6771814991
Marcin Sieńczyk Załącznik 2
Katepsyna G
Bardzo interesującą z punktu widzenia specyficzności substratowej jest katepsyna G, która u ludzi posiada podwójną — chymotrypsynową i trypsynową aktywność. Co ciekawe, dualny charakter wystąpił w ewolucji dopiero u człowieka, małp człekokształtnych i małp Starego Świata, natomiast u pozostałych ssaków obserwowana jest aktywność typu chymotrypsynowego.12 Pierwszą z przebadanych przeze mnie grupę inhibitorów katepsyny G stanowiły proste Cbz-blokowane fosfono-we analogi argininy, spośród których najsilniejszą zdolność hamowania aktywności proteolitycznej CatG wykazały Cbz-(4-NH2-Phg)p(OPh)2 (ko6s/I = 300 M-1s-1) oraz Cbz-(4-GuPhg)p(OPh)2 (1, k0&,j/I — 412 M-1s-1).13 Związek 1 wyselekcjonowałem do dalszych badań mających na celu określenie wpływu struktury grup estrowych na aktywność inhibitorową. Spośród przebadanych pochodnych najwyższą aktywnością wobec katepsyny G (k0{,s/I = 15 600 M-1s-1) charakteryzowała się pochodna zawierająca grupy fenylo-4-tiometylowe (2) jako ugrupowania estrowe. Nawiązana współpraca z prof. Krzysztofem Rolką i dr. hab. Adamem Lesnerem (Wydział Chemiczny, Uniwersytet Gdański) pozwoliła na przeprowadzenie dalszych badań nad optymalizacją struktury fosfono-wych inhibitorów katpsyny G. W oparciu o strukturę opracowanego przez Zespół prof. Rolki sub-stratu ludzkiej katepsyny G,14 do struktury związku 2 wprowadzono analogiczny N-końcowy łańcuch polipeptydowy. Otrzymany inhibitor (3, Ac-Phe-Val-Thr-(4-GuPhg)p(O-C6H4-4-S-CH3)2) wykazał ogromną siłę hamowania aktywności katepsyny G (k0(,s/I = 256 000 M_1s_1) będąc jednym z najsilniejszych nieodwracalnych inhibitorów katepsyny G opisanych do tej pory w literaturze. Ponadto związek 3 wykazał wysoką selektywność działania będąc około 253-razy mniej aktywnym wobec chymotrypsyny, 20-razy mniej wobec trypsyny i ponad 320-razy mniej wobec trombiny. Wykonane przeze mnie badania stanowią jedno z klasycznych podejść chemii medycznej stosowanych w poszukiwaniu nowych, aktywnych biologicznie związków chemicznych, gdzie na drodze kolejnych etapów optymalizacji wyjściowej struktury o relatywnie niskiej aktywności dochodzi się do wysoce specyficznego i selektywnego inhibitora docelowego enzymu (Rys.3).
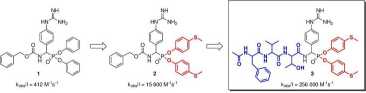
Rysunek 3: Strategia projektowania fosfonowych inhibitorów katepsyny G.
12 Raymond W.W., Trivedi N.N., Makarova A., Ray M., Craik C.S., Caughey G.H.: How immune peptidases change specificity: cathepsin G gained tryptic function but lost efficiency during primate euolution, J. Immunol., 2010, 185, 5360-5368.
13 [H.l] Sieńczyk M., Lesner A., Wysocka M., Łęgowska A., Pietrusewicz E., Rolka K., Oleksyszyn J.: New potent cathepsin G phosphonate inhibitors, Bioorg. Med. Chem., 2008, 16, 8863-8867.
14 Wysocka M., Łęgowska A., Bulak E., Jaśkiewicz A., Miecznikowska H., Lesner A., Rolka K.: New chromogenie substrates of human neutrophil cathepsin G containing non-natural aromatic amino acid residues in position P(l) selected by combinatorial chemistry methods, Mol. Divers., 2007, 11, 93-99.
Wyszukiwarka
Podobne podstrony:
Marcin Sieńczyk Załącznik 2 ryny na elektrofilowy atom fosforu inhibitora (Rys.2).9 Struktura estrów
Marcin Sieńczyk Załącznik 2 Wysoka zdolność związku 3 do inaktywacji katepsyny G oraz wysoka
Marcin Sieńczyk Załącznik 2 tygenów in vitro,17 podjęto próbę określenia roli katepsyny G w procesow
Marcin Sieńczyk Załącznik 2 dendrytyczne, mDCl) zastosowałem specyficzne inhibitory katepsyny G. W t
Marcin Sieńczyk Załącznik 2 W ramach kontynuacji podjętych badań dotyczących roli katepsyny G
Marcin Sieńczyk Załącznik 2 detekcji aktywnej katepsyny G w lizacie śledziony wołowej,23 potwierdzaj
Marcin Sieńczyk Załącznik 2 Rysunek 4: Niskocząsteczkowe sondy molekularne do detekcji neutrofilowyc
Marcin Sieńczyk Załącznik 2 Do niedawna w literaturze naukowej opisano zaledwie kilka przykładów
Marcin Sieńczyk Załącznik 2 która wykazała także prawie absolutną selektywność działania,
Marcin Sieńczyk Załącznik 2 Rysunek 5: Wzory ogólne otrzymanych na drodze kondensacji wieloskładniko
Marcin Sieńczyk Załącznik 2 Marcin Sieńczyk Załącznik 2 I Imię i Nazwisko: Marcin Sieńczyk H
Marcin Sieńczyk Załącznik 2 IV Wykaz osiągnięć naukowo-badawczych Tabela 1: Wykaz
Marcin Sieńczyk Załącznik 2 V Wskazanie osiągnięcia wynikającego z art. 16 ust. 2 ustawy z dnia 14 m
Marcin Sieńczyk Załącznik 2 H.6 Palesch D., Sieńczyk M.. Oleksyszyn J., Reich M., Wieczerzak E., Boe
Marcin Sieńczyk Załącznik 2 b) Omówienie celu naukowego/artystycznego ww. pracy/prac i osiągniętych
Marcin Sieńczyk Załącznik 2 Katepsyna G Proteinaza 3 NSP4 Rysunek 1: Struktura ludzkiej katepsyny G
Marcin Sieńczyk Załącznik 2 przyczyn śmierci na świecie.1 Zaobserwowano także, że neutrofilowa elast
Bardzo ważną z punktu widzenia zastosowań praktycznych grupę zagadnień sieciowych, stanowią zagadnie
więcej podobnych podstron