar@hijli.iitkgp.ernet.in
Phone: +91-3222-283754
Fax: +91-3222-255303
ranging from soil, water, stromatolites, and acid mine
drainage to ultra-deep mines [2, 3, 6–8]. Compared to
studies carried out over relatively broad spatial scales to
assess the microbial community structure in copper
and gold mines or in metal-/radionuclide-contaminated
environments, indigenous microbial communities in
uranium mines have been less explored [9]. Previous
studies on the microbial diversity in mine tailings,
mine wastes, mining-impacted sites, and other ura-
nium-contaminated subsurface environments revealed
the presence of physiologically and metabolically di-
verse populations organized in complex communities
[9–12]. In contrast to such extensive studies on diverse
uranium mine/uranium-contaminated samples, the mic-
robial community within subsurface uranium ore re-
mains unexplored and uncharacterized. Microorgan-
isms play important roles in mineral-ore formation and

Journal of Basic Microbiology 2011, 51, 372 – 384
Bacterial community within uranium ore
373
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
dissolution, by regulating critical geochemical reactions
[13]. Despite their inevitable presence and role in min-
eral biogeochemistry, the ecology of microorganisms in
such materials remains largely unresolved. Exploring
the microbial diversity and community composition
with metagenomic approaches is considered to be a real
advancement to a full understanding of how members
of a complex community interact and function within
their niches [8, 14]. Furthermore, understanding the
microbial community structure within the ore may pro-
vide the opportunity to exploit their potential in biomin-
eralization, bioremediation, biomining, and other re-
lated applications [15].
The Jaduguda uranium mine in India has been active
since 1968, and the current ore extraction is taking
place at a depth of about 850 m. As part of our attempt
to decipher the microbial diversity across different sites
of the Jaduguda U-mine area, the present paper deline-
ates the structure of the bacterial community from a
subsurface uranium ore. Both culture-dependent and
independent molecular approaches were adopted to
elucidate the bacterial communities. Total as well as
culturable bacterial communities were analyzed by
16SrRNA gene-based clone libraries, followed by dena-
turing gradient gel electrophoresis (DGGE). Isolation
and identification of bacteria of several pure cultures
from the ore samples were performed, along with a
characterization of their resistance properties to ura-
nium and other heavy metals.
Materials and methods
Sample collection and physicochemical analysis
A uranium ore sample was collected during April 2007
from an underground mine gallery (at a depth of
850 m) of the Jaduguda uranium mine (22°40′ N,
86°20′ E), India. The aseptically collected sample was
brought to the laboratory and stored at 4 °C for analy-
sis. About 200 g of subsample was ground using a ster-
ile mortar and pestle inside a laminar flow hood. The
powdered sample was suspended either in distilled
water or in 0.01 M CaCl
2
at a 1:10 ratio (w/v), and ORP,
salinity, conductivity and pH were measured with an
Orion multimeter. Heavy metals and actinide elements
were analyzed by ICP-MS (PerkinElmer Elan DRC)
and/or XRF (Philips PW 1480). Anions were estimated by
ion chromatography (Dionex DX-100) following their
extraction with deionized water [1:10 dilution (w/v)].
Total organic carbon, nitrogen, phosphorous and sulfur
(sulfate-S) were assessed with standard procedures [16].
Enumeration of microbial populations
The total microbial count was determined by fluores-
cent microscopy using 4′,6′-diamidino-2-phenylindole
(DAPI), following the protocol developed by Kepner and
Pratt [17]. Aerobic microbial populations were enumer-
ated by both dilution plate count and enrichment tech-
nique. For dilution plating, the powdered ore was sus-
pended in normal saline (0.9%) and mixed thoroughly
by continuous shaking (1 h, 200 rpm). The turbid sus-
pension was diluted in 10-fold steps, and 100 μl of sus-
pension from each dilution was plated in triplicate. For
the enumeration of heterotrophic neutrophilic bacte-
ria, R2A (pH 7.0) and pepton-trypton-yeast extract-glu-
cose (PTYG) agar media were used [18]. MGY medium
(pH 3.0) with 0.6% agarose was used for heterotrophic
acidophilic bacteria [19]. Moderately acidophilic thioba-
cilli were counted on Thiobacillus agar medium (pH 5.5)
(Himedia). For acidophilic iron- and sulfur-oxidizing
microorganisms, 9 K medium (pH 2.3) with FeSO
4
or
sodium-tetrathionate, respectively, was used [20]. The
cultures were incubated in the dark at 30 °C, and total
numbers of colony-forming units (CFU/g) were deter-
mined after 7 d of incubation. For Fe- or S-oxidizing
bacteria, the tubes were incubated for up to 3 weeks.
DNA extraction and amplification of 16S rRNA genes
The total metagenome from the ore sample was ex-
tracted using the MoBio Power soil
TM
DNA kit (MoBio,
Carlsbad, CA), according to the manufacturer’s instruc-
tions. The DNA of the culturable heterotrophic bacte-
rial community was extracted from the cell biomass
grown on R2A agar plates, using the DNeasy tissue kit
(Qiagen, Hilden, Germany) as per the manufacturer’s
instructions. For the cell biomass preparation, colonies
were flooded in sterile saline, scraped off the plate by a
sterile needle and collected in a centrifuge tube. All
DNA extractions were done in triplicate and the DNA
was pooled into one sample prior to use in PCR. PCR
amplification of the 16S rRNA genes was carried out
using the eubacterial forward primer 8F and the uni-
versal primer 1492R, using similar reaction and tem-
perature cycling conditions to those described by
Reardon et al. [21]. For DGGE, the primers GC-357F and
518R were used to amplify the V3 region of the bacte-
rial 16S rRNA gene, following the procedure described
by Muyzer et al. [22].
16S rRNA gene clone library construction and
screening by amplified ribosomal DNA restriction
analysis
The amplified PCR products (~1500 bp) were gel puri-
fied, cloned into the pGEM-T® Easy vector (Promega,

374
E. Islam and P. Sar
Journal of Basic Microbiology 2011, 51, 372 – 384
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Madison, WI, USA) and transformed into E. coli JM109,
following the manufacturer’s instructions. Two clone
libraries, J254TP and J254PW, were constructed from
the total metagenome and the culturable metagenome
(plate-washed DNA), respectively, with clones having
the proper length of the 16S rRNA gene fragment. For
screening of the clone libraries, the target 16S rRNA
gene fragments of the clones were amplified directly
from the fresh cell suspension using the vector-specific
primers SP6 and T7. The PCR-amplified product (5 μl)
from each clone was digested separately with HaeIII,
RsaI, AluI, and MspI in 20 μl reaction mixtures. All di-
gests were analyzed by 2.5% agarose gel electropho-
resis. Amplified ribosomal DNA restriction analysis
(ARDRA) patterns were checked manually, and each
ribotype or operational taxonomic unit (OTU) was de-
fined as group of clones that had similar enzyme re-
striction patterns. Ribotypes from the libraries J254TP
and J254PW were designated as G and R, respectively,
with a numeric value that is indicated in the ID of the
representative sequence. At least one representative
clone from each dominant OTU was selected for se-
quencing of the 16S rRNA gene insert. All the clones
were stored in 15% glycerol at –80 °C for future use.
Isolation and characterization of aerobic
heterotrophic bacteria
A number of morphologically distinct colonies grown
on R2A (pH 7.0) agar were selected, sub-cultured and
purified by repeated streaking on the same agar me-
dium. The Gram characteristics of all pure cultures
were determined. To avoid repeated selection of the
same isolate, the colonies were screened and grouped
by restriction fragment length polymorphism (RFLP) of
the 16S rRNA genes. Amplification and RFLP analysis of
the 16S rRNA genes were done according to Reardon
et al. [21]. Each RFLP group was designated by the letter
S plus a numeric value. At least one isolate from each
RFLP group was identified by 16S rRNA gene sequenc-
ing.
Denaturing gradient gel electrophoresis analysis
DGGE was performed using the D’code system (Bio-Rad,
Hercules). Polyacrylamde gels [8% (w/v) acrylamide/bi-
sacrylamide (37.5:1)] with a denaturing gradient from
35 to 65% [100% corresponds to 7 M urea and 40% (w/v)
formamide] were used. Following electrophoresis in
0.5 × Tris-acetate-EDTA (TAE) at a constant temperature
of 60 °C for 10 h at 70 V, the gel was stained with
ethidium bromide and the bands were visualized under
UV illumination. The bands of interest were excised
and the DNA was eluted in 50 μl PCR-grade water, fol-
lowing incubation overnight at 4 °C. The eluted DNA
was re-amplified using the GC-less 357F/518R primer
pair. The pGEM-T® Easy vector system (Promega, Madi-
son, WI, USA) was used for cloning of the re-amplified,
excised DGGE bands. Several clones from each DGGE
band were sequenced.
DNA sequencing, phylogenetic and statistical
analysis
Partial sequences of the 16S rRNA gene fragments were
determined on an ABI 3730XL machine. The sequences
were examined by the CHECK_CHIMERA program of
the Ribosomal Database Project (RDP-II). The sequence
data was compared with 16S rRNA gene sequences de-
posited in public databases, by using the BLAST (NCBI)
program, and an initial classification was made using
the classifier program in the Ribosomal Database Pro-
ject. The retrieved 16S rRNA gene sequences from Gen-
Bank were aligned along with the new sequences using
ClustalW, and phylogenetic analysis was done by MEGA
4.0 following neighbor-joining methods incorporating
the Jukes-Cantor distance correction [23]. Acidilobus
saccharovorans (AY350586) was selected as out-group.
1000 bootstrap analyses were conducted and >80% are
indicated at the nodes. Calculation of diversity indices
and rarefaction analysis were performed based on the
number and frequency of ribotypes identified in the
clone libraries. The Shannon diversity index (H), the
reciprocal of Simpson’s index of dominance (1/D), even-
ness (E), and percentage coverage of the clone libraries
were calculated as described earlier [21].
Resistance of isolates to uranium and other
heavy metals
The resistance to uranium and other heavy metals of 20
isolates covering all RFLP groups was tested, along with
the multiple metal-resistant Cupriavidus metallidurans
(DSMZ 2839) or E. coli JM109. U-resistance of the isolates
was tested following the procedure described by Suzuki
and Banfield [24]. Briefly, mid-exponential-phase cells
of each isolate were obtained, washed (0.9% NaCl) and
resuspended in saline at pH 7.0 and 4.0 and at pH 4.0
with U (80 ppm) under sterile conditions, and incubated
at 30 °C (with shaking at 150 rpm). Viable cell counts in
these three sets were monitored at different time inter-
vals up to 4 h by CFU counts on R2A agar plates
(pH 7.0). The metal resistance of the isolates was tested
by allowing bacterial growth in metal-supplemen-
ted R2A medium. Mid-log phase cells of each strain
were applied to R2A agar plates supplemented with
graded concentrations (0.1–10 mM) of different metals
[Cu(NO
3
)
2
⋅ 3 H
2
O, Cd(NO
3
)
2
⋅ 4 H
2
O, NiCl
2
⋅ 6 H
2
O, ZnCl
2
,

Journal of Basic Microbiology 2011, 51, 372 – 384
Bacterial community within uranium ore
375
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
and K
2
Cr
2
O
7
; Merck, Germany]. All plates were incu-
bated at 30 °C for 7 d and checked every 24 h for
growth.
Nucleotide sequence accession numbers
The nucleotide sequences obtained in the present study
were deposited in GenBank under the following acces-
sion numbers: FJ804427–FJ804452, FJ985719–FJ985739,
GQ901866–GQ901888, and GU723258–GU723272.
Results
Geochemical and microbiological characteristics
of the sample
The geochemical properties of the ore sample were
analyzed to assess the relevant physicochemical con-
ditions available to the inhabitant microbial commu-
nity (Table 1). The ore was found to be slightly acidic
(pH 6.3) and highly enriched in water-extractable SO
4
2–
(14 mM), NO
3
–
(1.4 mM), Cl
–
(14 mM), and total organic
carbon (136 mM). Along with uranium, the presence of
various heavy metals (V, Ni, Mn, Cu, Cr, etc.) at elevated
concentrations was noticed. The total microbial cell
counts as determined by fluorescence microscopy
yielded (9.5 ± 3.6) × 10
8
cells/g sample, whereas the
culturable aerobic cell counts were found to be more
than two orders of magnitude lower, indicating that
nearly 0.48% of the total cells were culturable under
the conditions tested. In our attempt to enumerate the
culturable bacteria of varied pH optima, maximum CFU
counts were obtained with R2A medium at pH 7.0
((4.6 ± 0.7) × 10
6
), indicating the predominance of het-
erotrophic neutrophilic organisms. Compared to this,
the CFU counts for acidophilic heterotrophs (in MGY
agarose medium) and moderately acidophilic sulfur-
oxidizing autotrophs (in Thiobacillus agar) were at least
two and three orders of magnitude lower, respectively.
For strictly acidophilic iron- or sulfur-oxidizing organ-
isms, no growth was observed using 9 K medium sup-
plemented with either FeSO
4
or Na-tetrathionate at pH
2.3.
Molecular analysis of the bacterial community
structure
Nearly full-length (~1.5 kb) 16S rRNA genes were PCR
amplified, and two clone libraries, J254TP and J254PW,
were constructed using 180 and 70 positive clones from
the total and culturable communities, respectively.
ARDRA with four restriction enzymes revealed the
assemblage of several distinct OTU within the total (68)
or culturable (38) communities, while the presence of
Table 1. Geo-microbial characteristics of the ore sample.
Parameter Value
pH 6.4
Conductivity (mS/cm)
0.2
ORP (mV)
247.0
Salinity (mg/L)
102.0
Total organic C (mM)
136.0
Total N (mM)
24.0
Total P (mM)
207.0
Total S (mM)
25.0
Elements (mM)
Co 0.9
Cr 4.3
Cu 6.0
Ni 10.6
Mn 9.0
Cd <0.005
As 0.5
Zn 0.4
Pb 0.4
Rb 1.6
V 12.0
Y 2.0
U 1.2
Th 0.04
La 1.8
Microbial counts
Total count/g
(9.5 ± 3.6) × 10
8
R
2
A medium (CFU/g)
(4.6 ± 0.7) × 10
6
PTYG medium (CFU/g)
(1.26 ± 0.4) × 10
4
MGY agarose, pH 3.0 (CFU/g)
0.3 × 10
3
Thiobacillus agar, pH 5.0 (CFU/g)
(1.08 ± 0.17) × 10
4
9K + FeSO
4
, pH 2.3
No growth
9K + Na-tetrathionate, pH 2.3
No growth
16S rRNA libraries
a
Number of clones analyzed
180,
69
Phylotype richness (no. of OTU)
68,
38
% Coverage
77.2,
59.4
Shannon diversity index (H) 3.6,
3.3
Evenness (E)
0.84,
0.9
Reciprocal of Simpsons (1/D) 18.4,
17.8
a
Values in normal face and in bold italics are for the clone
libraries J254TP and J254PW, respectively.
18 distinct RFLP groups was evident from 32 pure-
culture bacterial isolates (Table 1, see Fig. 3). Although
rarefaction analysis of the two clone libraries did not
show saturation (data not shown), coverage values indi-
cated satisfactory results for both the libraries (Table 1).
High Shannon and Simpson’s diversity indices along
with relatively low equitability values, particularly
within the total community, were observed (Table 1).
Phylogenetic analyses
Phylogenetic diversity was analyzed using the first
500–600 bp of the 16S rRNA gene sequence of one rep-
resentative clone from each major ribotype. From the
J254TP and J254PW libraries, 26 and 20 ribotypes cover-
ing nearly 78 and 72% of the communities, respecti-
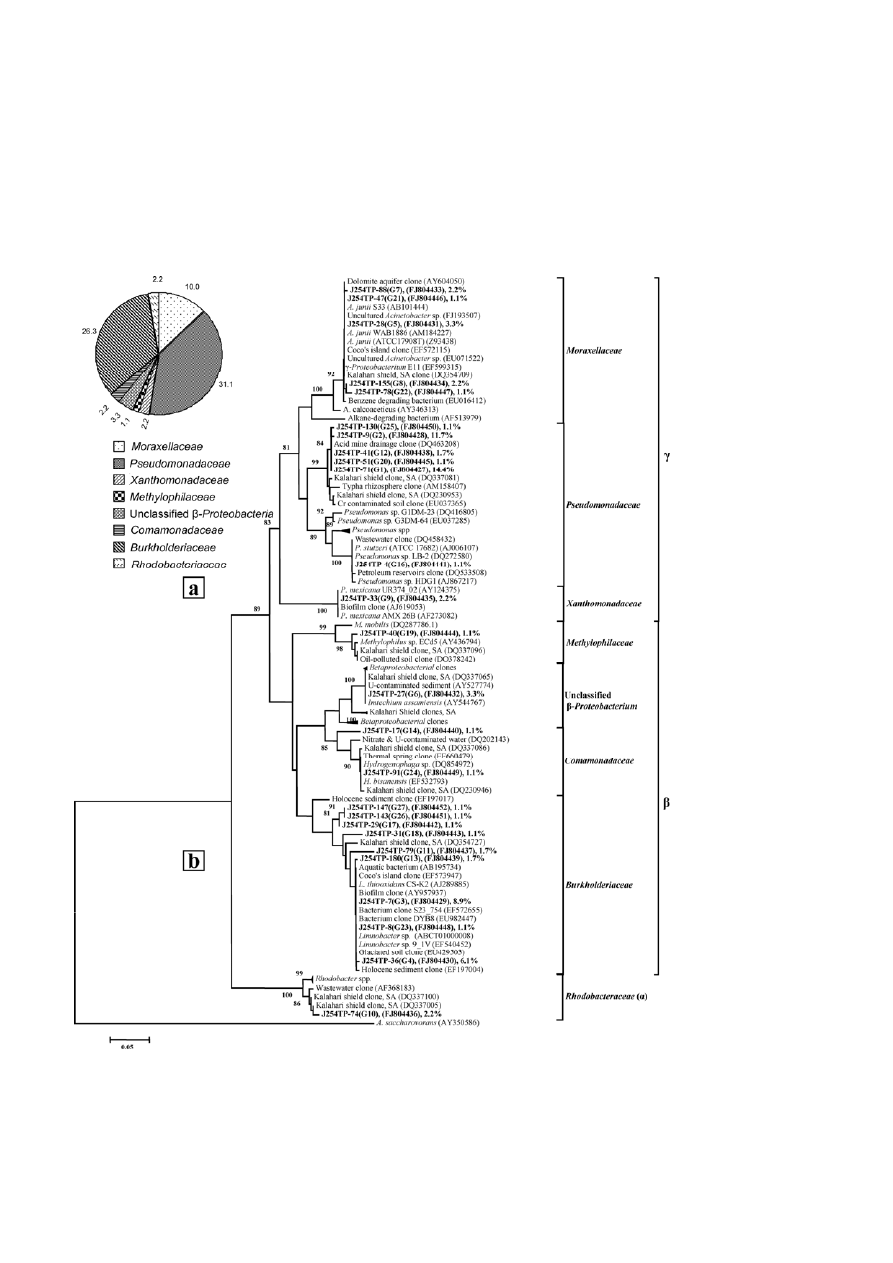
376
E. Islam and P. Sar
Journal of Basic Microbiology 2011, 51, 372 – 384
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
vely, were identified (Figs. 1a, b and 2a, b). Among the
pure culture isolates, the identities of all 18 RFLP
groups were ascertained. Apart from the similarity
search in databases, the phylogenetic lineage of all the
bacterial groups detected within the U-ore was ascer-
tained by retrieving similar sequences with an affi-
liation to uranium/metal contamination. As presented
in Figs. 1 and 2, the sequenced ribotypes from the two
clone libraries were affiliated to the phylum Proteobac-
teria. Within this phylum, members of the
γ
-Proteobac-
Figure 1. Relative abundance of bacterial groups detected in the total metagenome-derived library (J254TP) (a) and phylogenetic lineage of
the 16S rRNA gene sequences from the total metagenome-derived library (J254TP) with reference sequences in GenBank (b); clones with
designations including J254TP were analyzed in the present study. Sequence accession numbers are shown in parentheses, and the
relative abundance of each ribotype is shown with the representative sequence.
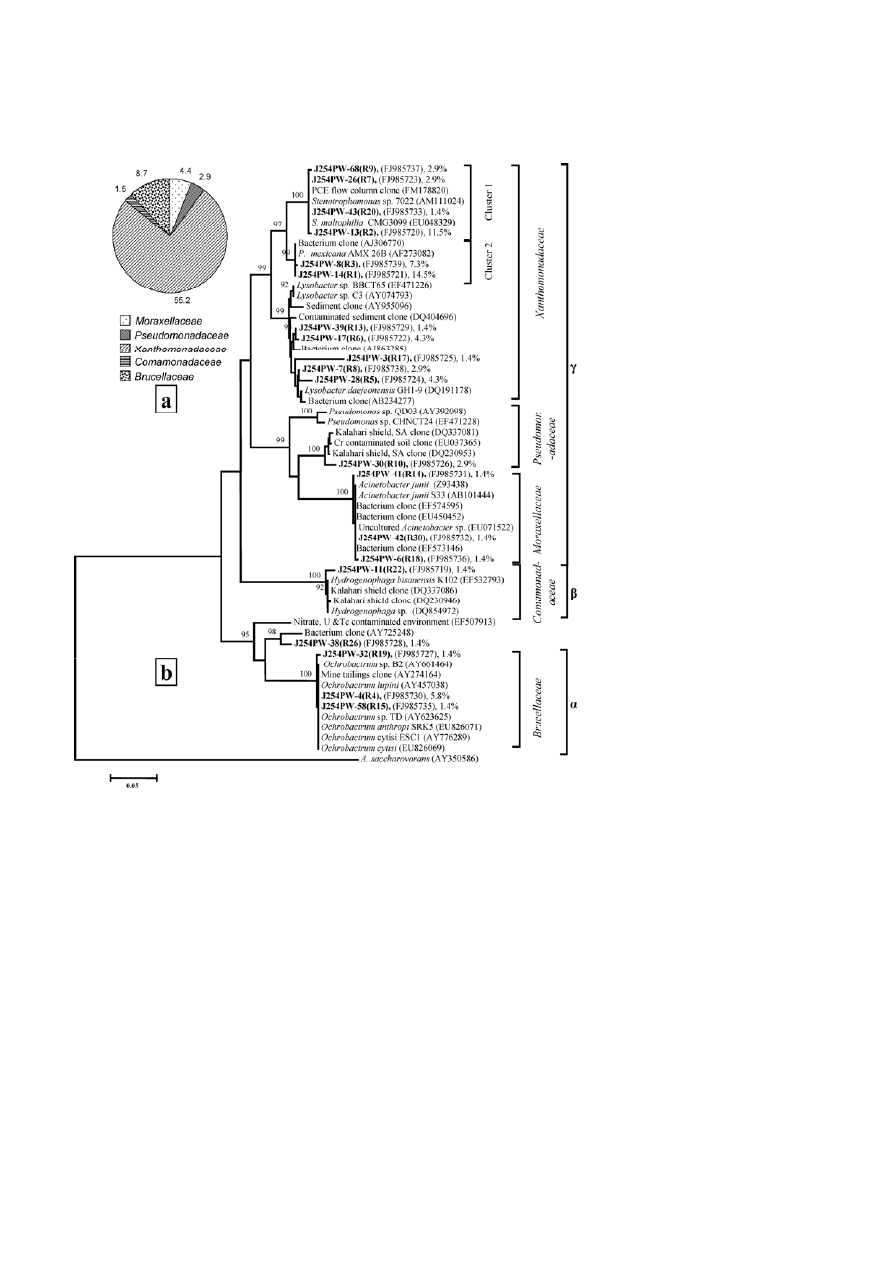
Journal of Basic Microbiology 2011, 51, 372 – 384
Bacterial community within uranium ore
377
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Figure 2. Relative abundance of bacterial groups detected in the plate-washed DNA-derived library (J254PW) (a) and phylogenetic lineage
of the 16S rRNA gene sequences from the plate-washed DNA-derived library (J254PW) with reference sequences in GenBank (b); clones
with designations including J254PW were analyzed in the present study. Sequence accession numbers are shown in parentheses. The
relative abundance of each ribotype in the clone library is given with the representative sequence.
teria were dominant in both libraries (43 and 62% of
J254TP and J254PW, respectively) as well as in the pure
culture isolates (74%), while
α
-Proteobacteria were
detected as less abundant (>2 and >8% of J254TP and
J254PW, respectively) bacterial groups. Members of the
β-Proteobacteria were detected in library J254TP (33%)
and the culturable isolates (9.5%). However, the bacte-
rial diversity was relatively wider among the pure cul-
ture isolates, with the presence of Actinobacteria and
Firmicutes along with
γ
- and
β
-Proteobacteria (Fig. 3a, b).
γ-Proteobacteria
Sequences representing major ribotypes or the isolates
were found to be affiliated to the Pseudomonadaceae,
Moraxellaceae, Xanthomonadaceae and Enterobacteri-
ceae of the
γ
-Proteobacteria. Pseudo-monadaceae mem-
bers were relatively more abundant in library J254TP
over the culturable counterparts (Figs. 1b, 2b and 4b).
Within library J254TP, six ribotypes, including the two
most dominant ones, together covering more than 30%
of the community, and a single ribotype from library
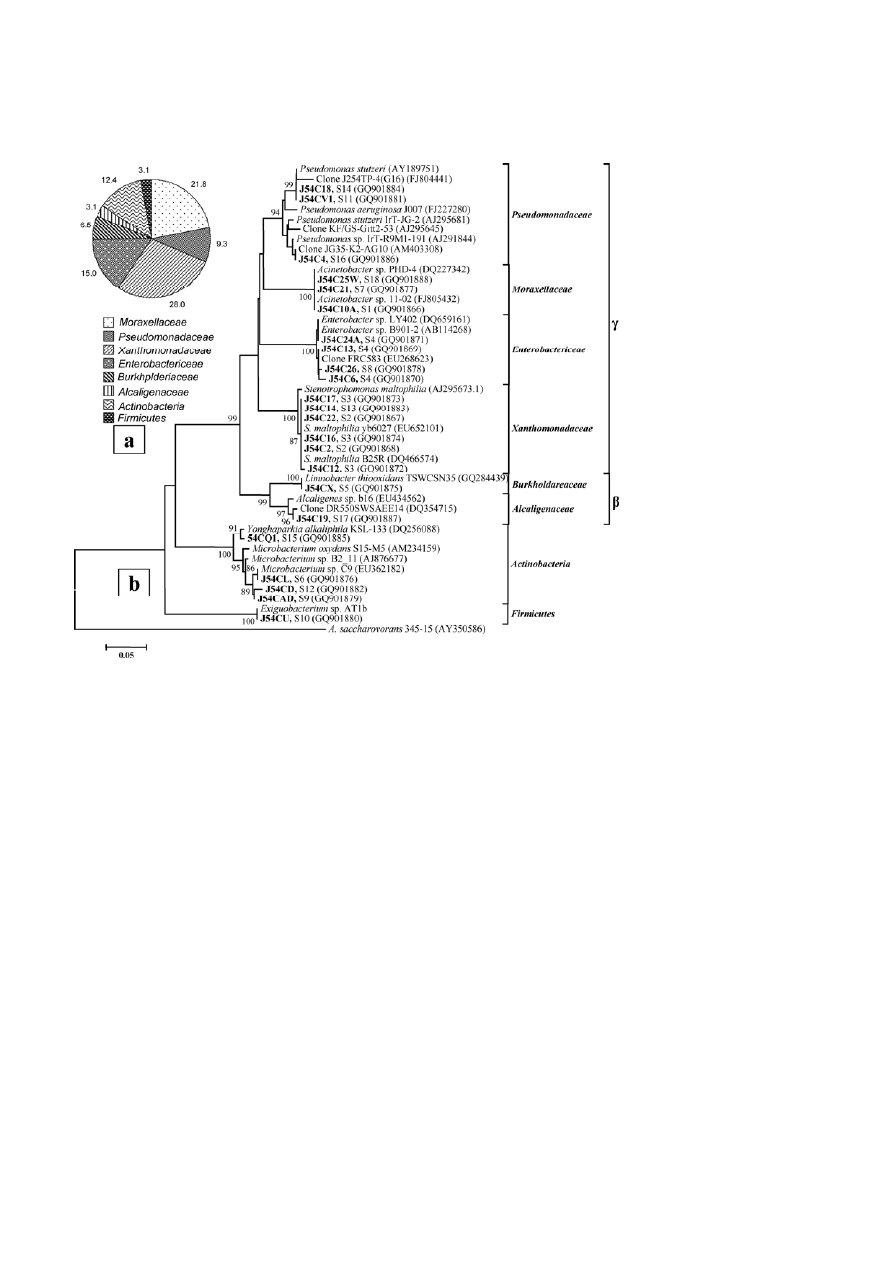
378
E. Islam and P. Sar
Journal of Basic Microbiology 2011, 51, 372 – 384
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Figure 3. Relative abundance of bacterial groups detected among the pure culture isolates (a) and phylogenetic lineage of the 16S rRNA
gene sequences from these isolates with reference sequences in GenBank (b); the isolates in the present study are shown in bold face and
the alpha-numeric value with the sequence ID indicates the RFLP group the particular isolate belongs to. Sequence accession numbers are
shown in parentheses.
J254PW showed high (≥97%) sequence identity to un-
cultured bacterial clones previously retrieved from the
Kalahari Shield (South Africa) and the Tong Lushan
copper mine (China) (Figs. 1b and 2b). Phylogenetic
analysis revealed a strong relationship (≥99% bootstrap
support) of all these ribotypes to the Pseudomonada-
ceae of the
γ
-Proteobacteria. Among the bacterial iso-
lates, relatively low-abundant RFLP groups represented
by strains J54CV1, J54C18 and J54C4 were affiliated to
Pseudomonas spp., showing close similarity to clones or
strains previously recovered from uranium waste/ore.
Of the plate-washed library, the only ribotype that be-
longs to the Pseudomonadaceae showed close relation-
ship to the most dominant ribotype (G1) from library
J254TP and was affiliated to Pseudomonas. Members of
the family Moraxellaceae represented the second most
abundant group among the
γ
-Proteobacteria within the
library J254TP and the pure culture isolates. As evident
from phylogenetic analysis, all the sequences affiliated
to this family showed close relationship to Acinetobacter
spp. (Figs. 1b, 2b and 4b). Particularly, the sequences
representing five ribotypes (G5, G7, G8, G21 and G22)
from J254TP presented a closer relation with A. juni and
uncultured clones from a dolomite aquifer, the Kala-
hari Shield, and hydrocarbon-degrading bacteria. The
family Xanthomonadaceae was well represented by
several ribotypes from the cultivable fraction (J254PW),
while only one ribotype from the total community
showed affiliation to this family. The two most abun-
dant ribotypes from J254PW and one less abundant
group from J254TP showed close relationship to the
Pseudoxanthomonas mexicana AMX 26B strain previously
retrieved from activated sludge (Figs. 1b and 2b). From
the plate wash community (J254PW), sequences repre-
senting a number of major and minor ribotypes formed
two distinct clades, one showing a strong relationship
to Stenotrophomonas species while the other was related
to Lysobacter species (Fig. 2b). Among the isolated bacte-
ria, six strains (J54C22, J54C17, J54C2, J54C16, J54C12,
and J54C14) representing three RFLP groups were found
Journal of Basic Microbiology 2011, 51, 372 – 384
Bacterial community within uranium ore
379
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
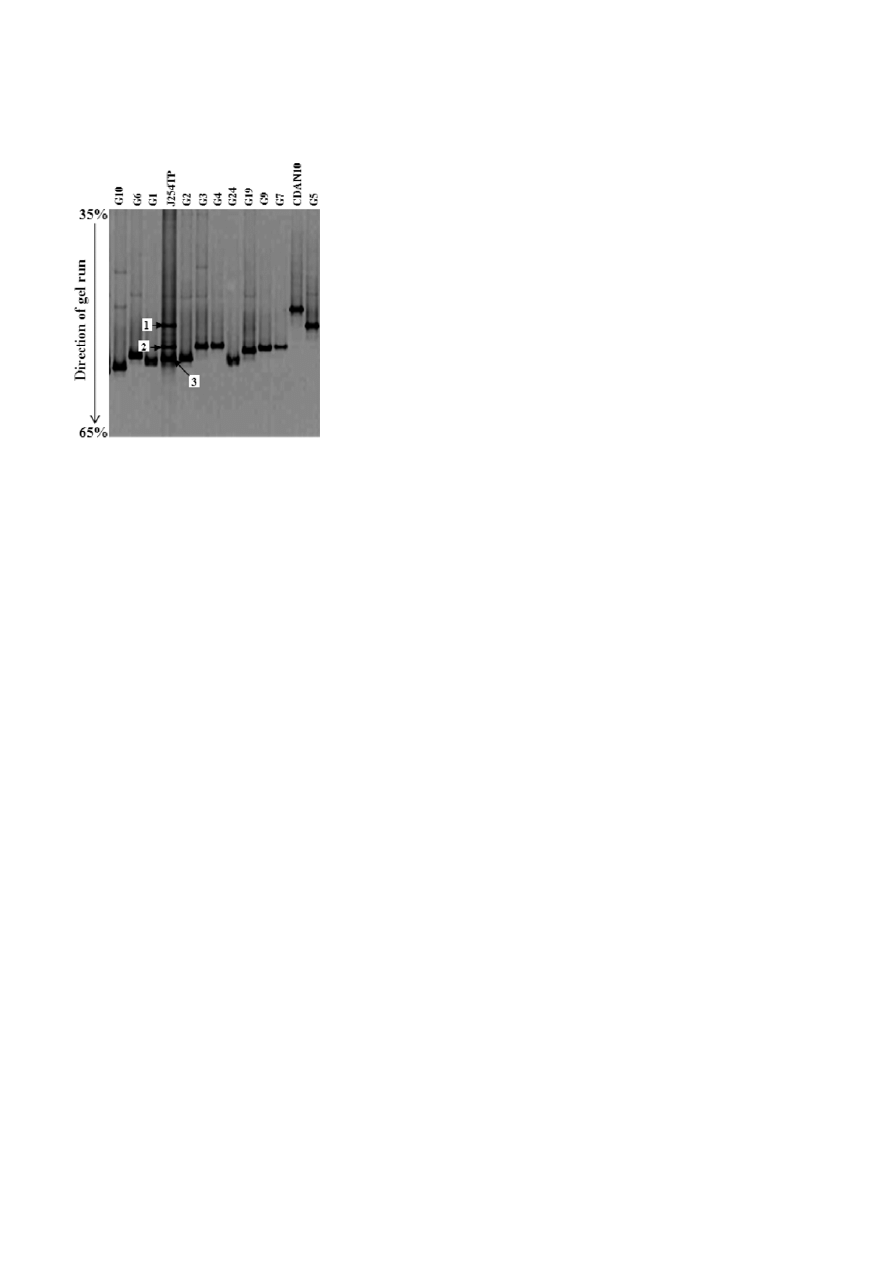
Figure 4. Negative image of a perpendicular DGGE gel with the
PCR products obtained with the primer set GC-357F and 518R. The
lane designated as J254TP was from the total metagenome, and
the adjoining lanes were from different clones representing the
dominant OTU (G1 to G10). Excised DGGE bands are indicated by
numbered arrows.
to be members of the Xanthomonadaceae, with high
affinity to S. maltophilia (Fig. 3b). The family Enterobac-
tericeae was represented only by four pure culture iso-
lates (two RFLP groups), affiliated with uncultured Ente-
robacter (clone FRC583, EU268623) recovered from a
uranium-contaminated subsurface under biostimulated
conditions with high nitrate and nickel pressure, and
with cultured biphenyl/polychlorinated biphenyl-de-
grading Enterobacter (clone LY402, DQ659161).
β-Proteobacteria
This class was mainly represented by ribotypes from
library J254TP. Thirteen ribotypes, covering >30% of
the total community, were affiliated to the
β
-Pro-teo-
bacteria, showing high nucleotide similarity only to the
uncultured clones previously obtained from diverse ex-
treme/oligotrophic habitats including a hot spring, a
Holocene sediment, and subsurface water, etc. (Fig. 1b).
Phylogenetic analysis indicated that, within this class,
nine ribotypes belonged to the family Burkholderiaceae.
Among these groups, the sequences representing a
number of abundant ribotypes showed distinct rela-
tionship to oligotrophic Limnobacter bacteria, along with
uncultured clones. The affinity with the genus Limno-
bacter was also observed for isolate J54CX showing strong
affinity to the thiosulfate-utilizing L. thiooxidans (Fig. 3b).
Isolate J54C19 showed affiliation to Alcaligenes sp. A
number of less abundant ribotypes from both the li-
braries showed relationship to hydrogen-oxidizing fac-
ultatively autotroph Hydrogenophaga spp., along with
other uncultured bacterial clones previously retrieved
from the Kalahari Shield and a thermal spring (Figs. 1b
and 2b). Among the relatively low-abundant groups
from library J254TP, the relationship to the methane-
metabolizing bacterium Methylophilus sp. and the unclas-
sified
β
-proteobacterium Imtechium assamiensis (AY54-
4767) was noticed (Fig. 1b).
α-Proteobacteria
A member of the class
α
-Proteobacteria was relatively
more abundant in the plate-washed community
(J254PW). Three ribotypes from this library showed
close relationship to denitrifying Ochrobactrum spp. (Fig.
2b). From the library J254TP, only one ribotype that
showed high sequence identity to uncultured clones
from the Kalahari Shield indicated its relationship to
metal- (Te, Se and Rh) and nitrate-reducing Rhodobacter
spp. (Fig. 1b).
Actinobacteria and Firmicutes
Members of these two phyla were detected only within
the pure culture isolates. Four isolates, namely J54CQ1,
J54CL, J54CAD and J54CD, belong to the Microbacteri-
aceae family. The first isolate showed a strong relation
with Yonghaparkia alkaliphila KSL-133 (DQ256088), a no-
vel member of the family, while the other three strains
showed close relationship to Microbacterium spp. previ-
ously isolated from a chromium-contaminated site. The
only isolate within the phylum Firmicutes, J54CU,
showed relationship to Exiguobacterium AT1b, isolated
from a Yellowstone National Park sample, with 99%
sequence identity.
Community analysis by DGGE
DGGE-based analysis of the total bacterial community
was done to validate our clone library-derived findings.
The total community metagenome as well as plasmid
DNA containing the cloned 16S rRNA genes from the
major ribotypes were used to amplify the V3 region.
The DGGE profile for the total metagenome yielded
three major bands, while all plasmids (containing clon-
ed 16S rRNA genes representing the major ribo-types
from library J254TP) yielded single bands (Fig. 4). As
evident from Fig. 4, the first, second and third band
from the community DNA co-migrated, at the same
position, with the bands from ribotypes representing
the bacterial genera Acinetobacter, P. mexicana, and un-
cultured Pseudomonadaceae clones, respectively. Sequen-
ce analysis of individual DGGE bands further revealed
that, while the first band is composed of both α- and
γ
-Proteobacteria, the second and third bands represen-
ted only
β
- and
γ
-Proteobacteria, respectively (Table 2).
Particularly, out of the five clones sequenced from the

380
E. Islam and P. Sar
Journal of Basic Microbiology 2011, 51, 372 – 384
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Table 2. Taxonomic affiliation of the 16S rRNA gene clones retrieved from the major DGGE bands.
DGGE band no. Band clone
(Acc. No.)
BLAST match (Acc. No.)
Identity
(%)
Putative groups
B11 (GU723258)
Clone JUMSS254TP-155(G8) (FJ804434)
100
γ
-Proteobacteria
B12 (GU723259)
Uncultured α-proteobacterium (AM408957)
99
α
-Proteobacteria
B13 (GU723260)
Uncultured Acinetobacter sp. (GQ390224)
99
γ
-Proteobacteria
B14 (GU723261)
Uncultured Acinetobacter sp. (GQ390224)
100
γ
-Proteobacteria
1
B15 (GU723262)
Uncultured α-proteobacterium (AM408957)
98
α
-Proteobacteria
B21 (GU723263)
Clone J254TP-29(G17) (FJ804442)
99
β
-Proteobacteria
B22 (GU723264)
Clone J254TP-29(G17) (FJ804442)
99
β
-Proteobacteria
B23 (GU723265)
Clone J254TP-29(G17) (FJ804442)
100
β
-Proteobacteria
B24 (GU723266)
Clone J254TP-29(G17) (FJ804442)
100
β
-Proteobacteria
2
B25 (GU723267)
Clone J254TP-29(G17) (FJ804442)
94
β
-Proteobacteria
B31 (GU723268)
Clone JUMSS254TP-9(G2) (FJ804428)
99
γ
-Proteobacteria
B32 (GU723269)
Clone JUMSS254TP-9(G2) (FJ804428)
98
γ
-Proteobacteria
B34 (GU723270)
Clone JUMSS254TP-9(G2) (FJ804428)
99
γ
-Proteobacteria
B35 (GU723271)
Clone JUMSS254TP-71(G1) (FJ804427)
98
γ
-Proteobacteria
3
B36 (GU723272)
Clone JUMSS254TP-71(G1) (FJ804427)
98
γ
-Proteobacteria
first-band library, two were similar to the uncultured
α
-Proteobacteria clone B716 while the other three
showed high identity (100%) to the sequence represent-
ing ribotype G8, which is closely affiliated to Acinetobac-
ter or uncultured Moraxellaceae of the
γ
-Proteobacteria.
For the second band, all sequences showed high iden-
tity to the sequence of the ribotype G17, which is affi-
liated to uncultured Burkholderiaceae. Of note, se-
quences representing the third band showed high
(≥98%) identity only to the most dominant ribotypes,
G1 and G2, of J254TP, thus indicating their affiliation to
uncultured Pseudomonadaceae. Based on this DGGE
data, we inferred that the community is mainly com-
posed of uncultured Pseudomonadaceae, Burkholderi-
aceae, and Moraxellaceae members, overall substantiat-
ing our clone library-based findings.
Resistance to uranium and other heavy metals
of major culturable heterotrophic bacteria
Uranium resistance studies using acidic NaCl solution
indicated that, except for Pseudomonas spp. J54C4 and
J54C18 and Yonghaparkia J54CQ1, all strains were able
to tolerate both uranium and an acidic nutrient-
deprived state (Table 3). Strains that could survive very
well in the presence of U for 1 h were mostly belonging
to Stenotrophomonas, Microbacterium, Enterobacter and
Pseudomonas. Among these isolates, Stenotrophomonas
spp. J54C22 and J54C12, Microbacterium spp. J54CAD and
J54CD, Pseudomonas sp. J54C18 and Acinetobacter sp.
J54C25W could withstand U even for up to 4 h. Under
similar conditions, E. coli cells showed strong sensitivity
to uranium in low pH, with two orders of magnitude
lower CFU counts following only 1 h of exposure to U,
confirming that sensitivity to the chemical toxicity of
uranium was readily detected in our experiment. Our
results indicate that uranium resistance is well distrib-
uted among the diverse microbial groups and that
>60% of the isolated strains tested were resistant to
uranium for at least 1 h.
In addition to uranium resistance, the ability of a
number of isolates to withstand considerably high con-
centrations of other heavy metals (Ni, Zn and Cu) was
noticed (Table 3). Ni and Zn resistance was found to be
more frequent, followed by resistance to Cu, Cr and Cd.
Eleven strains related to Pseudomonas, Stenotrophomonas,
Alcaligenes or Acinetobacter were found to be resistant to
Zn, with a maximum tolerable concentration (MTC) of
2 mM. Most of the Zn-resistant isolates were also able
to withstand Ni (2 mM) and Cr (0.4–0.5 mM). Compared
to this, only few strains (Alcaligenes sp. J54C19, Yongha-
parkia sp. J54CQ1, Acinetobacter sp. J54C21, and Entero-
bacter sp. J54C13) showed Cu or Cd resistance up to 1 or
1.5 mM, respectively. The metal resistance capacities of
our isolates were found to be mostly comparable (ex-
cept for Cd and Zn) to the multiple-metal-resistant
C. metallidurans bacteria tested along with the isolates.
Discussion
The present study describes the bacterial diversity from
a subsurface U-ore and the ability of the culturable
bacteria to withstand uranium and other metals. This
investigation creates opportunities to examine the mi-
crobial community structure and ecology within a geo-
chemically distinct habitat, and explores the potential
of microbial resources for their possible exploitation in
relevant bioprocesses (bioremediation, biomining, bio-
mineralization, and bioprospecting).
The uranium ore sample used in this study showed
characteristic chemical properties (high contents of
uranium and other metals, near neutral pH, moderate

Journal of Basic Microbiology 2011, 51, 372 – 384
Bacterial community within uranium ore
381
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
Table 3.
Uranium and other heavy metal resistance properties of isolated bacterial strains.
CFU 1 h
CFU 4 h
Metal io
n used (mM)
Bacterial
strains
CFU (washed)
a
without U
with U
without U
with U
Cu
Cr
Cd
Ni
Zn
GC
Acinetobacter
sp. J54C10A
(5.53 ± 0.52)
× 10
6
(4.6 ± 1.02)
× 10
6
(2.26
±
0.55)
× 10
5
(3.67
±
0.8)
× 10
6
(1.33
±
0.57)
× 10
4
0.5
0.5 <0.1
<0.5
0.5 –ve
Stenotrophomonas
sp. J54C22
(3.48 ± 0.49)
× 10
7
(1.91
±
0.31)
× 10
7
(3.69
±
1.04)
× 10
7
(2.64
±
1.04)
× 10
7
(3.09
±
0.56)
× 10
7
0.5
0.4
0.3
2
2
–ve
Enterobacter
sp. J54C13
(5.36 ± 1.52)
× 10
7
(3.07
±
0.51)
× 10
7
(5.33
±
1.53)
× 10
6
(4.73
±
0.35)
× 10
7
(4.00
±
1.73)
× 10
6
0.5
0.4
1.5
1
2
–ve
Enterobacter
sp. J54C6
(5.58 ± 2.28)
× 10
8
(3.91
±
0.27)
× 10
8
(3.79
±
0.54)
× 10
7
(3.41
±
1.78)
× 10
7
(1.57
±
0.73)
× 10
6
0.5
0.4
0.3
2
2
–ve
Enterobacter
sp. J54C24A
(9.92 ± 3.24)
× 10
7
(5.13
±
0.35)
× 10
7
(4.20
±
2.52)
× 10
7
(1.05
±
0.17)
× 10
7
(4.30
±
0.86)
× 10
7
0.5
0.2 0.5
0.5
0.5 –ve
Stenotrophomonas
sp. J54C12
(1.44 ± 0.05)
× 10
9
(1.15
±
0.06)
× 10
9
(1.66
±
0.06)
× 10
9
(2.15
±
0.05)
× 10
9
(1.81
±
0.04)
× 10
9
0.5
0.4
0.3
1
2
–ve
Microbacterium
sp. J54CL
(8.26 ± 0.62)
× 10
8
(7.78
±
0.80)
× 10
8
(2.86
±
0.10)
× 10
8
(5.82
±
0.44)
× 10
8
(3.00
±
0.87)
× 10
6
0.2
0.4
0.1
0.5
<0.5
+ve
Acinetobacter
sp. J54C21
(4.40 ± 1.15)
× 10
7
(5.00
±
0.78)
× 10
7
6.67
±
1.53
× 10
6
(4.87
±
0.90)
× 10
7
(6.33
±
0.52)
× 10
6
1
0.5 <0.1
<0.5
0.5 +ve
Enterobacter
sp. J54C26
(6.57 ± 0.49)
× 10
7
(5.57
±
0.72)
× 10
7
(1.68
±
0.20)
× 10
7
(8.15
±
0.27)
× 10
7
(3.00
±
0.46)
× 10
6
0.5
<0.2
<0.1
2
2
–ve
Microbacterium
sp. J54CAD
(6.76 ± 0.04)
× 10
9
(6.46
±
0.3)
× 10
9
(6.33
±
0.21)
× 10
9
(4.54
±
0.25)
× 10
9
(4.19
±
0.12)
× 10
9
0.5
<0.2 <0.1
0.5
<0.5 +ve
Exiguobacterium
sp. J54CU
(5.60 ± 1.05)
× 10
7
(3.70
±
0.36)
× 10
7
(1.07
±
0.51)
× 10
7
(3.50
±
0.61)
× 10
7
<1
× 10
6
0.5
0.4
0.1
1
2
+ve
Pseudomonas
sp. J54CV1
(4.48 ± 0.87)
× 10
6
(1.20
±
0.27)
× 10
6
(8.17
±
3.69)
× 10
4
<1 × 10
4
<1
× 10
4
0.2
0.2
<0.1
0.5
<0.5
–ve
Microbacterium
sp. J54CD
(3.41 ± 0.14)
× 10
8
(3.63
±
0.06)
× 10
8
(2.28
±
0.15)
× 10
8
(2.88
±
0.17)
× 10
8
(2.49
±
0.14)
× 10
8
0.2
0.4
0.1
0.5
<0.5
+ve
Stenotrophomonas
sp. J54C14
(2.16 ± 0.27)
× 10
7
(1.76
±
0.07)
× 10
7
(4.37
±
0.73)
× 10
6
(1.68
±
0.36)
× 10
7
(1.33
±
058)
× 10
6
0.5
0.4
0.3
0.5
2
–ve
Pseudomonas
sp. J54C18
(3.79 ± 0.48)
× 10
7
(6.59
±
1.12)
× 10
6
(3.56
±
0.55)
× 10
6
(2.93
±
0.34)
× 10
6
(1.47
±
0.19)
× 10
6
0.5
<0.2
0.1
2
2
–ve
Yonghaparkia
sp
. 54CQ1
(5.21 ± 0.98)
× 10
8
(4.10
±
0.36)
× 10
8
(3.27
±
0.45)
× 10
8
(6.26
±
0.51)
× 10
7
(4.47
±
0.75)
× 10
7
1
0.2
0.1
2
2
–ve
Pseudomonas
sp. J54C4
(1.22 ± 0.24)
× 10
10
(5.55
±
0.38)
× 10
9
(1.77
±
0.35)
× 10
7
(5.73
±
0.25)
× 10
8
(8.44
±
1.5)
× 10
6
0.2
<0.2
0.1
0.5
0.5
–ve
Alcaligenes
sp. J54C19
(2.88 ± 0.43)
× 10
7
(2.99
±
0.13)
× 10
7
(1.03
±
0.16)
× 10
7
(1.83
±
0.10)
× 10
7
(9.23
±
0.61)
× 10
5
1
<0.2
0.3
2
2
–ve
Acinetobacter
sp. J54C25W
(8.57 ± 1.16)
× 10
7
(3.20
±
0.26)
× 10
7
(2.77
±
0.25)
× 10
7
(2.20
±
0.98)
× 10
7
(1.03
±
0.15)
× 10
7
0.5
<0.2
0.1
2
2
–ve
D. radiodurans
(6.37 ± .06)
× 10
8
(1.06
±
0.15)
× 10
7
(2.5 ± 0.5)
× 10
6
(7.13
±
0.12)
× 10
6
<1
× 10
5
ND
ND ND
ND
ND +ve
C. metallidurans
ND
ND ND
ND ND
1
0.4
3.5
2
5
–ve
E. coli
. JM109
(1.87±0.15)
× 10
8
(9.17
±
0.65)
× 10
7
(7.33
±
0.57)
× 10
6
(6.05
±
0.15)
× 10
6
(4.83
±
2.02)
× 10
5
<0.2
<0.2 <0.1
<0.5
<0.5 –ve
a
CFU count at 0 h. GC = Gram characteristics, ND = Not determined.

382
E. Islam and P. Sar
Journal of Basic Microbiology 2011, 51, 372 – 384
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
total organic carbon content) along with a significant
presence of microbial biomass as evident from the mi-
crobial counts. Compared to the total cell counts report-
ed previously for other metal radionuclide-contaminat-
ed mine/subsurface environments [10, 25], the count
within the ore was found to be relatively high, al-
though it was lower (by one order of magnitude) than
the value normally obtained from other pristine habi-
tats [26]. To gain insight into the bacterial community
structure present within the sample, both total- and
culturable-community DNA were subjected to clone
library studies, followed by DGGE analysis and charac-
terization of the aerobic heterotrophic bacteria in pure
culture form. The high diversity indices along with the
relatively low equitability as ascertained through clone
library analysis, particularly within the total commu-
nity, indicated a diverse and uneven community struc-
ture. Compared to a recent report on diversity and
biogeography of bacterial communities from various
habitats, the observed values indicated a significantly
diverse bacterial assemblage within the ore [27]. Al-
though heavy metal and radionuclide contamination
have been shown to limit the microbial diversity in
several metal- and uranium-rich environments [9, 11],
the present observation indicating the assemblage of
different bacterial species (as ribotypes/RFLP groups)
perhaps represented a distinct phenomenon. In spite of
the presence of several toxic metals along with ura-
nium, the observed bacterial abun-dance and Shannon
diversity index could possibly be correlated with the
higher total organic carbon content and the circum-
neutral pH of the sample. Both these environmental
factors are known to have a strong influence on micro-
bial diversity, by regulating the bioavailability of metal
ions and/or by enhancing the microbial potential to
withstand metal toxicity [9, 27].
With respect to the bacterial community composi-
tion, the most abundant bacterial groups within the
ore were affiliated to the Proteobacteria (
γ
>
β
>
α
sub-
divisions), Actinobacteria and Firmicutes. Although the
presence of Proteobacteria, Actinobacteria and Firmi-
cutes was previously noticed within various metal- and
radionuclide-contaminated sites [9, 10, 28], the observed
predominance of the
γ
-Proteobacteria in the present
sample is noteworthy. In contrast to previous reports
on frequent detection of
α
- and
β
-Proteobacteria from
various subsurface habitats including radioactive waste
repositories and gold mines [2, 10], the prevalence of
the
γ
-Proteobacteria in the present sample corroborates
well with earlier findings that report the dominance of
bacterial members affiliated to the
γ
-Proteobacteria at a
uranium depository site (Gunnison, CO, USA) and in
uranium mill tailings (Shiprock, NM, USA) [12]. The
observed correlation between the abundance of
γ
-Pro-
teobacteria within the U-ore (or ore-related materials)
could be attributed to their capability to survive and
flourish in the specific geochemical environment with-
in the U-ore. The detailed phylogenetic analysis of the
16S rRNA genes representing the ribotypes and isolates
not only provided their taxonomic relationship but
also, more importantly, indicated their strong related-
ness to microorganisms with environmentally signifi-
cant attributes. In general, most of the members of
each phylogenetic group were shown to be related to
organisms having metabolic properties highly relevant
to microbial life and their role within the mineral ore.
Among the
γ
-Proteobacteria members, the predomi-
nance of the Pseudomonadaceae was noticeable. Al-
though there is no previous report on the abundance of
Pseudomonadaceae members within mineral ore, their
prevalence in the test sample is not surprising, since
these groups of bacteria are highly ubiquitous with
great metabolic and genetic diversity [29]. Following the
Pseudomonadaceae, the family Moraxellaceae repre-
sented the second most abundant group within the
γ
-
Proteobacteria, with strong affiliation of its members
to Acinetobacter spp. The observed relation of all the
Moraxellaceae members to Acinetobacter seems to be
environmentally significant, since this genus is report-
ed to have the abilities to synthesize polyphosphate
bodies, sequester heavy metals and degrade organic
compounds [30]. Bacterial members affiliated to P. mexi-
cana were detected as most abundant group within the
plate wash community, although they were not repre-
sented by the pure culture isolates. However, the other
major bacterial genus, S. maltophilia, detected in the
plate wash community represented the maximum
number of isolates as well. Both the P. mexicana and
S. maltiphilia strains were previously reported from U-
contaminated environments and found to have versa-
tile abilities to interact with metallic contaminants [11,
21]. In line with previous investigators, we have also
observed elevated uranium resistance abilities of the
Stenotrophomonas spp. isolated from the ore sample (re-
fer to the section on resistance to uranium and other
metals). Within the class
β
-Proteobacteria, our analysis
revealed a closer relationship of its members with Lim-
nobacter sp., Hydrogenophaga sp., Methylophilus sp., and
Alcaligenes sp., previously recovered from various oli-
gotrophic environments, with potential in thiosulfate
oxidation, metabolism of molecular hydrogen, methane
and heavy metals sequestration, respectively. Represen-
tatives of the
α
-Proteobacteria were affiliated to Rhodo-
bacter sp. and Ochrobactrum sp. Rhodobacter is known to

Journal of Basic Microbiology 2011, 51, 372 – 384
Bacterial community within uranium ore
383
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
reduce telluride, selenite, rhodium and nitrate, while
Ochrobactrum is a denitrifying bacterium capable of
tolerating high levels of reactive nitrogen oxides and
could sequester heavy metals by exopolysaccharides.
The presence of Microbacterium (Actinobacteria) and
Exiguobacterium (Firmicutes) within the uranium ore
corroborates well with previous reports on their ability
to occupy diverse and extreme ecological niches rang-
ing from volcanic deposits, deep sea sediments, radioac-
tive waste depositories, and hot springs to Siberian
permafrost and glacial ice, etc. [31, 32].
One of the most important physiological properties
necessary for the survival of inhabitant microbes with-
in the uranium- and other toxic metal-rich environ-
ments of U-ore is their ability to withstand such toxic-
ity. As evident from the present study, a large pro-
portion of the bacteria, affiliated to diverse taxonomic
groups, are resistant to one or multiple metals includ-
ing uranium. In our opinion, these resistance proper-
ties might have evolved as survival strategies within the
inhabitant microorganisms. A mechanism (viz. horizon-
tal gene transfer (HGT)) that facilitates acquisition of
the metal resistance determinant by relatively vulner-
able species is well reported in metal- and radionuclide-
contaminated habitats [18] and, in this regard, bacterial
isolates from the uranium ore-rich environment of
Jaduguda have shown the incidence of such HGT for
the metal resistance genes nik and copA [33]. Compared
to other heavy metals, uranium resistance by bacteria
is less well studied and there are only few strains (Mi-
crobacterium sp., Arthrobacter sp., Sphingomonas sp. and
Bacillus sp.) identified from uranium mines/uranium-
contaminated sites that showed resistance to this
highly toxic element at low pH [18, 24, 31]. In contrast
to previous reports that showed the Gram-positive acti-
nobacteria (Microbacterium sp. and Arthrobacter sp.) as
predominant U-resistant organisms, our findings on the
dominance of Gram-negative members like Stenotropho-
monas, Acinetobacter, Enterobacter and Pseudomonas along
with Microbacterium presented a characteristic observa-
tion. High metal resistance by most of these bacterial
genera has previously been observed, and in many cases
the metal resistance properties are well characterized
[18, 31].
Acknowledgements
This work was supported by grants from BRNS, De-
partment of Atomic Energy, Government of India. The
authors are grateful to the Uranium Corporation of
India Ltd., Jaduguda, for kind support. Ekramul Islam
gratefully acknowledges the financial assistance re-
ceived from the Council of Scientific and Industrial
Research, India.
References
[1] Dong, H., 2008. Microbial life in extreme environments:
Linking geological and microbiological processes. In: Di-
lek, Y., Furnes, H., Muehlenbachs, K., (eds.), Links be-
tween Geological Processes, Microbial Activities and Evo-
lution of Life., Springer, Berlin, pp. 237–280.
[2] Rastogi, G., Osman, S., Kukkadapu, R., Engelhard, M. et al.,
2010. Microbial and mineralogical characterizations of
soils collected from the deep biosphere of the former
Homestake gold mine, South Dakota. Microb. Ecol., DOI:
10.1007/s00248-010-9657-y.
[3] Wanger, G., Onstott, T.C., Southam, G., 2008. Stars of the
terrestrial deep subsurface: A novel ‘star-shaped’ bacterial
morphotype from a South African platinum mine. Geo-
biology, 6, 325–30.
[4] Southam, G., Saunders, J.A., 2005. The geomicrobiology of
ore deposits. Econ. Geol., 100, 1067–1084.
[5] Pfiffner, S.M., Cantu, J.M., Smithgall, A., Peacock, A.D.
et al., 2006. Deep subsurface microbial biomass and com-
munity structure in Witwatersrand Basin mines. Geomic-
robiol. J., 23, 431–442.
[6] Fukuda, A., Hagiwara, H., Ishimura, T., Kouduka, M.,
Ioka, S., Amano, Y., Tsunogai, U., Suzuki, Y., Mizuno, T.,
2010. Geomicrobiological properties of ultra-deep granitic
groundwater from the Mizunami Underground Research
Laboratory (MIU), central Japan. Microb. Ecol., 60, 214–225.
[7] Papineau, D., Walker, J.J., Mojzsis, S.J., Pace, N.R., 2005.
Composition and structure of microbial communities
from stromatolites of Hamelin Pool in Shark Bay, Wes-
tern Australia. Appl. Environ. Microbiol., 71, 4822–4832.
[8] Valenzuela, L., Chi, A., Beard, S., Orell, A. et al., 2006.
Genomics, metagenomics and proteomics in biomining
microorganisms. Biotechnol. Adv., 24, 197–211.
[9] Rastogi, G., Osman, S., Vaishampayan, P.A., Andersen,
G.L., Stetler, L.D., Sani, R.K., 2010. Microbial diversity in
uranium mining-impacted soils as revealed by high-
density 16S microarray and clone library. Microb. Ecol.,
59, 94–108.
[10] Akob, D.M., Mills, H.J., Kostka, J.E., 2007. Metabolically
active microbial communities in uranium-contaminated
subsurface sediments. FEMS Microbiol. Ecol., 59, 95–107.
[11] Merroun, M.L., Selenska-Pobell, S., 2008. Bacterial interac-
tions with uranium: An environmental perspective. J.
Contam. Hydrol., 102, 285–295.
[12] Radeva, G., Selenska-Pobell, S., 2005. Bacterial diversity in
water samples from uranium wastes as demonstrated by
16S rDNA and ribosomal intergenic spacer amplification
retrievals. Can. J. Microbiol., 51, 910–923.
[13] Gadd, G.M., Metals, minerals and microbes: Geomicrobio-
logy and bioremediation. Microbiology, 156, 609–643.
[14] Oremland, R.S., Capone, D.G., Stolz, J.F., Fuhrman., J.,
2005. Whither or wither geomicrobiology in the era of

384
E. Islam and P. Sar
Journal of Basic Microbiology 2011, 51, 372 – 384
© 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.jbm-journal.com
‘community metagenomics’. Nat. Rev. Microbiol., 3, 572–
578.
[15] Rawlings, D.E., Johnson, D.B., 2007. The microbiology of
biomining: development and optimization of mineral-oxi-
dizing microbial consortia. Microbiology, 153, 315–324.
[16] Jackson, M.L., 1967. Soil Chemical Analysis. Prentice Hall
of India Pvt. Ltd., New Delhi.
[17] Kepner, R.L, Pratt, J.R., 1994. Use of fluorochromes for
direct enumeration of total bacteria in environmental
samples: Past and present. Microbiol. Rev., 58, 603–615.
[18] Martinez, R.J., Wang, Y., Raimondo, M.A., Coombs, J.M.,
Barkay, T., Sobecky, P.A., 2006. Horizontal gene transfer
of PIB-type ATPases among bacteria isolated from radio-
nuclide- and metal-contaminated subsurface soils. Appl.
Environ. Microbiol., 72, 3111–3118.
[19] Bhattacharyya, S., Chakrabarti, B.K., Das, A., Kundu, P.N.,
Banerjee, P.C., 1991. Acidiphilium symbioticum sp. nov., an
acidophilic heterotrophic bacterium from Thiobacillus fer-
rooxidans cultures isolated from Indian mines. Can. J. Mi-
crobiol., 37, 78–85.
[20] Olson, G.J., McFeters, G.A., Temple, K.L., 1981. Occurrence
and activity of iron- and sulfur-oxidizing microorganisms
in alkaline coal strip mine spoils. Mirob. Ecol., 7, 39–50.
[21] Reardon, C.L., Cummings, D.E., Petzke, L.M., Kinsall, B.L.
et al., 2004. Composition and diversity of microbial com-
munities recovered from surrogate minerals incubated in
an acidic uranium-contaminated aquifer. Appl. Environ.
Microbiol., 70, 6037–6046.
[22] Muyzer, G., de Waal, E.C., Uitterlinden, A.G., 1993. Profil-
ing of complex microbial populations by denaturing gra-
dient gel electrophoresis analysis of polymerase chain re-
action-amplified genes coding for 16S rRNA. Appl.
Environ. Microbiol., 59, 695–700.
[23] Tamura, K., Dudley, J., Nei, M., Kumar, S., 2007. MEGA4:
Molecular Evolutionary Genetics Analysis (MEGA) soft-
ware version 4.0. Mol. Biol. Evol., 24, 1596–1599.
[24] Suzuki, Y., Banfield, J.F., 2004. Resistance to, and accumu-
lation of, uranium by bacteria from a uranium-conta-
minated site. J. Geomicrobiol., 21, 113–121.
[25] Kieft, T.L., Kovacik, W.P., Ringelberg, D.B., White, D.C.,
Haldeman, D.L., Amy, P.S., Hersman, L.E., 1997. Factors
limiting microbial growth and activity at a proposed high-
level nuclear repository, Yucca Mountain, Nevada. Appl.
Environ. Microbiol., 63, 3128–3133.
[26] Janssen, P.H., Yates, P.S., Grinton, B.E., Taylor, P.M., Sait,
M., 2002. Improved culturability of soil bacteria and isola-
tion in pure culture of novel members of the divisions
Acidobacteria, Actinobacteria, Proteobacteria, and Verru-
comicrobia. Appl. Environ. Microbiol., 68, 2391–2396.
[27] Fierer, N., Jackson, R.B., 2006. The diversity and biogeo-
graphy of soil bacterial communities. Proc. Natl. Acad.
Sci. USA, 103, 626–631.
[28] Fields, M.W., Yan, T., Rhee, S.K., Carroll, S.L. et al., 2005.
Impacts on microbial communities and cultivable isola-
tes from groundwater contaminated with high levels of
nitric acid-uranium waste. FEMS Microbiol. Ecol., 53,
417–428.
[29] Peix, A., Ramirez-Bahena, M.H., Velazquez, E., 2009. His-
torical evolution and current status of the taxonomy of
genus Pseudomonas. Infect. Genet. Evol., 9, 1132–1147.
[30] OECD, 2008. Consensus document on information used in
the assessment of environmental applications involving
Acinetobacter. Series on Harmonization of Regulatory Over-
sight in Biotechnology No. 46. Organisation for Economic
Co-operation and Development (OECD), Paris.
[31] Nedelkova, M., Merroun, M.L., Rossberg, A., Hennig, C.,
Selenska-Pobell, S., 2007. Microbacterium isolates from the
vicinity of a radioactive waste depository and their inter-
actions with uranium. FEMS Microbiol. Ecol., 59, 694–
705.
[32] Chen, S., Shao, Z., 2009. Isolation and diversity analysis of
arsenite-resistant bacteria in communities enriched from
deep-sea sediments of the Southwest Indian Ocean Ridge.
Extremophiles, 13, 39–48.
[33] Chaudhary, S., 2010. Uranium and other heavy metal
resistance and accumulation in a Pseudomonas aeruginosa
strain: Potential in bioremediation. Ph.D. thesis, Indian
Institute of Technology Kharagpur, Kharagpur, India.
((Funded by
• BRNS, Department of Atomic Energy, Government of
India))
Wyszukiwarka
Podobne podstrony:
jobm 201000013
jobm 201000298
jobm 201000191
jobm 201000321
jobm 201000018
jobm 201000214
jobm 201000067
jobm 201000037
jobm 201000074
jobm 201000280
jobm 201000385
jobm 201000198
jobm 201000458
jobm 201000147
jobm 201000520
jobm 201000342
jobm 201000420
jobm 201000364
jobm 201000317
więcej podobnych podstron