6771814999
Marcin Sieńczyk Załącznik 2
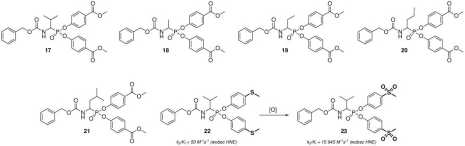
która wykazała także prawie absolutną selektywność działania, pozostając nieaktywną wobec chy-motrypsyny, trypsyny czy świńskiej elastazy trzustkowej. Ogólnie, dla wszystkich z otrzymanych analogów najwyższą aktywność inhibitorową zaobserwowałem dla pochodnych zawierających podstawnik karboksymetylowy w pozycji para fenylowych pierścieni estrowych. Przykładowo, najsilniejszym inhibitorem HNE spośród analogów alaniny był związek 18 (ka/K* = 7 722 M_1s_1), wśród analogów Abu — pochodna 19 (ka/Kj = 22 031 M-1s-1), natomiast dla analogów nor-waliny — 20 (ka/K* = 17 858 M-1s-1). Fosfonowe analogi norleucyny praktycznie pozbawione były aktywności inhibitorowej w badanym zakresie stężeń. Podobnie w przypadku fosfonowych analogów leucyny, które nie wykazały aktywności inhibitorowej wobec elastazy, okazały się jednak silnymi inhibitorami chymotrypsyny, spośród których najsilniejsza była pochodna 21 (ka/K, = 17 858 M_1s_1). Najbardziej interesującym okazał się fakt „aktywacji” nieaktywnej wobec HNE pochodnej Cbz-Valp(0-C6H4-4-SCH3)a (22) do Cbz-Valp(0-C6H4-4-S02CH3)2 (23, ka/K* = 15 945 M_1s_1). Utlenienie grup -S-CH3 do -SOa_CH3 prowadzące do jego aktywacji może być wykorzystane jako swoisty przełącznik molekularny in vivo, przykładowo w stanach zapalnych czy rozwoju nowotworów, którym towarzyszy wysoki potencjał utleniający. W badaniach in mtro przeprowadzonych na wybranych liniach komórek nowotworowych (A549, Lovo, LovoDX, MCF7) najsilniejszy efekt antyproliferacyjny wykazała pochodna 17, która okazała się znacznie bardziej aktywna od Sivelestatu, jedynego dotychczas zatwierdzonego do użytku inhibitora elastazy. Co więcej, efekt antyproliferacyjny był proporcjonalny do zdolności hamowania elastazy przez testowane związki w badaniach in vitro.
Kolejny aspekt realizowanych przeze mnie w ostatnich latach prac badawczych stanowił niejako naturalne rozwinięcie opracowanych w trakcie realizacji pracy doktorskiej metod otrzymywania fosfonowych peptydomimetyków.29 W zakresie realizowanych prac naukowych zaprojektowałem struktury, jak i samą koncepcję otrzymywania bibliotek fosfonowych inhibitorów ludzkiej neutrofi-1 a) Sieńczyk M., Kliszczak M., Oleksyszyn J.: Synthesis of isocyanide derivatives of a-aminoalkylphosphonate diphenyl esters, Tetrahedron Lett., 2006, J,7, 4209-4211; b) Sieńczyk M., Kliszczak M., Oleksyszyn J.: Nowe estry difenylowe kwasów l-izocyjanoalkanofosjonowych i sposób ich wytwarzania, Patent Polski 209628; c) Sieńczyk M., Kliszczak M., Maliszewska I., Zboińska E., Oleksyszyn J.: Sposób wytwarzania fosfonowych analogów pseudopeptydów i ich zastosowanie jako inhibitory enzymów piateolitycznych i czynników przeciwbakteryjnych, Patent Polski 212735.
Wyszukiwarka
Podobne podstrony:
Marcin Sieńczyk Załącznik 2 przyczyn śmierci na świecie.1 Zaobserwowano także, że neutrofilowa elast
Marcin Sieńczyk Załącznik 2 ryny na elektrofilowy atom fosforu inhibitora (Rys.2).9 Struktura estrów
Marcin Sieńczyk Załącznik 2Katepsyna G Bardzo interesującą z punktu widzenia specyficzności
Marcin Sieńczyk Załącznik 2 Wysoka zdolność związku 3 do inaktywacji katepsyny G oraz wysoka
Marcin Sieńczyk Załącznik 2 tygenów in vitro,17 podjęto próbę określenia roli katepsyny G w procesow
Marcin Sieńczyk Załącznik 2 dendrytyczne, mDCl) zastosowałem specyficzne inhibitory katepsyny G. W t
Marcin Sieńczyk Załącznik 2 W ramach kontynuacji podjętych badań dotyczących roli katepsyny G
Marcin Sieńczyk Załącznik 2 detekcji aktywnej katepsyny G w lizacie śledziony wołowej,23 potwierdzaj
Marcin Sieńczyk Załącznik 2 Rysunek 4: Niskocząsteczkowe sondy molekularne do detekcji neutrofilowyc
Marcin Sieńczyk Załącznik 2 Do niedawna w literaturze naukowej opisano zaledwie kilka przykładów
Marcin Sieńczyk Załącznik 2 Rysunek 5: Wzory ogólne otrzymanych na drodze kondensacji wieloskładniko
Marcin Sieńczyk Załącznik 2 Marcin Sieńczyk Załącznik 2 I Imię i Nazwisko: Marcin Sieńczyk H
Marcin Sieńczyk Załącznik 2 IV Wykaz osiągnięć naukowo-badawczych Tabela 1: Wykaz
Marcin Sieńczyk Załącznik 2 V Wskazanie osiągnięcia wynikającego z art. 16 ust. 2 ustawy z dnia 14 m
Marcin Sieńczyk Załącznik 2 H.6 Palesch D., Sieńczyk M.. Oleksyszyn J., Reich M., Wieczerzak E., Boe
Marcin Sieńczyk Załącznik 2 b) Omówienie celu naukowego/artystycznego ww. pracy/prac i osiągniętych
Marcin Sieńczyk Załącznik 2 Katepsyna G Proteinaza 3 NSP4 Rysunek 1: Struktura ludzkiej katepsyny G
więcej podobnych podstron