
1
Chem. Fiz. TCH II/05
1
Podstawowe równania –
podsumowanie (1)
Ts
u
f
−
=
w
q
u
+
=
dw
dq
du
+
=
sdT
Tds
du
df
−
−
=
dq
ds
=
T
:
go
odwracalne
procesu
dla
sdT
dq
du
df
−
−
=
:
zatem
dw
dq
du
=
−
:
go)
odwracalne
procesu
(dla
kolei
z
a
sdT
dw
df
−
=
:
wtedy
a
( )
0
:
na
izochorycz
jest
przemiana
jednak
gdy
=
T,V
df
PdV
dw
−
=
( )
dw
df
T
=
:
izotermy
dla
a
To wszystko jest prawdziwe, gdy w układzie nie występuje praca
nie objętościowa!!!
( )
dw
df
dq
Tds
T
<
>
i
:
lnego
nieodwraca
procesu
dla
a
( )
0
:
oraz
<
T,V
df

2
Chem. Fiz. TCH II/05
2
Podstawowe równania –
podsumowanie (2)
Ts
h
g
−
=
PV
u
h
+
=
VdP
PdV
du
dh
+
+
=
sdT
Tds
dh
dg
−
−
=
dq
ds
=
T
:
go
odwracalne
procesu
dla
sdT
VdP
PdV
dw
dg
−
+
+
=
:
zatem
dw
dq
du
=
−
:
i
( )
0
:
ej
izobaryczn
przemiany
dla
,
=
+
=
PdV
dw
dg
P
T
PdV
dw
−
=
:
ponadto
a
( )
VdP
PdV
dw
dg
T
+
+
=
:
izotermy
dla
Prawdziwe, gdy w układzie nie występuje praca nie objętościowa!!!
( )
VdP
PdV
dw
dg
dq
Tds
T
+
+
<
>
i
:
lnego
nieodwraca
procesu
dla
a
( )
0
:
oraz
<
T,P
dg
sdT
Tds
VdP
PdV
du
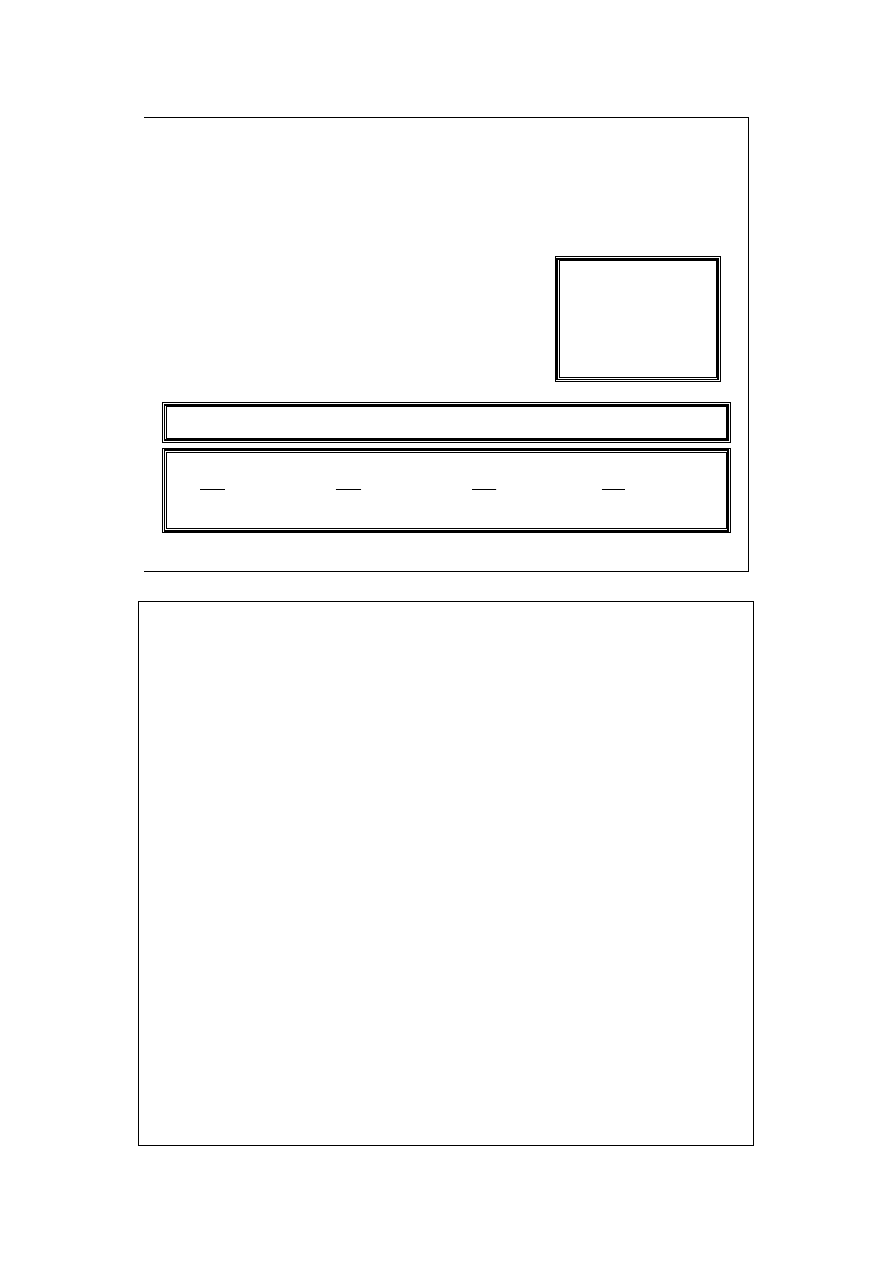
dg
−
−
+
+
=
3
Chem. Fiz. TCH II/05
3
Podstawowe równania –
podsumowanie (3)
(odwr.)
dw
Tds
du
+
=
PdV
Tds
du
−
=
obj.
tylko
praca
gdy
a
Pole powyżej zawiera podstawowe równania termodynamiki.
Ts
h
g
−
=
PV
u
h
+
=
dq
dw
du
+
=
(odwr.)
q
Tds
=
Ts
u
f
−
=
VdP
PdV
du
dh
+
+
=
:
zatem
VdP
Tds
dh
+
=
:
i
sdT
Tds
du
df
−
−
=
:
podobnie
PdV
sdT
df
−
−
=
:
i
sdT
Tds
dh
dg
−
−
=
:
raz
jeszcze
VdP
sdT
dg
+
−
=
:
i
s
T
f
V
−
=
∂
∂
P
V
f
T
−
=
∂
∂
s
T
g
P
−
=
∂
∂
V
P
g
T
=
∂
∂
A w polu różowym są ich konsekwencje ! (równania w żółtym polu są bowiem
różniczkami zupełnymi).
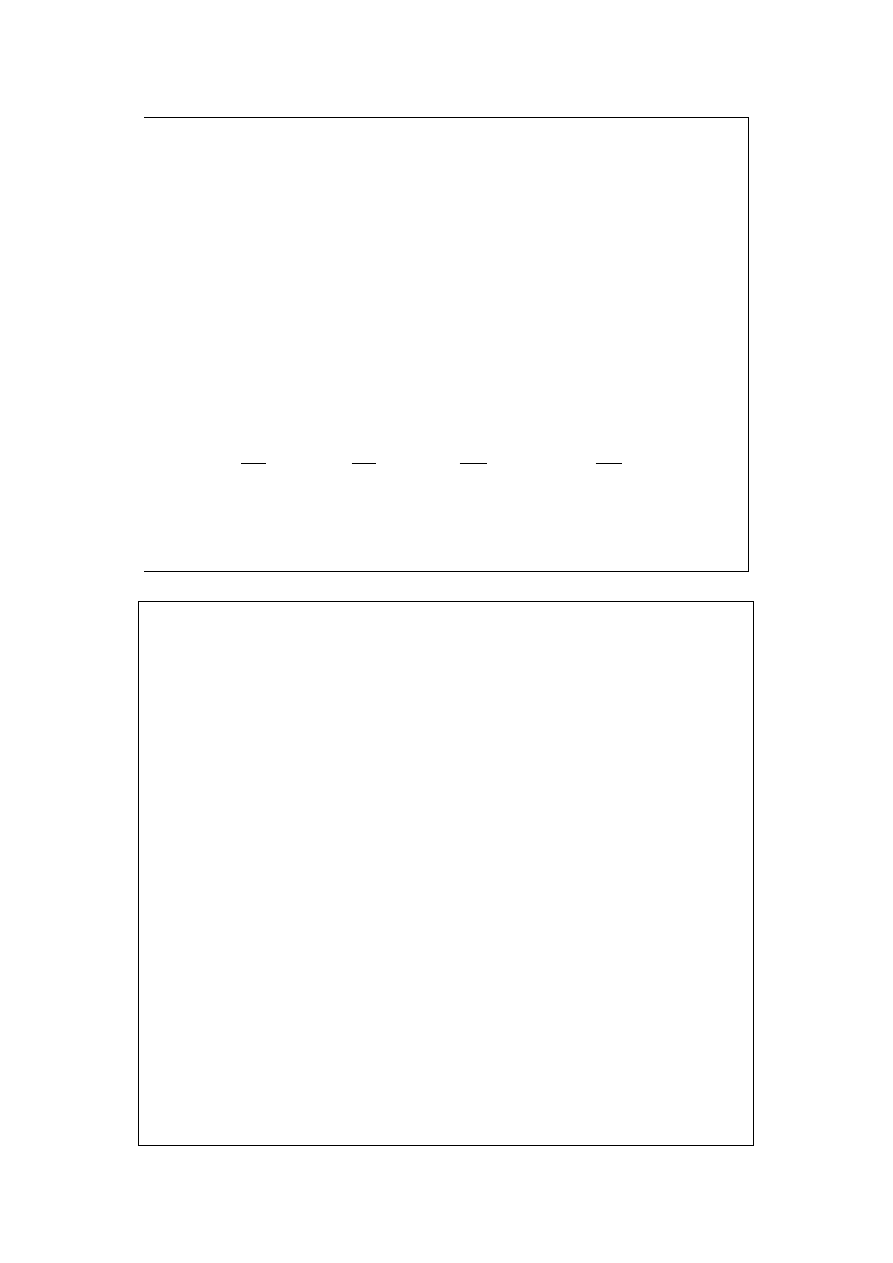
4
Chem. Fiz. TCH II/05
4
Termodynamika układów
otwartych
W układach otwartych, tzn. wymieniających z otoczeniem także
materię, zmienia się ich skład.
Jeśli zawierają one więcej niż jeden, maksymalnie zaś k składników, to
dowolna funkcja stanu musi zależeć od parametrów stanu układu i jego
składu.
)
,...
,...
,
,
,
(
2
1
k
i
n
n
n
n
T
P
f
y
=
W konsekwencji:
i
n
P
T
i
n
P
T
n
T
n
P
dn
dn
dy
dn
dn
dy
dP
dP
dy
dT
dT
dy
dy
i
j
j
i
i
≠
≠
+
+
+
=
,
,
1
,
,
1
,
,
1
Ostatni człon wyrażenia na różniczkę zupełną jest kluczowy dla zrozumienia
istoty zagadnienia. Oznacza on infinitezymalną zmianę stężenia (zawartości)
składnika i, przy niezmiennych stężeniach (zawartościach) pozostałych
składników układu.

5
Chem. Fiz. TCH II/05
5
Cząstkowe molowe
wielkości (1)
Wielkości określone pochodną:
i
j
n
P
T
i
dn
dy
≠
,
,
nazywamy cząstkowymi molowymi wielkościami:
i
j
n
P
T
i
i
dn
dy
Y
≠
=
,
,
za pomocą których możemy opisać zmiany stanu układu otwartego:
k
k
i
i
n
T
n
P
dn
Y
dn
Y
dn
Y
dP
dP
dy
dT
dT
dy
dy
i
i
+
+
+
+
+
+
=
...
...
1
1
,
,
W szczególności, w warunkach
izotermiczno-izobarycznych:
∑
=
=
k
i
i
i
dn
Y
dy
1
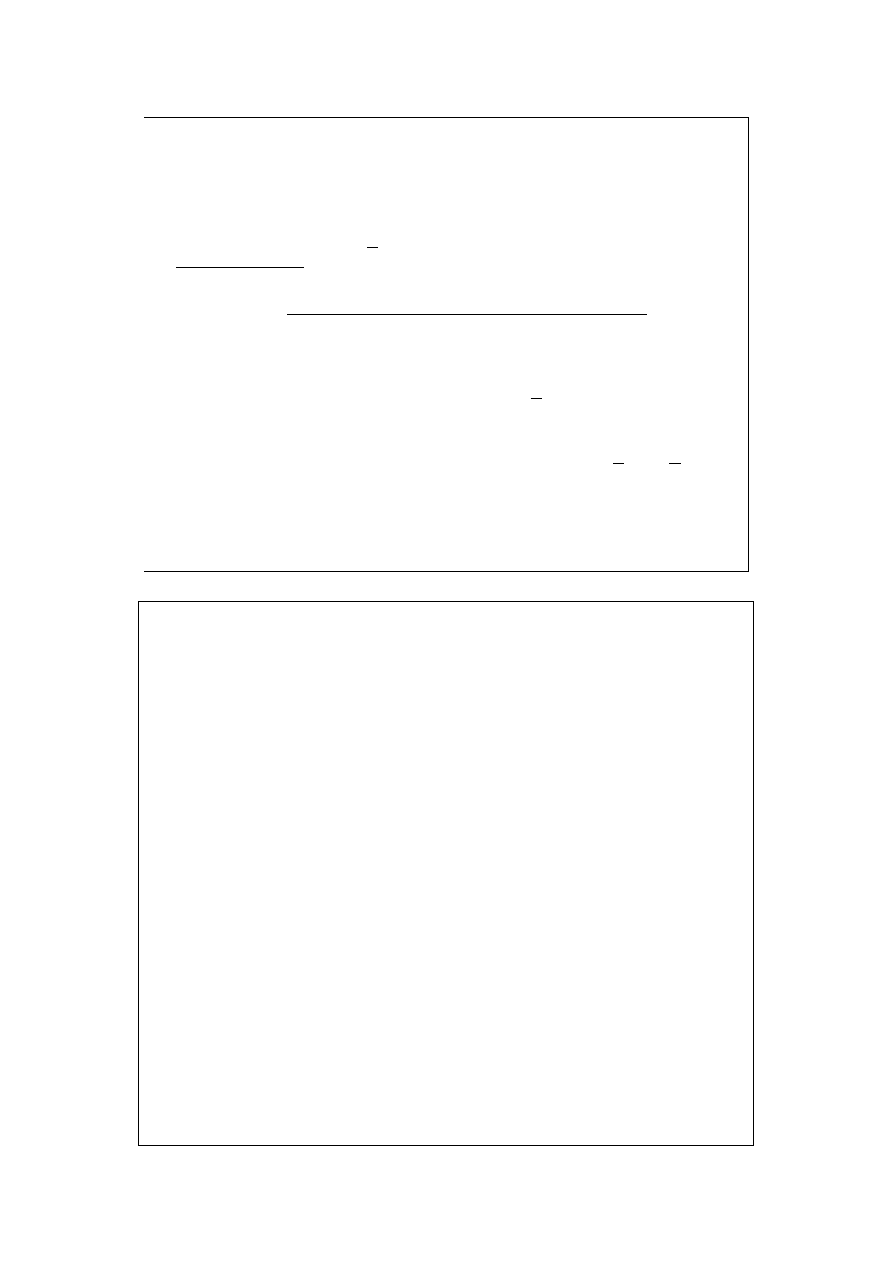
6
Chem. Fiz. TCH II/05
6
Cząstkowe molowe
wielkości (2)
Można ją więc scałkować (po dowolnej drodze) od stanu
n
i
=0 do stanu n
i
(końcowe). Droga ta może być taka, aby:
const
Y
i
=
_
W tych warunkach:
∑
=
k
i
i
i
dn
Y
1
jest różniczką zupełną funkcji y
ergo, funkcją stanu układu, zależną od jego składu.
W wyniku czego otrzymujemy:
∑
=
=
k
i
i
i
Y
n
y
1
∑
∑
=
=
+
=
k
i
i
i
k
i
i
i
dn
Y
Y
d
n
dy
1
1
zaś najogólniejsze wyrażenie na jej
różniczkę zupełną dane jest równaniem:
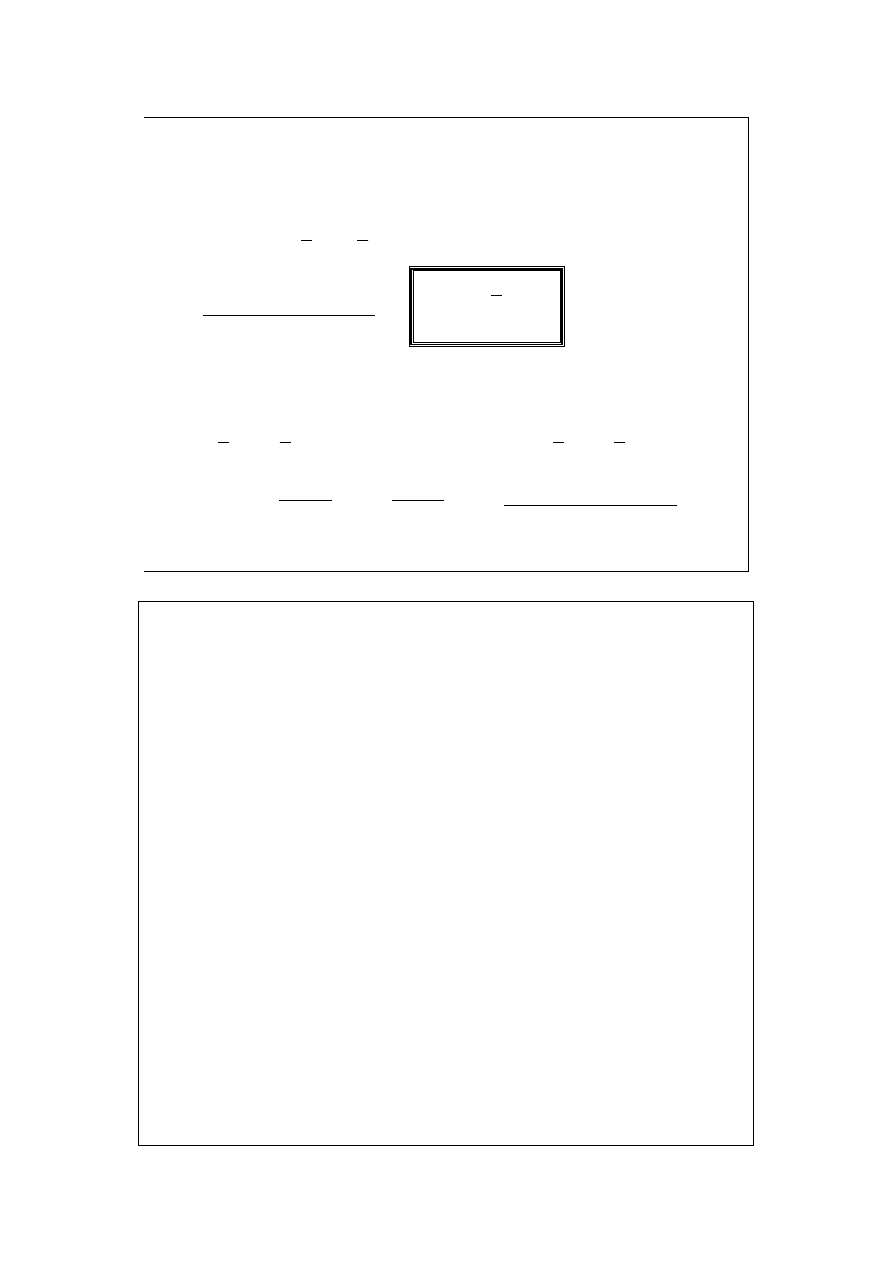
7
Chem. Fiz. TCH II/05
7
Równanie Gibbsa-Duhema
Ostatnie wyrażenie nosi nazwę równania Gibbsa-Duhema.
Jeśli:
∑
∑
=
=
+
=
k
i
i
i
k
i
i
i
dn
Y
Y
d
n
dy
1
1
a równocześnie:
∑
=
=
k
i
i
i
dn
Y
dy
1
_
ergo, musi być prawdziwe:
0
1
=
∑
=
k
i
i
i
Y
d
n
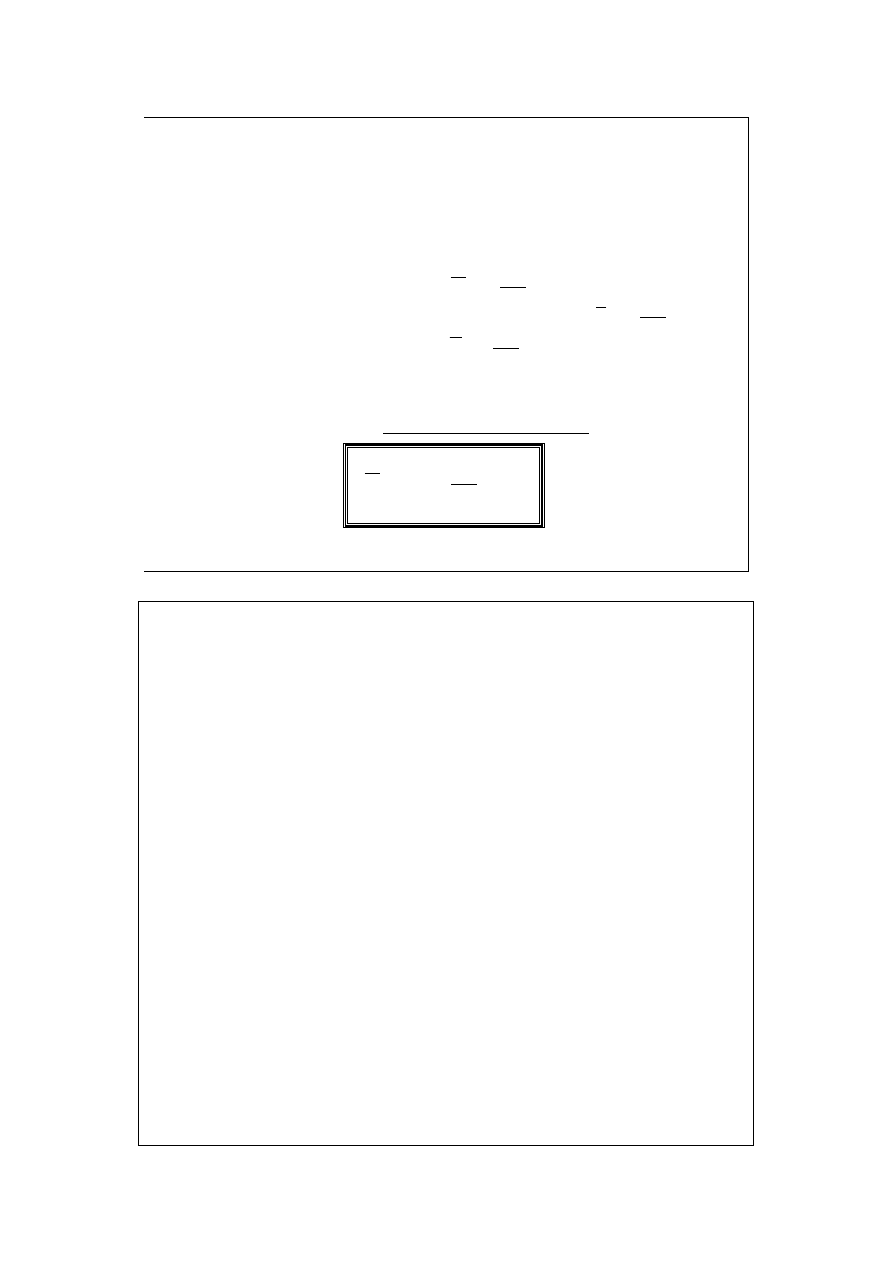
Dla układu dwuskładnikowego, można je wyrazić:
0
2
2
1
1
=
+
Y
d
n
Y
d
n
0
2
2
1
1
=
+
Y
d
x
Y
d
x
lub:
gdzie:
2
1
2
2
2
1
1
1
;
n
n
n
x
n
n
n
x
+
=
+
=
są ułamkami molowymi
składników 1 i 2.
8
Chem. Fiz. TCH II/05
8
Cząstkowe molowe
wielkości (3)
Najczęściej stosowane cząstkowe molowe wielkości to:
cząstkowa molowa entalpia:
i
j
n
P
T
i
i
n
h
H
≠
∂
∂
=
,
,
cząstkowa molowa entropia:
i
j
n
P
T
i
i
n
s
S
≠
∂
∂
=
,
,
cząstkowa molowa objętość:
i
j
n
P
T
i
i
n
v
V
≠
∂
∂
=
,
,
i najważniejsza z nich, cząstkowa molowa entalpia swobodna,
zwana też potencjałem chemicznym:
i
j
n
P
T
i
i
i
n
g
G
≠
∂
∂
=
=
,
,
µ

9
Chem. Fiz. TCH II/05
9
Cząstkowe molowe
wielkości (4)
Zależności pomiędzy cząstkowymi molowymi wielkościami są
analogiczne do zachodzących pomiędzy h, s, v i g.
Np., różniczkując względem n
i
wyrażenie:
Ts
h
g
−
=
otrzymujemy:
=
−
=
=
∂
∂
−
∂
∂
=
∂
∂
i
i
i
i
i
i
S
T
H
n
s
T
n
h
n
g
µ
a wychodząc z zależności:
s
T
g
P
−
=
∂
∂
otrzymamy:
S
T
n
s
n
T
g
i
i
i
−
=
∂
∂
=
∂
∂
−
=
∂
∂
∂
µ
2
10
Chem. Fiz. TCH II/05
10
Cząstkowe molowe
wielkości (5)
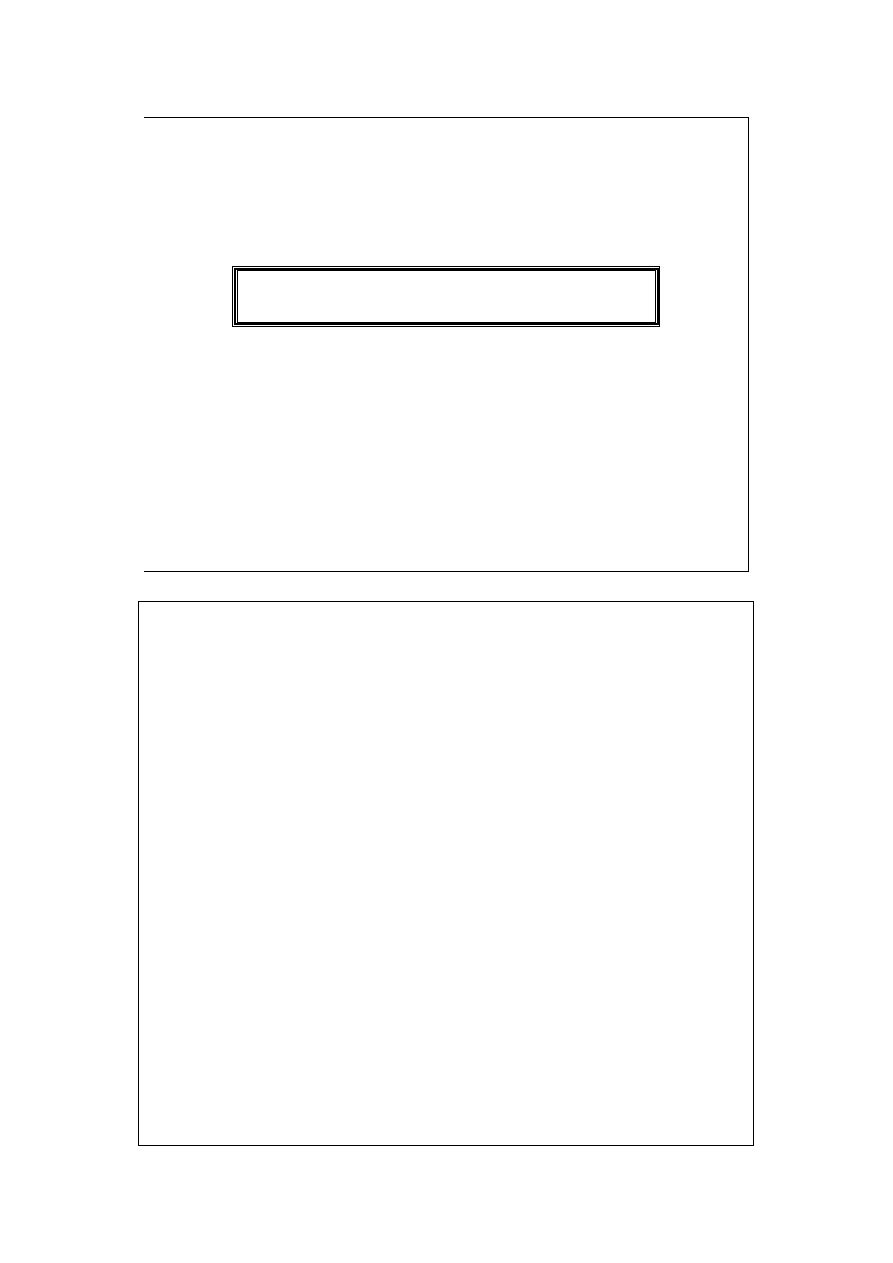
Najważniejsza zależność termodynamiki chemicznej:
k
k
i
i
dn
dn
dn
SdT
VdP
dG
µ
µ
µ
+
+
+
+
+
−
=
...
...
1
1
Pod stałym ciśnieniem i w stałej temperaturze:
k
k
i
i
dn
dn
dn
dG
µ
µ
µ
+
+
+
+
=
...
...
1
1
A wtedy, praca nieobjętościowa może być spowodowana zmianą
składu chemicznego układu. Np. w ogniwie galwanicznym zmiana
jego składu chemicznego (od substratów do produktów) prowadzi
do uzyskania pracy elektrycznej.
11
Chem. Fiz. TCH II/05
11
Gazy
Zagadnienia omawiane w tej części, przynajmniej w
pewnym zakresie, traktuję jako powtórkę.
Niektóre przeźrocza w tej serii są zatem jedynie
materiałem pomocniczym!
Większość omawianych tutaj zagadnień można
(i należy) powtórzyć sobie z dowolnego podręcznika
chemii fizycznej lub fizyki.

12
Chem. Fiz. TCH II/05
12
Gaz doskonały(1).
Prawa gazowe
Boyle’a
izoterma
const
T
const
PV
=
=
dla
;
Charlesa
izobara
const
P
T
V
=
∝
dla
;
Gay-Lussaca
izochora
const
V
T
P
=
∝
dla
;
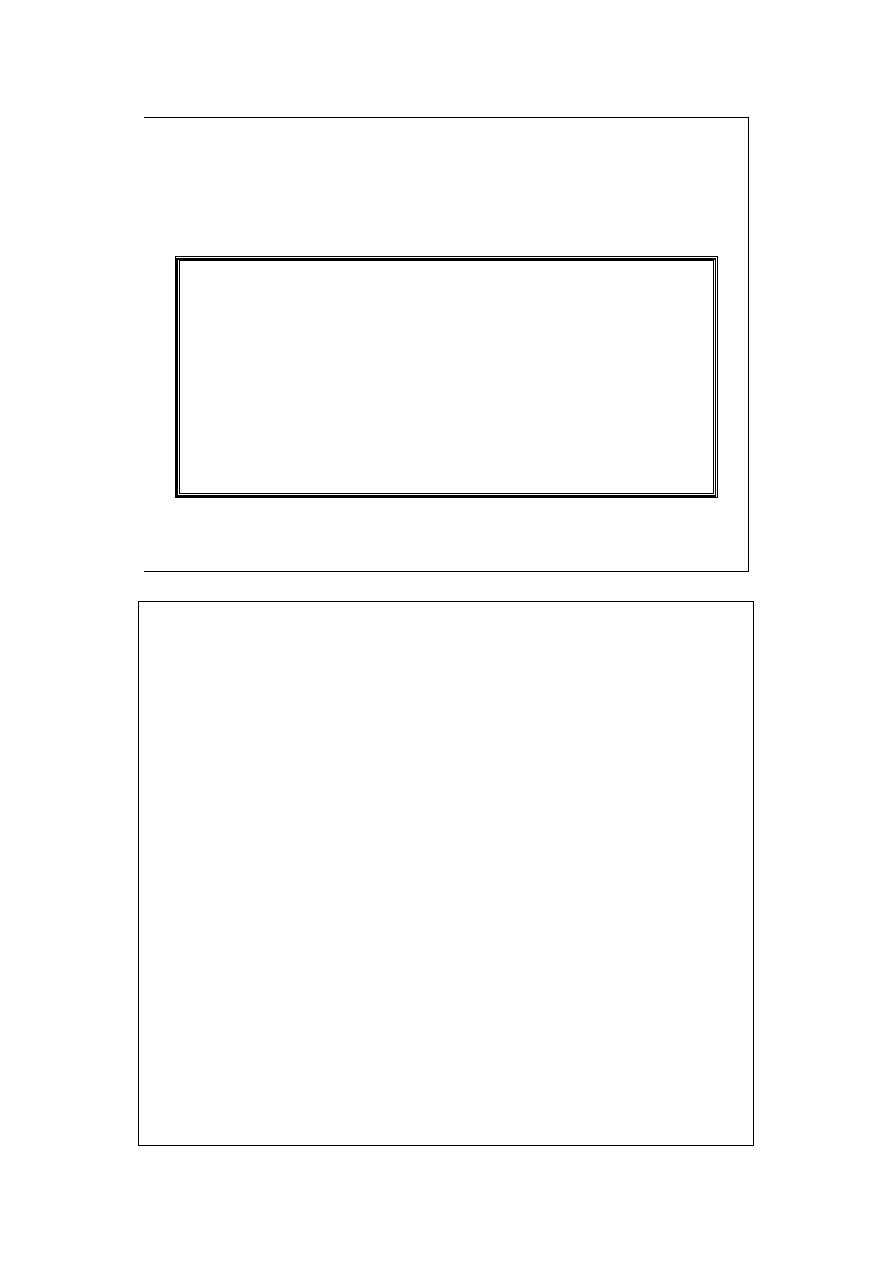
Równanie Clapeyrona
nRT
PV
=
T
PV
R
m
P 0
lim
→
=
Jeśli ktoś chce „wyprowadzać” równanie Clapeyrona, to może to uczynić
wykorzystując fakt, że dwa dowolne punkty na płaszczyźnie P-V można zawsze
połączyć jedną izotermą i jedną izochorą (lub izobarą).
W odniesieniu do gazów rzeczywistych – równanie Clapeyrona (podobnie jak
cząstkowe prawa gazowe) ma charakter graniczny! Dlatego te ostatnie
nazywamy często „prawami gazów rozrzedzonych”.
13
Chem. Fiz. TCH II/05
13
Gaz doskonały(2).
Prawa gazowe, c.d.
Avogadro
n
const
V
⋅
=
Daltona
;
;
1
i
i
k
i
i
x
P
P
P
P
⋅
=
=
∑
=
∑
=
=
k
i
i
i
i
n
n
x
1
Założenia kinetycznego modelu gazu doskonałego:
•
Cząsteczki gazu, o masie m znajdują się w ciągłym, chaotycznym
ruchu.
•
Jedyne oddziaływanie pomiędzy cząsteczkami, jak również
między nimi a ścianką zbiornika to zderzenia idealnie sprężyste.
•
Rozmiary cząsteczek są pomijalnie małe w porównaniu ze średnią
drogą pomiędzy zderzeniami (średnią drogą swobodną).
Od prawa Avogadro są odstępstwa. Tylko gaz doskonały ma objętość molową
dokładnie 22,4 dm
3
/mol w 273,15K, pod normalnym ciśnieniem.
14
Chem. Fiz. TCH II/05
14
Gaz doskonały(3).
Ciśnienie wg modelu kinetycznego
1
2
mv
mv
t
F
−
=
∆
Druga zasada dynamiki Newtona:
V
nN
t
Av
l
A
x
∆
=
2
1
Liczba cząsteczek uderzających w ściankę o pow.
A
w czasie
∆
t
(zmiana szybkości z mv
x
na – mv
x
):
Ich masa (masa jednej cząsteczki to M/N
A
) :
Ich całkowita zmiana pędu :
V
nM
t
Av
lm
x
∆
=
2
1
V
nM
t
Av
t
F
x
∆
=
∆
2
Wywierane ciśnienie (F/A) :
V
nM
v
P
x
2
=
Ponieważ :
2
2
2
2
z
y
x
v
v
v
c
+
+
=
To ostatecznie :
nM
c
PV
2
3
1
=
n oznacza liczbę moli w objętości V.
Składowe szybkości są równe we wszystkich kierunkach (x, y i z).
Otrzymaliśmy więc prawo Boyle’a (dla stałej T, średnia szybkość kwadratowa
jest stała, jak zobaczymy z rozkładu Maxwella).
Słuszność ostatniej zależności potwierdzono doświadczalnie (Stern) mierząc c.
Wielkość c nazywamy średnią szybkością kwadratową.
W ten sposób otrzymaliśmy - na drodze rozważań z fizyki klasycznej – prawo
Boyle’a.
15
Chem. Fiz. TCH II/05
15
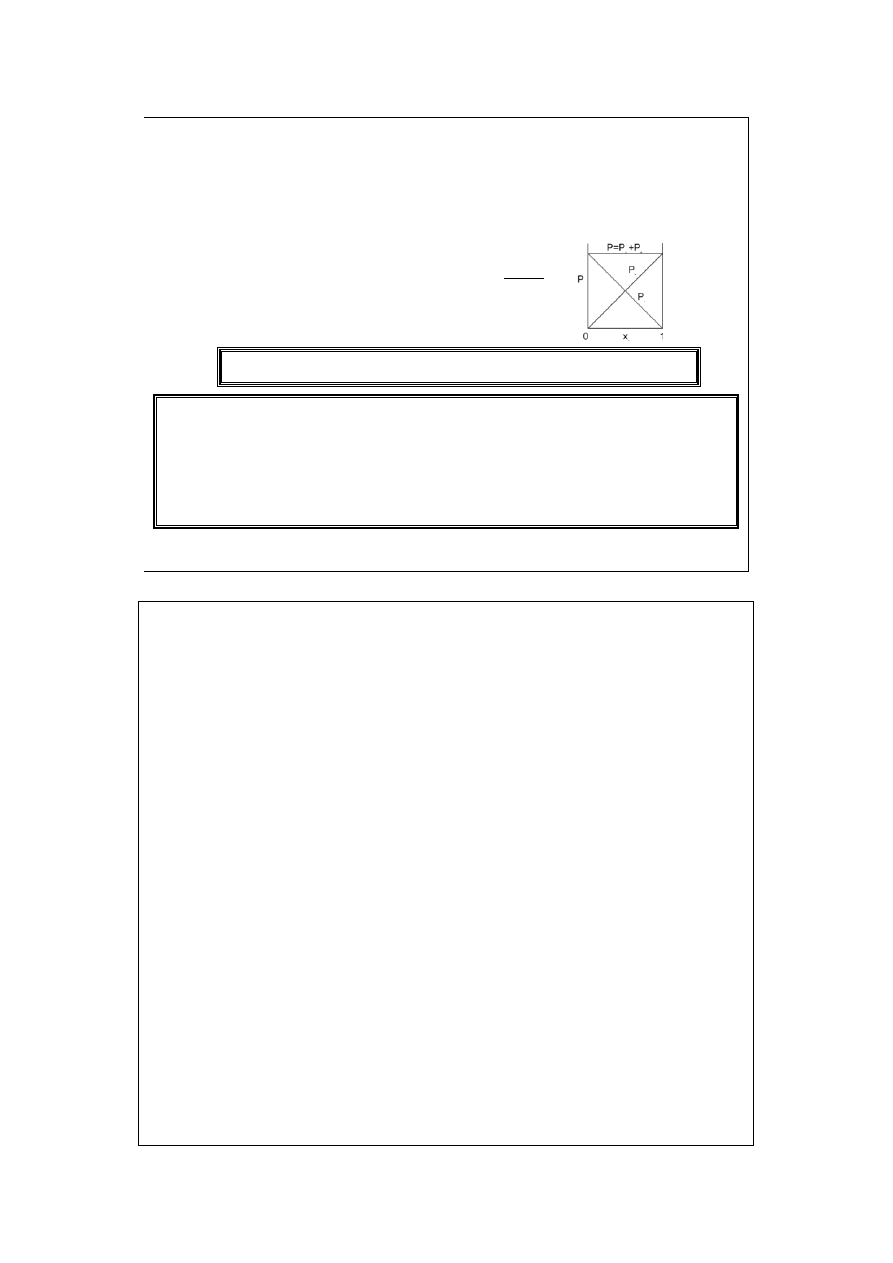
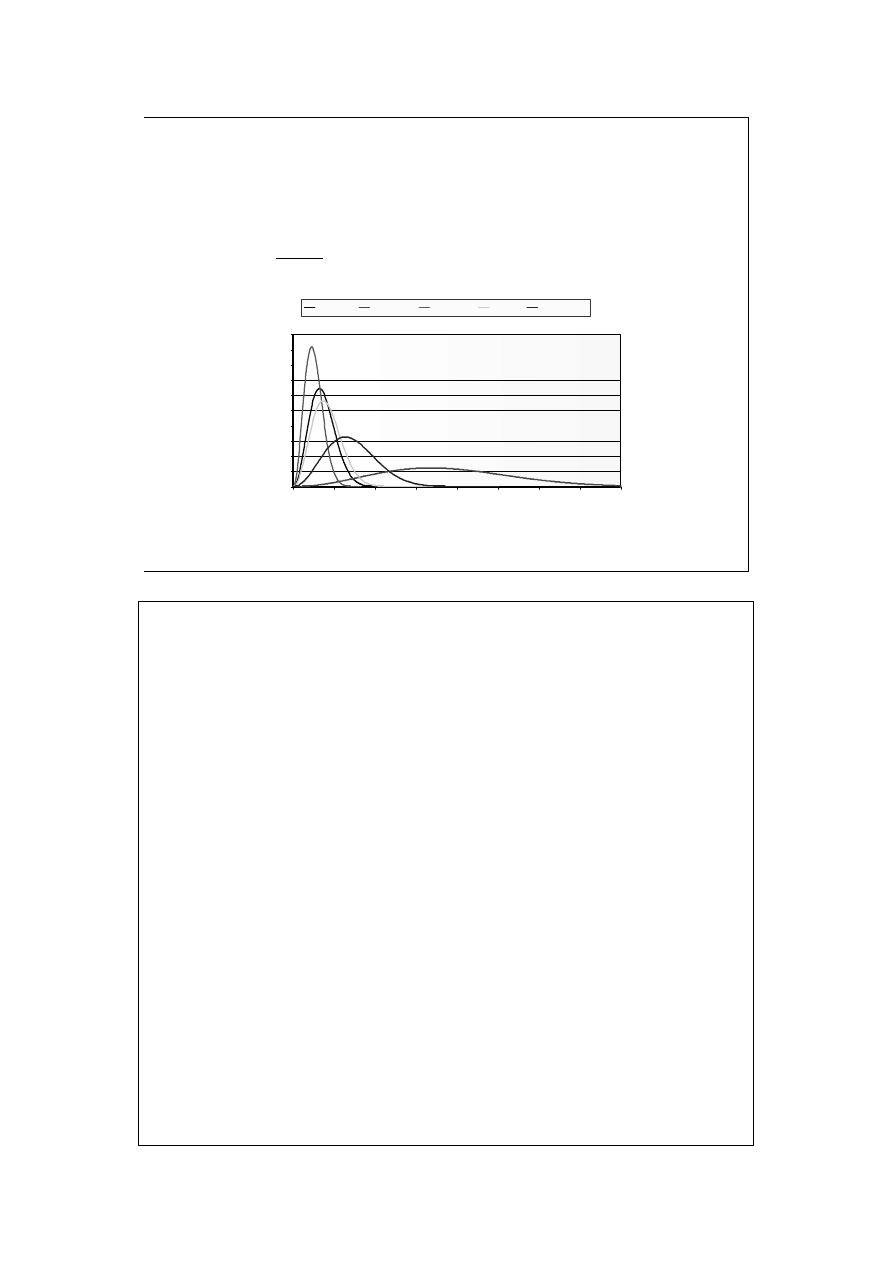
Rozkład Maxwella (1)
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
0
10
20
30
40
50
60
70
80
s zybkoś ć, m/s
wz
g
lę
dna
l
ic
z
ba
c
z
ą
st
e
c
z
e
k
He; 10 K
He; 273 K
Rn; 273 K
Kr; 273
Azot;273 K
)
2
/(
2
2
/
3
2
2
4
)
(
RT
Mv
e
v
RT
M
v
f
−
=
π
π
bez wyprowadzania
Z wykresu widzimy jakie szybkości osiągają cząsteczki w temperaturze 0
o
C
Wtedy widzimy zależność od masy molowej.
Na przykładzie helu pokazano też zależność od temperatury.
Oś y jest właściwie osią gęstości prawdopodobieństwa. Całka z rozkładu
Maxwella od 0 do
∞ wynosi 1.
Maksimum rozkładu wskazuje nam szybkość najbardziej prawdopodobną.
Wysokość powyżej której pole równa się polu poniżej - odpowiada szybkości
średniej (jednakowe prawdopodobieństwo większych i mniejszych szybkości,
połowa cząsteczek ma szybkość wyższą, połowa niższą).

16
Chem. Fiz. TCH II/05
16
Rozkład Maxwella (2)
2
/
1
3
=
M
RT
c
Wykorzystując rozkład Maxwella można także wyznaczyć
takie własności cząsteczek gazu jak:
szybkość średnia kwadratowa :
szybkość średnia :
szybkość najbardziej prawdopodobna:
średnia szybkość względna:
częstość zderzeń:
średnia droga swobodna:
2
/
1
8
=
M
RT
c
π
2
/
1
2
*
=
M
RT
c
2
/
1
8
2
=
⋅
=
πµ
kT
c
c
wzgl
kT
P
c
V
N
c
z
wzgl
wzgl
⋅
=
⋅
=
σ
σ
P
kT
z
c
σ
λ
2
=
=
Wzór na średnią szybkość kwadratową wynika już z samego modelu
kinetycznego. Jeśli ostateczną postać wyrażenia na PV porównamy z równaniem
gazu doskonałego, to jej prawa strona musi równać się nRT (i stąd wzór).
Średnia szybkość względną to szybkość cząsteczek gazu względem siebie
(zbliżania się lub oddalania).
Wielkość
µ to masa zredukowana dwóch mijających się cząsteczek:
µ=m
1
m
2
/(m
1
+m
2
).
Wielkość
σ to przekrój czynny na zderzenie: σ = πd
2
.
17
Chem. Fiz. TCH II/05
17
Gazy rzeczywiste. Równanie
stanu Van der Waalsa
nRT
PV
=
Udoskonalenie (urealnienie) równania Clapeyrona:
na 1 mol gazu:
Poprawka na objętość mola cząsteczek gazu, b
m
V
RT
P
=
b
V
RT
P
m
−
=
Poprawka na oddziaływania międzycząsteczkowe, a
2
m
m
V
a
b
V
RT
P
−
−
=
• same cząsteczki gazu zajmują pewną objętość, b
• atraktywne (przyciągające) oddziaływania międzycząsteczkowe zmniejszają pęd
(zatem siłę) cząsteczek zmierzających ku ściance, a także częstość ich zderzeń ze
ścianką, dodatkowo zmniejszają więc ciśnienie (do kwadratu stężenia cząsteczek).
Wyprowadzenie równania van der Waalsa:
Poprawka b, ma zatem wymiar m
3
/mol (najmniejsza jest dla Ne < He < H
2
).
Oddziaływania międzycząsteczkowe zależą od stężenia cząsteczek gazu, które
jest odwrotnością objętości molowej. Z tego względu, druga poprawka musi
mieć wymiar Pa·m
-6
·mol
2
. Poprawka a najmniejsza jest dla He < Ne < H
2
.
Johannes Diederik van der Waals otrzymał nagrodę Nobla z fizyki w 1910 roku.
18
Chem. Fiz. TCH II/05
18
Izotermy Van der Waalsa
(1)
punkt krytyczny
b
V
c
3
=
2
27b
a
P
c
=
bR
a
T
c
27
8
=
Związki między
współczynnikami r-nia Van
der Waalsa a parametrami
krytycznymi:
Związki te łatwo wyprowadzić przekształcając równanie Van der Waalsa do
postaci wielomianu trzeciego stopnia względem V
c
i przyrównując go z
rozwinięciem wielomianu (V-V
c
)
3
=0.
Ten ostatni wynika z warunku, że dla punktu krytycznego wszystkie trzy
pierwiastki równania powinny być identyczne (jeden potrójny pierwiastek).
Znaczenie temperatury krytycznej. Powyżej tej temperatury nie da się skroplić
gazu, choćby stosując największe ciśnienia.
Równanie Van der Waalsa nadszacowuje objętość krytyczną.
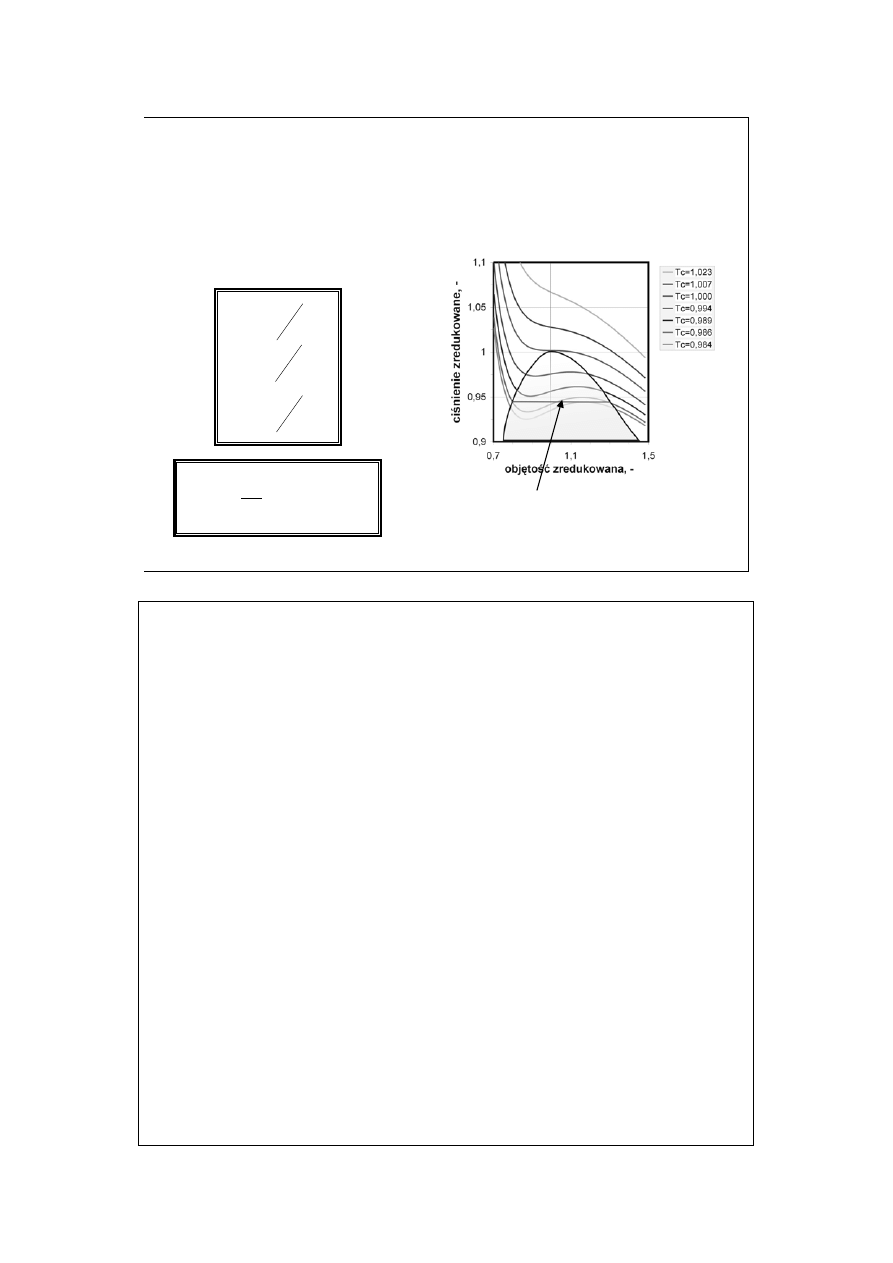
19
Chem. Fiz. TCH II/05
19
Izotermy Van der Waalsa
(2)
c
P
P
=
π
c
V
V
=
φ
c
T
T
=
θ
rzeczywisty przebieg
izotermy podczas skraplania
zredukowane parametry stanu
i równanie Van der Waalsa
(
)
θ
φ
φ
π
8
1
3
3
2
=
−
+
Obszar cieniowany wskazuje rejon współistnienia fazy ciekłej i gazowej.
„Pętle Van der Waalsa” są artefaktami, prawdziwy przebieg izotermy pokazuje
poziomy odcinek prostoliniowy. Prowadzi się go tak, aby powierzchnie pod
dodatnią i nad ujemną częścią pętli były jednakowe.
Znaczenie gazu w stanie nadkrytycznym w analityce (ekstrakcja, chromatografia
w stanie nadkrytycznym łącząca zalety cieczowej i gazowej).
20
Chem. Fiz. TCH II/05
20
Wirialne równania stanu
...)
'
'
'
1
(
3
2
+
+
+
+
=
P
D
P
C
P
B
RT
PV
m
+
+
+
+
=
...
1
3
2
m
m
m
m
V
D
V
C
V
B
RT
PV
Zaproponował je Kamerlingh Onnes (dwie formy):
Współczynniki B, C, D (B’, C’, D’) noszą nazwę
współczynników wirialnych (zależą od temperatury).
Często stosuje się jedynie drugi współczynnik wirialny:
P
B
RT
PV
m
"
+
=
Z powodu niedoskonałości równania Van der Waalsa zaproponowano szereg
innych równań stanu (Berthelota, Dietericiego, Beattiego-Bridgemana).
Niedogodność równania wirialnego (zależność współczynników od T).
Zastosowane tutaj podejście (rozwijanie w szereg potęgowy) jest często
spotykane w chemii fizycznej. Pierwszy człon daje równanie gazu doskonałego.
21
Chem. Fiz. TCH II/05
21
Gazy rzeczywiste w tych samych warunkach zredukowanej objętości
i temperatury wywierają takie samo ciśnienie zredukowane.
Zasada stanów
odpowiadających sobie
Wynika ze zredukowanego r-nia Van der Waalsa (znikają w nim
bowiem charakterystyczne dla poszczególnych gazów współczynniki
a
i b). Inne równania stanu też ją zawierają.
Zasada ta zawodzi, gdy cząsteczki gazu są nie sferyczne lub polarne.
(
)
θ
φ
φ
π
8
1
3
3
2
=
−
+

22
Chem. Fiz. TCH II/05
22
Skraplanie gazów. Efekt
Joule’a-Thomsona (1).
Klasyczne metody sprężania poniżej w temperaturze T<T
c
często nie
wystarczały do skroplenia gazów o coraz niższych T
c
.
q = 0
adiabatyczne
rozprężanie
-w = P
2
V
2
– P
1
V
1
James Joule
rura izolowana
termicznie
przegroda
porowata
P
1
,T
1
P
2
,T
2
U
2
– U
1
= P
2
V
2
– P
1
V
1
U
2
+ P
2
V
2
= U
1
+ P
1
V
1
H
2
= H
1
proces izoentalpowy
William Thomson to późniejszy lord Kelvin.
Efekt J-T jest podstawą skraplania gazów metodą Lindego.
Przegroda porowata, to tzw. dławik.
Uwaga! Praca jest wykonywana na układzie, gaz bowiem jest
PRZETŁACZANY przez dławik.

23
Chem. Fiz. TCH II/05
23
Skraplanie gazów. Efekt
Joule’a-Thomsona (2).
Współczynnik Joule’a – Thomsona:
H
JT
P
T
∂
∂
=
µ
Można dowieść, że :
−
∂
=
V
dT
V
T
C
P
P
JT
1
µ
Dla gazu doskonałego (V=RT/P):
0
;
0
=
=
−
∂
JT
P
V
dT
V
T
µ
Dla każdego gazu rzeczywistego istnieje tzw. temperatura inwersji, T
inw
.
Gdy T < T
inw
,
µ
JT
>0
, gdy T > T
inw
,
µ
JT
<0
, gdy T = T
inw
,
µ
JT
=0
.
T
inw
zależy od ciśnienia (mogą być dwie T
inw
– dolna i górna).
Dowód proszę przeprowadzić sobie samemu, wychodząc z różniczki zupełnej
entalpii (względem ciśnienia i temperatury) oraz używając różniczki
dH=TdS+VdP, a także uwzględniając, że pochodna cząstkowa entropii po
ciśnieniu (w stałej T) jest równa pochodnej cząstkowej objętości po temperaturze
przy stałym ciśnieniu.
Przy okazji pytanie. Dlaczego proste rozprężanie adiabatyczne gazu doskonałego
prowadzi jednak do obniżenia jego temperatury?
24
Chem. Fiz. TCH II/05
24
Na każdy stopień swobody ruchu translacyjnego (na każdy człon
kwadratowy energii kinetycznej) cząsteczki przypada identyczna
energia równa ½kT
Zasada ekwipartycji energii
Prawidłowość ta jest spełniona tylko dla helu (i innych gazów
jednoatomowych).
Najwyraźniej, cząsteczki gazu posiadają jeszcze inną energię.
Gdyby, jak wynika z modelu kinetycznego, energia translacyjna była
jedyną energią cząsteczek gazu, to:
R
C
dT
du
V
V
2
3
=
=
a ze znanych względów
R
C
P
2
5
=
Ekwipartycja ( z łac. equipartitio) znaczy „równy podział”.
Trzy stopnie swobody ruchu translacyjnego, to ruch wzdłuż każdej z trzech osi
przestrzennych, x, y, z.

25
Chem. Fiz. TCH II/05
25
Cząsteczki mogą także wykonywać ruch rotacyjny (obrót wokół osi
symetrii). Dla cząsteczek dwuatomowych lub o budowie liniowej (2
osie, moment bezwładności wokół trzeciej – głównej osi cząsteczki
liniowej – jest pomijalny), istnieją dwa stopnie swobody rotacji, zatem
Pojemności cieplne gazów
(1)
gdzie J jest momentem bezwładności.
Dla cząsteczek przestrzennych, o trzech momentach bezwładności
RT
J
E
rot
=
=
2
2
1
ω
RT
E
rot
2
3
=
Niektóre lekkie gazy słabo wzbudzają się rotacyjnie w niskich temperaturach,
stąd wspomniane w wykładzie 4 odstępstwa od reguły Troutona.

26
Chem. Fiz. TCH II/05
26
Zatem, dla cząsteczek liniowych:
Pojemności cieplne gazów
(2)
Stwierdzono jednak, że w wysokich temperaturach, krzywa
ogrzewania gazów wieloatomowych wykazuje jeszcze większe
pojemności cieplne. Dochodzi wtedy do wzbudzenia oscylacyjnego.
a dla nieliniowych (przestrzennych):
R
C
V
2
5
=
R
C
P
2
7
=
R
C
V
3
=
R
C
P
4
=
Chodzi o krzywą T=f(H), pokazaną na wykładzie 4.

27
Chem. Fiz. TCH II/05
27
a na każde drganie przypadają dwa stopnie swobody (energia
potencjalna i kinetyczna). Zatem w wysokich temperaturach, dla
gazów dwuatomowych
Liczba drgań normalnych wynosi, dla cząsteczek :
nieliniowych liniowych
Pojemności cieplne gazów
(3)
Ogólnie, energia wewnętrzna gazów dana jest równaniem:
5
3
−
N
6
3
−
N
R
C
V
2
7
=
R
C
P
2
9
=
const
RT
RT
RT
E
E
E
E
U
nukl
el
osc
rot
tr
+
+
+
=
+
+
+
=
2
3
.
.,
Wzrost C
V
ponad wartość 5/2RT zachodzi nie skokowo, ale płynnie.
Energia elektronów i jąder atomowych nie zależy od temperatury, zatem nie
znajduje odbicia w pojemnościach cieplnych.

28
Chem. Fiz. TCH II/05
28
Ściśliwość gazów (1)
RT
PV
Z
m
=
0
=
dP
dZ
Współczynnik ściśliwości gazów dany jest wzorem:
Dla gazu doskonałego wynosi on zawsze 1 zaś pochodna:
'
lim
...
'
2
'
0
B
dP
dZ
C
B
dP
dZ
P
=
+
+
=
→
Dla gazów rzeczywistych:
Jednak B nie musi być równe zeru, ponadto zależy od temperatury.
Istnieje temperatura, zwana temperaturą Boyle’a, w której B = 0
dla P
→ 0, czyli gazy rzeczywiste zachowują się w niej naprawdę
jak gaz doskonały (w niskich ciśnieniach).
Z faktu, że Z jest dla gazów doskonałych równe 1 wynika, że jego wartość może
być miarą odchyleń od prawa Boyle’a.
To, że dla niższych ciśnień równanie wirialne przechodzi w równanie Clapeyrona
(pomijamy drugi i wyższe współczynniki wirialne) nie znaczy, że gazy
rzeczywiste stają się doskonałymi, a jedynie, że równanie Clapeyrona dobrze
opisuje ich zachowanie.

29
Chem. Fiz. TCH II/05
29
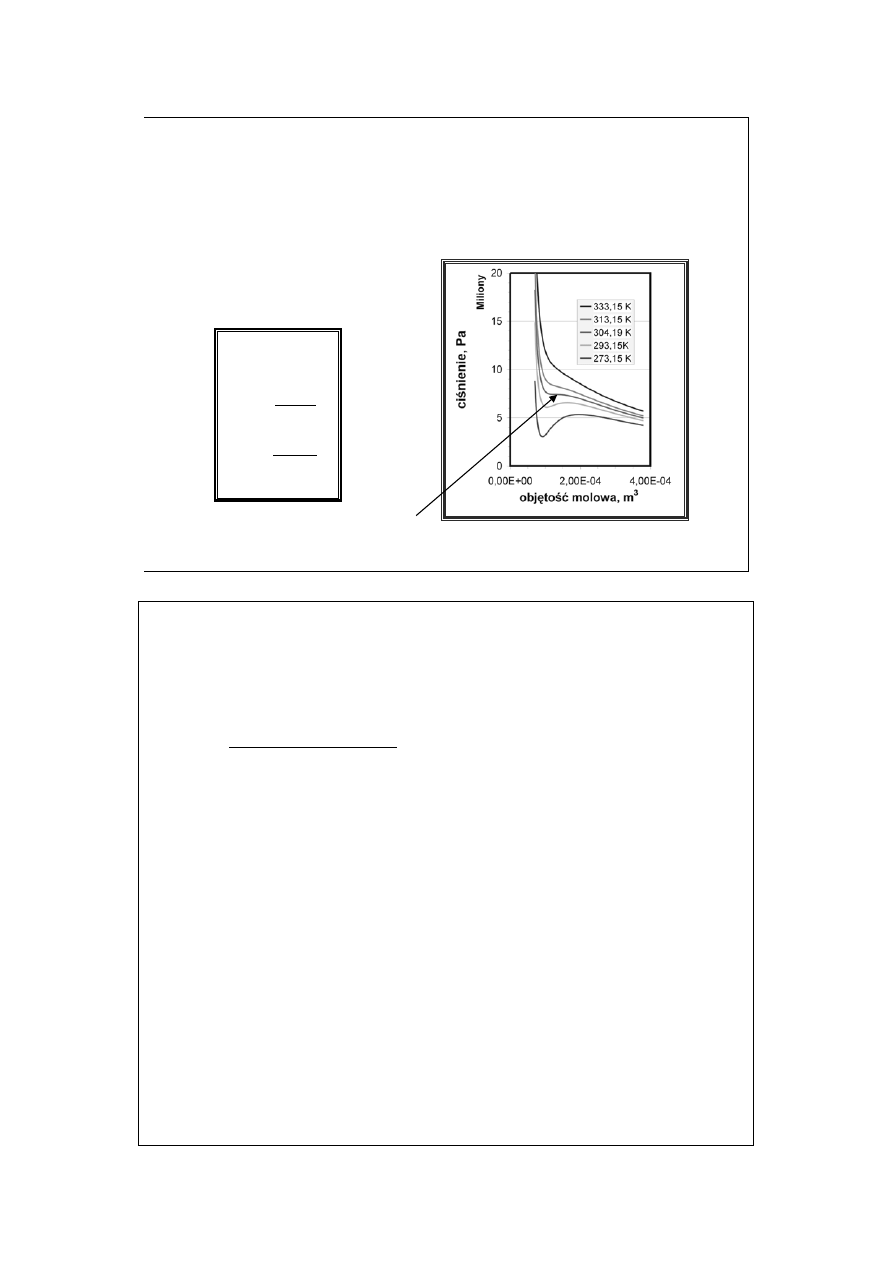
Powodem są oddziaływania międzycząsteczkowe odpychające
(bliskiego zasięgu) i przyciągające (dalszego zasięgu).
Ściśliwość gazów (2)
gaz doskonały

30
Chem. Fiz. TCH II/05
30
Lepkość gazów (1)
(1)
;
A
dx
dv
F
η
=
(2)
;
8
4
τ
η
π
l
∆P
r
v
=
Współczynnik tarcia wewnętrznego,
η
, czyli lepkość, możemy
rozpatrywać w kategoriach teorii kinetycznej gazów, jako wymianę pędu
przez cząsteczki sąsiadujących warstw poruszającego się gazu.
(tylko 1/3 wymienia pęd wzdłuż osi x); N=N
A
·
λ
A/V
m
λ
v
v+
λ
·dv/dx
Jedna cząsteczka przenosi pęd:
dx
dv
m
p
λ
=
1
Jeżeli w V=
λ
A
znajduje się N cząsteczek:
dx
dv
Nm
p
t
λ
3
1
=
dx
dv
A
p
t
2
3
1
ρλ
=
Równanie (1) jest równaniem definiującym lepkość, gdzie F jest siłą tarcia
między warstwami o powierzchni styku A, przy gradiencie szybkości w kierunku
normalnym do płaszczyzny warstw równym dv/dx.
Równanie (2) jest równaniem Poiseuille’a, gdzie r jest promieniem rurki
kapilarnej, l – jej długością,
∆
P – różnicą ciśnień między wylotem a wlotem,
τ
-
czasem wypływu, zaś v – objętością gazu jaka przepłynęła przez rurkę.
Na szkicu
λ
jest średnią drogą swobodną cząsteczek gazu.
Oś x jest tu osią pionową.
Ostatnie równanie zawiera gęstość,
ρ
, która jest równa m·N
A
/V
m
.

31
Chem. Fiz. TCH II/05
31
Lepkość gazów (2)
Porównując ostatnie równanie z r-niem Poiseuille’a:
Jeżeli wszystko dzieje się w czasie
τ
=1/z
:
τ
ρλ
1
2
3
1
dx
dv
A
F
=
a ponieważ
λ
/
τ
=ĉ
:
A
dx
dv
c
F
λ
ρ
3
1
=
c
ρλ
η
3
1
=
który to wynik możemy dowolnie komplikować podstawiając doń
wielkości uzyskane z rozkładu Maxwella.
Wnioski:
(sprawdzające się doświadczalnie)
• lepkość nie zależy od ciśnienia,
• lepkość zależy od pierwiastka kwadrato-
wego temperatury
Wzór Sutherlanda:
T
c
T
+
=
1
0
η
η
Odnośnie niektórych symboli – patrz slajd „Rozkład Maxwella (2)”.
We wzorze Sutherlanda (półempirycznym) stałe
η
0
i c są wielkościami
charakterystycznymi dla danej substancji.
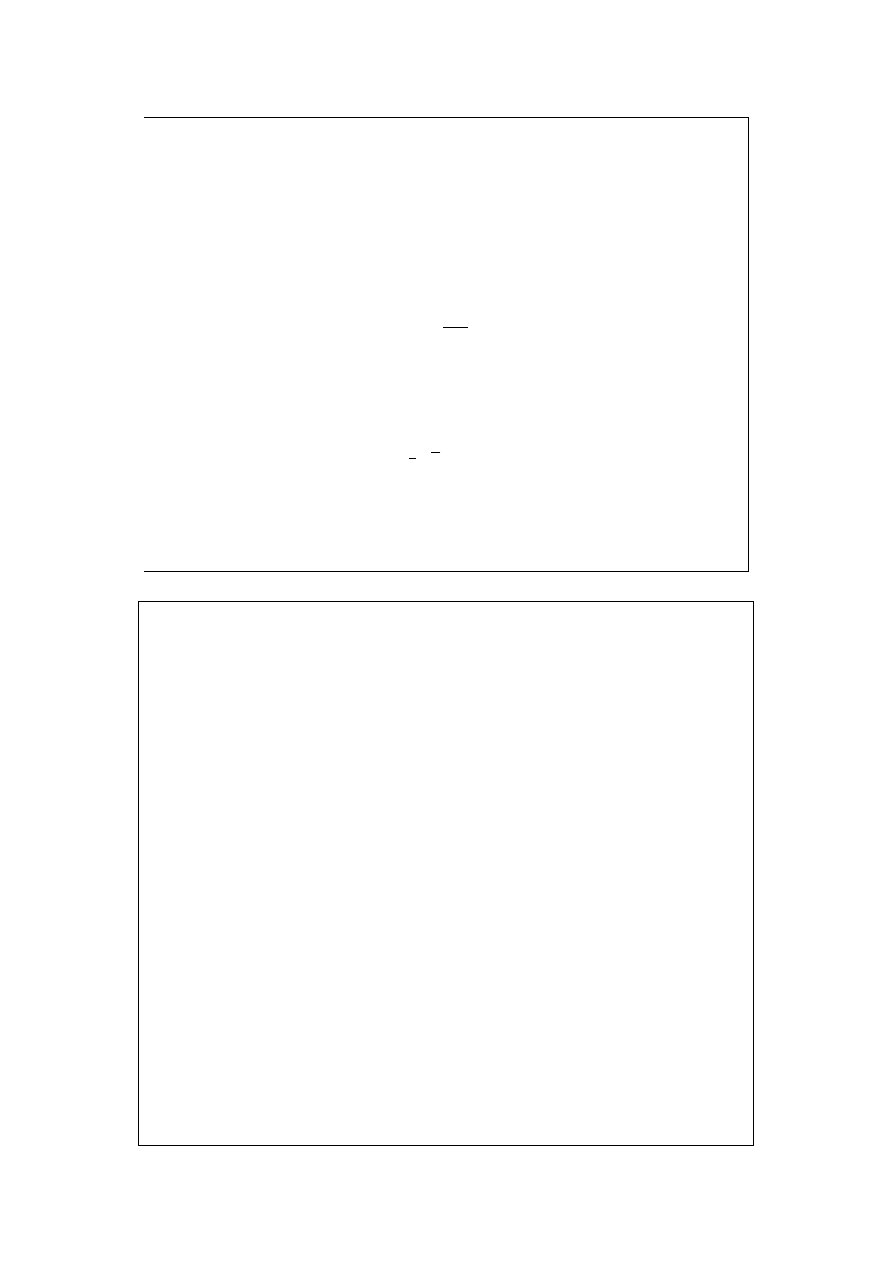
32
Chem. Fiz. TCH II/05
32
Przewodnictwo cieplne
gazów
τ
χ
Ad
dx
dT
dq
=
V
V
c
c
c
η
λ
ρ
χ
=
=
3
1
Przepływ ciepła zależy od gradientu temperatury dT/dx. Ilość ciepła
przechodząca przez prostopadłą do gradientu temperatury
powierzchnię A, w czasie d
τ
wynosi:
Przewodnictwo cieplne gazu,
χ
, polega na przenoszeniu energii
kinetycznej przez cząsteczki pomiędzy sąsiadującymi warstwami.
Rozumując analogicznie jak w przypadku lepkości, otrzymujemy:
Przewodnictwo cieplne gazów jest niezmiernie ważne w
niektórych metodach detekcji gazów i par (np. w GC).
Wielkość c
V
jest ciepłem właściwym (pojemnością cieplną na gram) przy stałej
objętości.

33
Chem. Fiz. TCH II/05
33
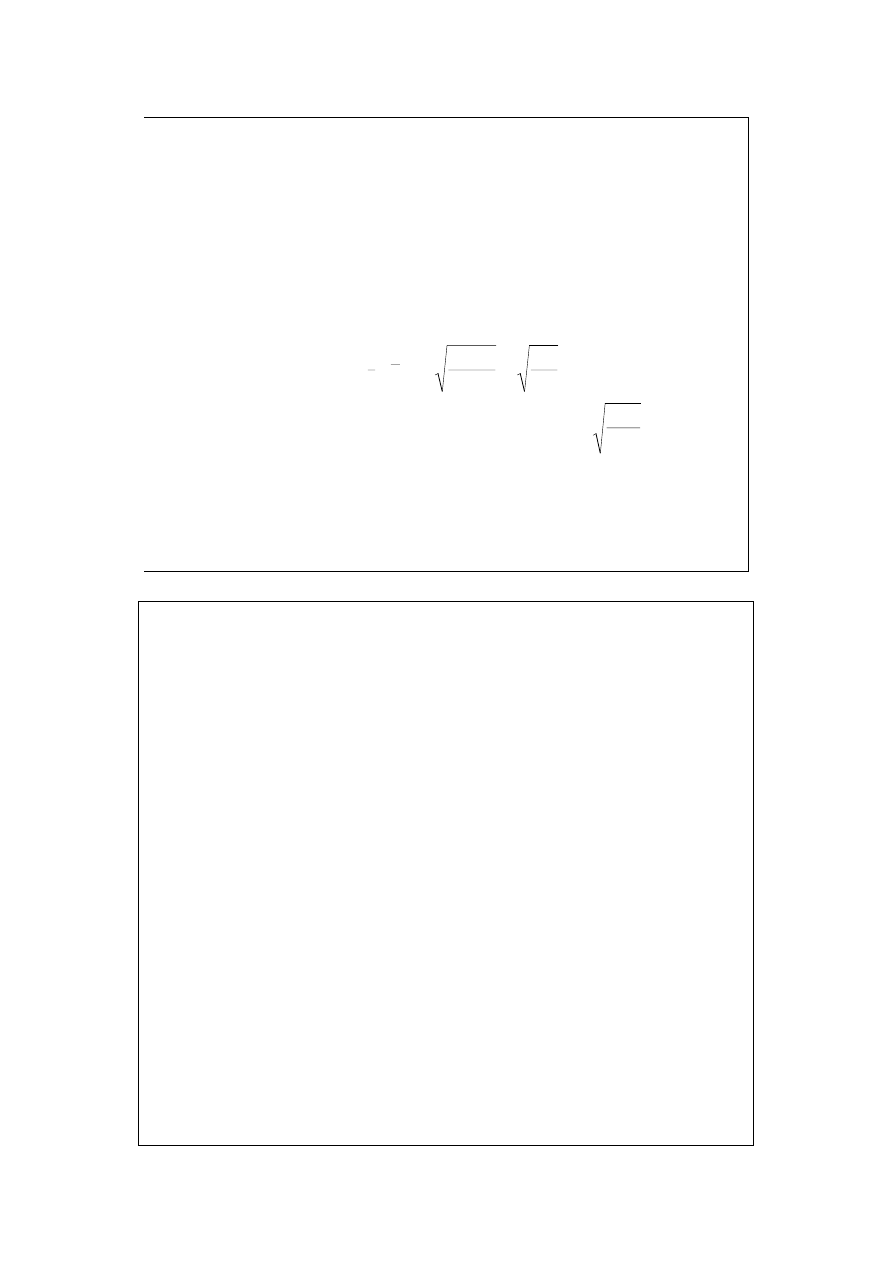
Dyfuzja
τ
Ad
dx
dc
D
dm
=
c
D
λ
3
1
=
Dyfuzja zależy od gradientu stężeń dc/dx. Masa substancji
przechodząca przez prostopadłą do gradientu stężenia powierzchnię
A
, w czasie d
τ
wynosi (II prawo Ficka):
Współczynnik dyfuzji, D, jest charakterystyczny dla substancji i tem-
peratury. Rozumując analogicznie jak w poprzednich przypadkach, gdy
autodyfuzja polega na ruchu termicznym cząsteczek gazu, otrzymujemy:
34
Chem. Fiz. TCH II/05
34
Efuzja
π
ρ
π
ρ
µ
2
2
4
1
P
RT
M
P
c
=
=
=
πρ
2
P
v
=
Efuzja polega na wypływie gazu z naczynia pod ciśnieniem przez otwór
(lub otwory) o wielkości mniejszej od średniej drogi swobodnej.
Strumień masy gazu przechodzący przez takie otwory
(masa na 1 cm
2
na 1 sekundę) wynosi:
Efuzja ma ogromne znaczenie praktyczne. Opisuje przepływ przez
przegrody porowate (np. separacja izotopów).
Objętościowo zaś (cm
3
/(cm
2
s)):
Ostatni wzór to tzw. prawo Grahama, odkryte już doświadczalnie w 1829 roku.

35
Chem. Fiz. TCH II/05
35
Entalpia swobodna a
lotność gazów (1)
V
dP
G
T
=
∂
Z części termodynamicznej wykładu pamiętamy, że:
Dla izotermy zatem:
VdP
dG
=
oraz:
∫
=
−
2
1
1
2
P
P
VdP
G
G
1
2
1
2
ln
2
1
P
P
RT
P
dP
RT
G
G
P
P
=
=
−
∫
Dla gazu doskonałego:
Jeżeli P
1
= P
0
, a odpowiadającą mu G oznaczymy G
0
, to dla innego
ciśnienia P:
0
0
ln
P
P
RT
G
G
+
=
Równanie to spełnione jest dla gazów rzeczywistych jedynie
ze wszystkimi znanymi ograniczeniami.
Pierwsza pochodna cząstkowa jest wynikiem przekształcenia równania na
slajdzie „Podstawowe równania. Podsumowanie(3)”.
P
0
= 1 atm = 1,01325·10
5
Pa.
G
0
nazywamy standardową entalpią
swobodną.

36
Chem. Fiz. TCH II/05
36
Entalpia swobodna a
lotność gazów (2)
Definicja lotności (ciśnienia efektywnego):
0
0
ln
P
f
RT
G
G
+
=
P
f
φ
=
gdzie
φ
jest współczynnikiem lotności.
Stan standardowy gazu rzeczywistego jest hipotetycznym stanem, w
którym gaz znajdujący się pod ciśnieniem P
0
zachowuje się jak gaz
doskonały.
Ogólnie zatem:
φ
ln
ln
0
0
RT
P
P
RT
G
G
+
+
=
Można dowieść, że:
(
)
∫
−
=
P
P
dP
Z
0
1
ln
φ
a także:
...
'
'
ln
2
2
1
+
+
=
P
C
P
B
φ
Są uzasadnione powody, aby tak właśnie
zdefiniować stan standardowy.
Gdyby przyjąć, że w stanie
standardowym f = P
0
, to stany
standardowe różnych gazów byłyby
różne i skomplikowane.
Gdyby zaś założyć, że stan standardowy
odpowiada P = 0, to powstaje kłopot,
gdyż G
→
-
∞
, gdy P
→
0.
Związek Z z równaniem wirialnym –
patrz slajd „Ściśliwość gazów (1)”.
Można także obliczać lotność
korzystając z równania Van der Waalsa.

37
Chem. Fiz. TCH II/05
37
Potencjał chemiczny
mieszanin gazowych (1)
i
n
T
i
V
dP
i
=
∂
,
µ
Dla mieszanin obowiązuje analogicznie:
Jeżeli mieszanina spełnia prawo Daltona:
i
i
dP
V
d
=
µ
i
i
i
P
RT
n
V
=
Dla mieszaniny gazów doskonałych:
a zatem:
i
n
T
P
i
i
i
P
RT
n
V
V
i
j
=
∂
∂
=
≠
,
,
oraz
i
i
i
P
dP
RT
d
=
µ
Ostatecznie:
0
0
ln
P
P
RT
i
i
i
+
=
µ
µ
Gdzie
µ
i
0
jest standardowym potencjałem chemicznym składnika i
(odpowiadającemu jego ciśnieniu równemu standardowemu).

38
Chem. Fiz. TCH II/05
38
Potencjał chemiczny
mieszanin gazowych (2)
Uprzednie równania spełniane są przez gazy rzeczywiste ze
wszystkimi znanymi ograniczeniami.
Musimy więc stosować lotności.
'
"
'
"
ln
i
i
i
i
f
f
RT
µ
µ
−
=
Oznaczając przez
µ
i
0
potencjał chemiczny składnika i
w stanie standardowym
0
0
ln
P
f
RT
i
i
i
+
=
µ
µ
Używamy dla stanów oznaczeń ’ i ”
ponieważ oznaczenia cyfrowe
zarezerwowane są dla składników.
Wyszukiwarka
Podobne podstrony:
chf tch I wykl 010Ac
chf tch I wykl 004c
chf tch I wykl 010a
chf tch I wykl 007c
chf tch I wykl 001c
chf tch I wykl 012c
chf tch I wykl 001c
chf tch I wykl 005b
chf tch I wykl 004b
chf tch I wykl 006b
chf tch I wykl 008c
chf tch I wykl 009c
chf tch I wykl 003c
chf tch I wykl 002c
chf tch I wykl 010c
chf tch I wykl 006c
chf tch I wykl 011c
chf tch I wykl 010Ac
więcej podobnych podstron