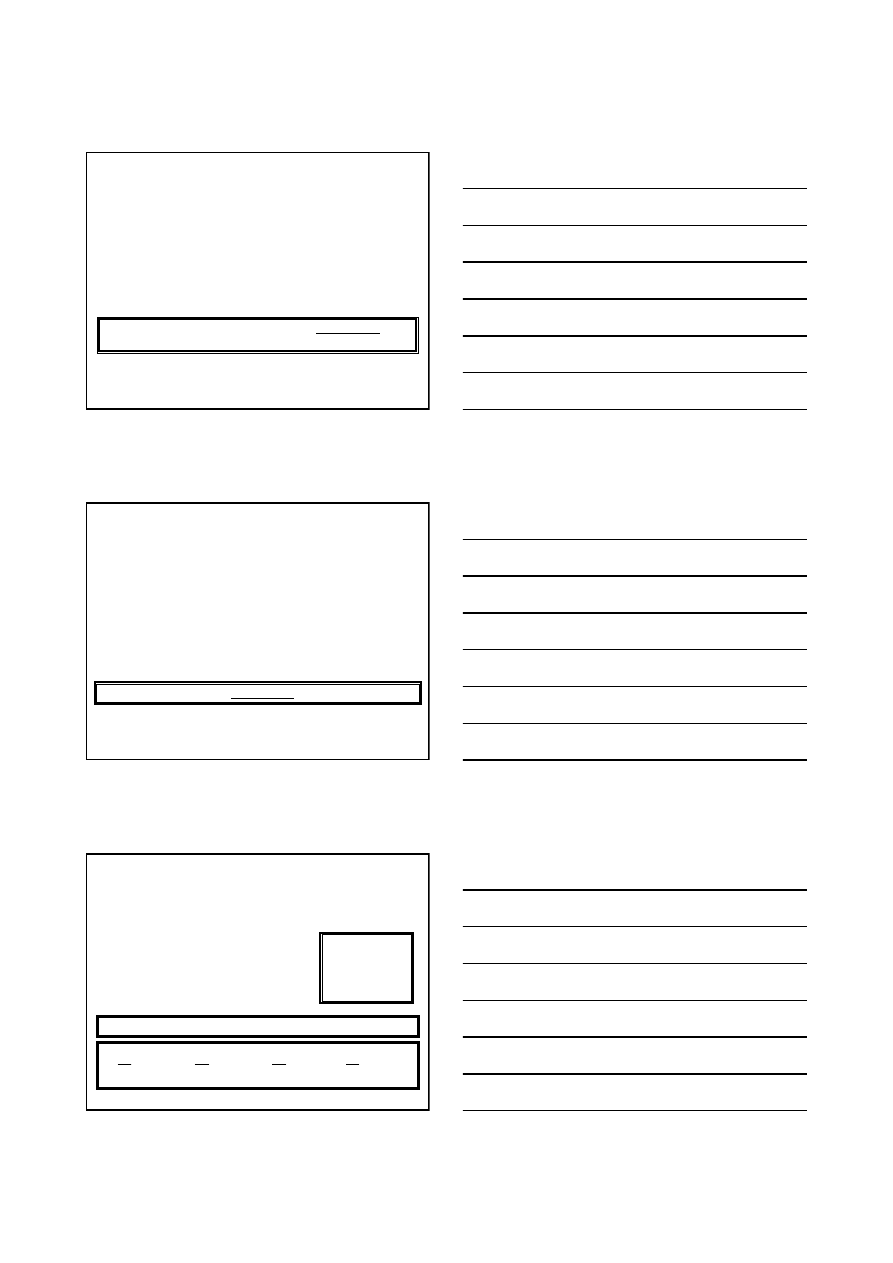
•1
Chem. Fiz. TCH II/05
1
Podstawowe równania –
podsumowanie (1)
Ts
u
f
−
=
w
q
u
+
=
dw
dq
du
+
=
sdT
Tds
du
df
−
−
=
dq
ds
=
T
:
go
odwracalne
procesu
dla
sdT
dq
du
df
−
−
=
:
zatem
dw
dq
du
=
−
:
go)
odwracalne
procesu
(dla
kolei
z
a
sdT
dw
df
−
=
:
wtedy
a
( )
0
:
na
izochorycz
jest
przemiana
jednak
gdy
=
T,V
df
PdV
dw
−
=
( )
dw
df
T
=
:
izotermy
dla
a
To wszystko jest prawdziwe, gdy w układzie nie występuje praca
nie objętościowa!!!
( )
dw
df
dq
Tds
T
<
>
i
:
lnego
nieodwraca
procesu
dla
a
( )
0
:
oraz
<
T,V
df
Chem. Fiz. TCH II/05
2
Podstawowe równania –
podsumowanie (2)
Ts
h
g
−
=
PV
u
h
+
=
VdP
PdV
du
dh
+
+
=
sdT
Tds
dh
dg
−
−
=
dq
ds
=
T
:
go
odwracalne
procesu
dla
sdT
VdP
PdV
dw
dg
−
+
+
=
:
zatem
dw
dq
du
=
−
:
i
( )
0
:
ej
izobaryczn
przemiany
dla
,
=
+
=
PdV
dw
dg
P
T
PdV
dw
−
=
:
ponadto
a
( )
VdP
PdV
dw
dg
T
+
+
=
:
izotermy
dla
Prawdziwe, gdy w układzie nie występuje praca nie objętościowa!!!
( )
VdP
PdV
dw
dg
dq
Tds
T
+
+
<
>
i
:
lnego
nieodwraca
procesu
dla
a
( )
0
:
oraz
<
T,P
dg
sdT
Tds
VdP
PdV
du
dg
−
−
+
+
=
Chem. Fiz. TCH II/05
3
Podstawowe równania –
podsumowanie (3)
(odwr.)
dw
Tds
du
+
=
PdV
Tds
du
−
=
obj.
tylko
praca
gdy
a
Pole powyżej zawiera podstawowe równania termodynamiki.
Ts
h
g
−
=
PV
u
h
+
=
dq
dw
du
+
=
(odwr.)
q
Tds
=
Ts
u
f
−
=
VdP
PdV
du
dh
+
+
=
:
zatem
VdP
Tds
dh
+
=
:
i
sdT
Tds
du
df
−
−
=
:
podobnie
PdV
sdT
df
−
−
=
:
i
sdT
Tds
dh
dg
−
−
=
:
raz
jeszcze
VdP
sdT
dg
+
−
=
:
i
s
T
f
V
−
=
∂
∂
P
V
f
T
−
=
∂
∂
s
T
g
P
−
=
∂
∂
V
P
g
T
=
∂
∂
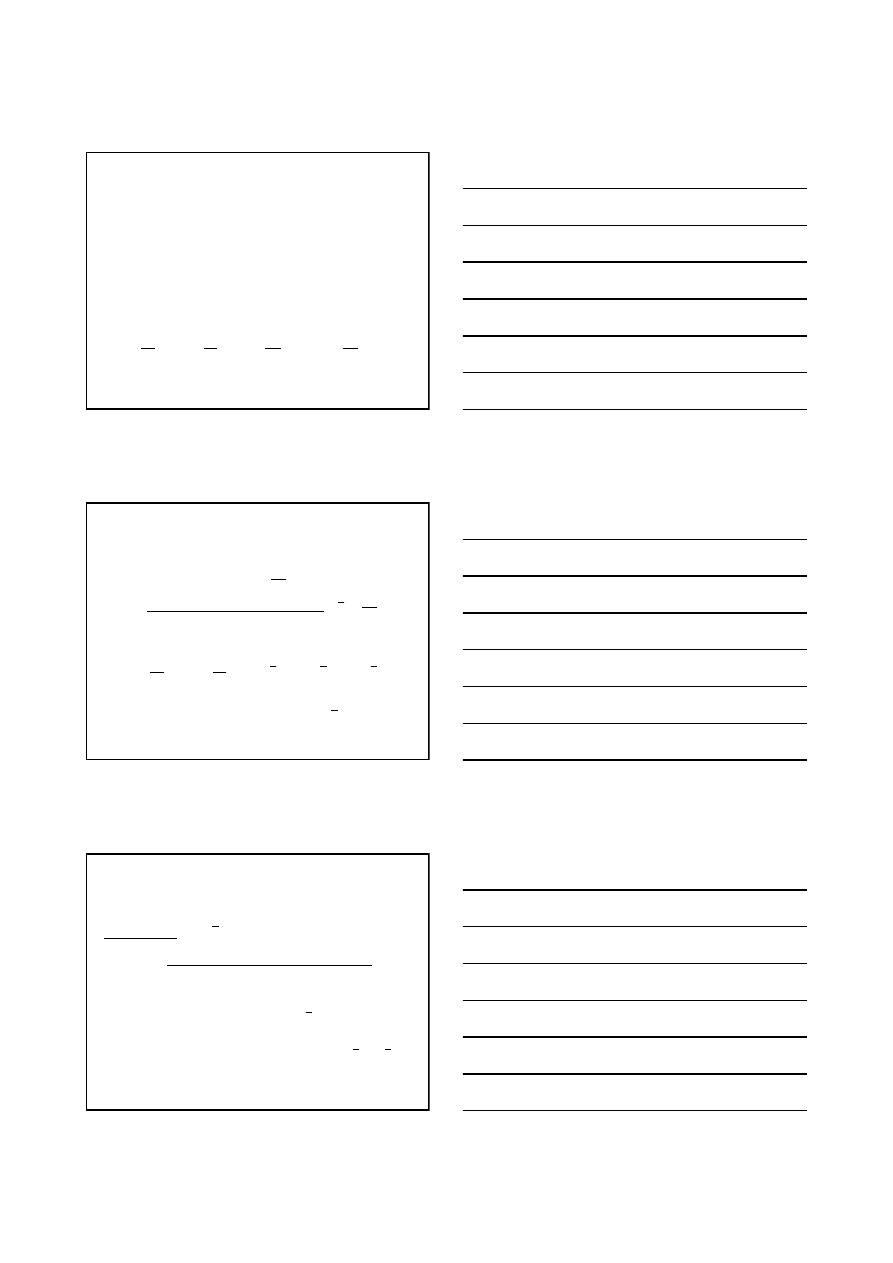
•2
Chem. Fiz. TCH II/05
4
Termodynamika układów
otwartych
W układach otwartych, tzn. wymieniających z otoczeniem także
materię, zmienia się ich skład.
Jeśli zawierają one więcej niż jeden, maksymalnie zaś k składników, to
dowolna funkcja stanu musi zależeć od parametrów stanu układu i jego
składu.
)
,...
,...
,
,
,
(
2
1
k
i
n
n
n
n
T
P
f
y
=
W konsekwencji:
i
n
P
T
i
n
P
T
n
T
n
P
dn
dn
dy
dn
dn
dy
dP
dP
dy
dT
dT
dy
dy
i
j
j
i
i
≠
≠
+
+
+
=
,
,
1
,
,
1
,
,
1
Chem. Fiz. TCH II/05
5
Cząstkowe molowe
wielkości (1)
Wielkości określone pochodną:
i
j
n
P
T
i
dn
dy
≠
,
,
nazywamy cząstkowymi molowymi wielkościami:
i
j
n
P
T
i
i
dn
dy
Y
≠
=
,
,
za pomocą których możemy opisać zmiany stanu układu otwartego:
k
k
i
i
n
T
n
P
dn
Y
dn
Y
dn
Y
dP
dP
dy
dT
dT
dy
dy
i
i
+
+
+
+
+
+
=
...
...
1
1
,
,
W szczególności, w warunkach
izotermiczno-izobarycznych:
∑
=
=
k
i
i
i
dn
Y
dy
1
Chem. Fiz. TCH II/05
6
Cząstkowe molowe
wielkości (2)
Można ją więc scałkować (po dowolnej drodze) od stanu
n
i
=0 do stanu n
i
(końcowe). Droga ta może być taka, aby:
const
Y
i
=
_
W tych warunkach:
∑
=
k
i
i
i
dn
Y
1
jest różniczką zupełną funkcji y
ergo
, funkcją stanu układu, zależną od jego składu.
W wyniku czego otrzymujemy:
∑
=
=
k
i
i
i
Y
n
y
1
∑
∑
=
=
+
=
k
i
i
i
k
i
i
i
dn
Y
Y
d
n
dy
1
1
zaś najogólniejsze wyrażenie na jej
różniczkę zupełną dane jest równaniem:
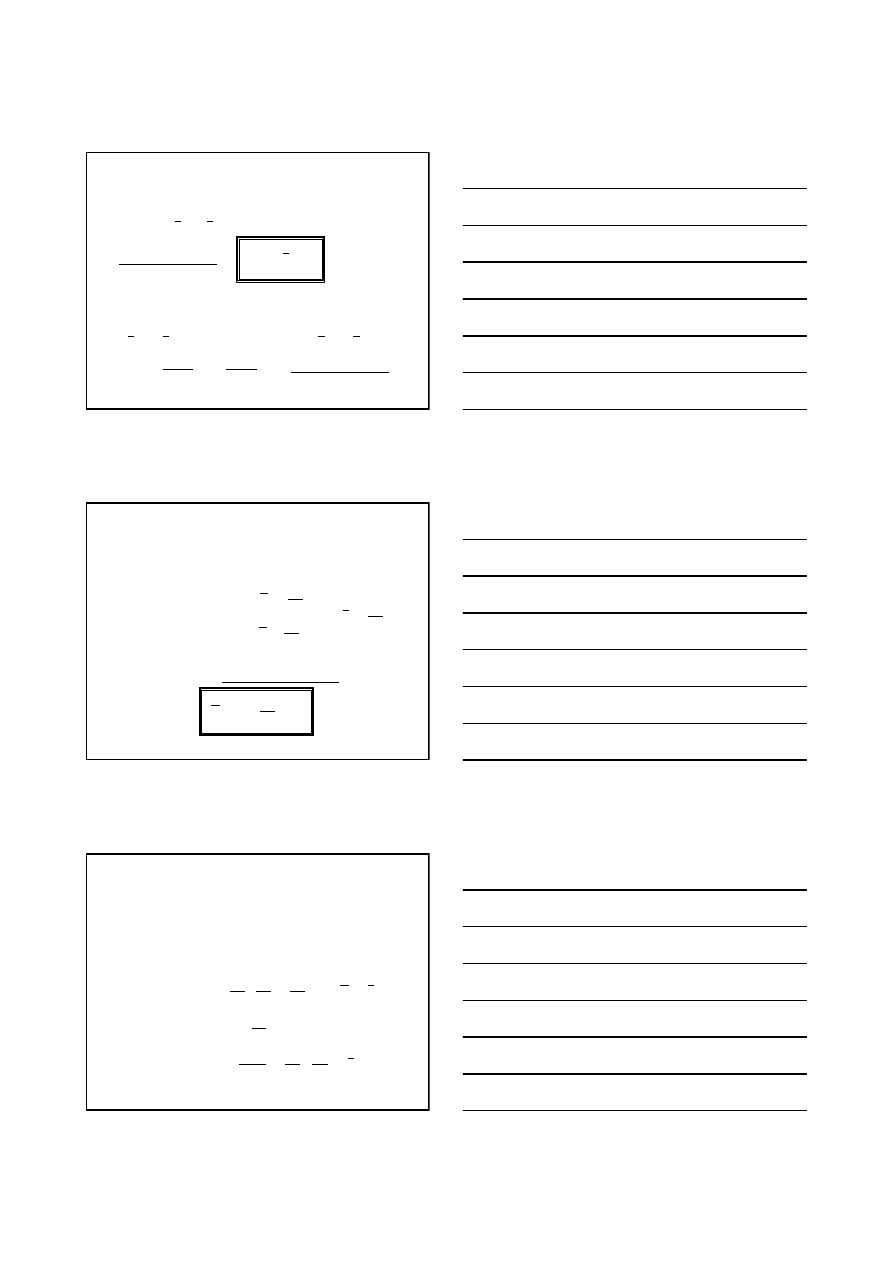
•3
Chem. Fiz. TCH II/05
7
Równanie Gibbsa-Duhema
Ostatnie wyrażenie nosi nazwę równania Gibbsa-Duhema.
Jeśli:
∑
∑
=
=
+
=
k
i
i
i
k
i
i
i
dn
Y
Y
d
n
dy
1
1
a równocześnie:
∑
=
=
k
i
i
i
dn
Y
dy
1
_
ergo
, musi być prawdziwe:
0
1
=
∑
=
k
i
i
i
Y
d
n
Dla układu dwuskładnikowego, można je wyrazić:
0
2
2
1
1
=
+
Y
d
n
Y
d
n
0
2
2
1
1
=
+
Y
d
x
Y
d
x
lub:
gdzie:
2
1
2
2
2
1
1
1
;
n
n
n
x
n
n
n
x
+
=
+
=
są ułamkami molowymi
składników 1 i 2.
Chem. Fiz. TCH II/05
8
Cząstkowe molowe
wielkości (3)
Najczęściej stosowane cząstkowe molowe wielkości to:
cząstkowa molowa entalpia:
i
j
n
P
T
i
i
n
h
H
≠
∂
∂
=
,
,
cząstkowa molowa entropia:
i
j
n
P
T
i
i
n
s
S
≠
∂
∂
=
,
,
cząstkowa molowa objętość:
i
j
n
P
T
i
i
n
v
V
≠
∂
∂
=
,
,
i najważniejsza z nich, cząstkowa molowa entalpia swobodna,
zwana też potencjałem chemicznym:
i
j
n
P
T
i
i
i
n
g
G
≠
∂
∂
=
=
,
,
µ
Chem. Fiz. TCH II/05
9
Cząstkowe molowe
wielkości (4)
Zależności pomiędzy cząstkowymi molowymi wielkościami są
analogiczne do zachodzących pomiędzy h, s, v i g.
Np., różniczkując względem n
i
wyrażenie:
Ts
h
g
−
=
otrzymujemy:
=
−
=
=
∂
∂
−
∂
∂
=
∂
∂
i
i
i
i
i
i
S
T
H
n
s
T
n
h
n
g
µ
a wychodząc z zależności:
s
T
g
P
−
=
∂
∂
otrzymamy:
S
T
n
s
n
T
g
i
i
i
−
=
∂
∂
=
∂
∂
−
=
∂
∂
∂
µ
2
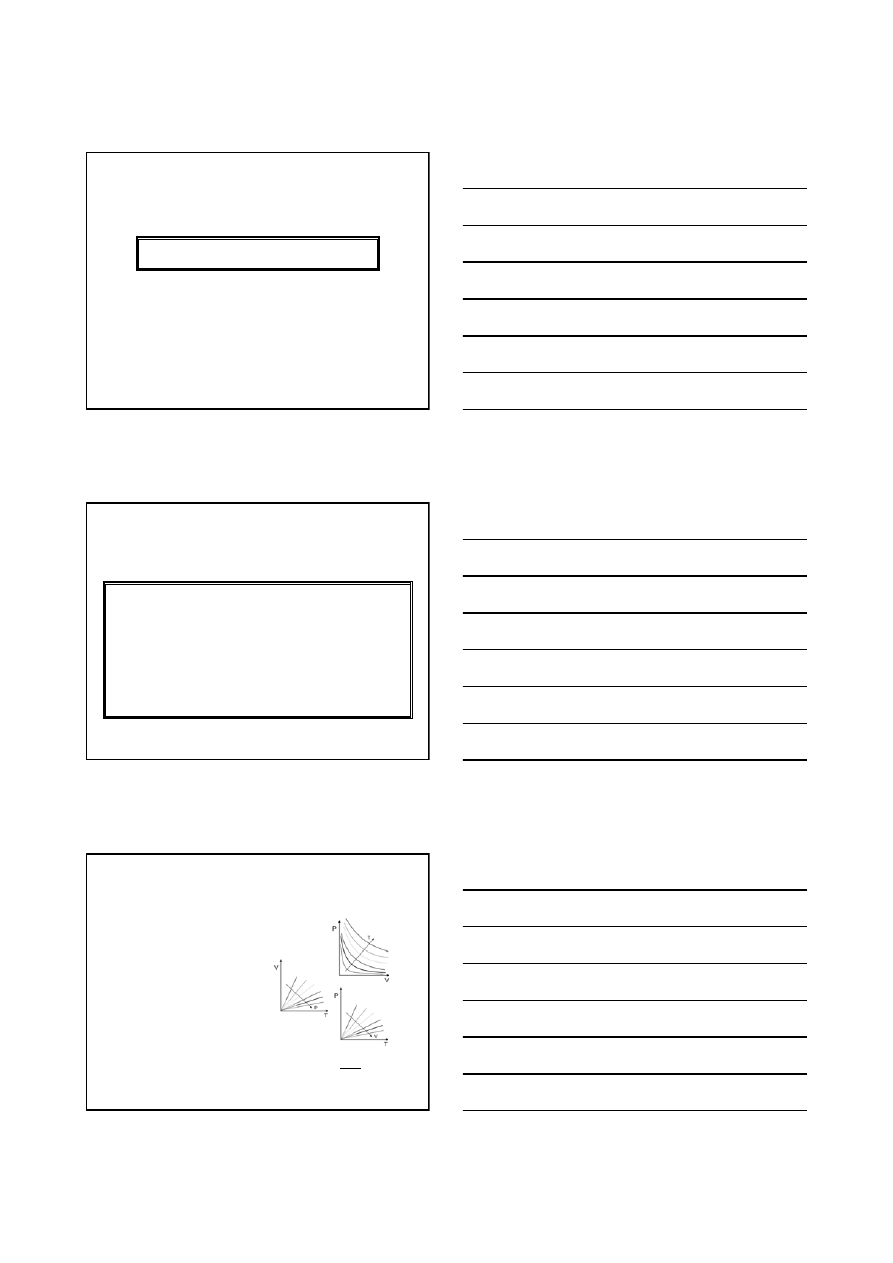
•4
Chem. Fiz. TCH II/05
10
Cząstkowe molowe
wielkości (5)
Najważniejsza zależność termodynamiki chemicznej:
k
k
i
i
dn
dn
dn
SdT
VdP
dG
µ
µ
µ
+
+
+
+
+
−
=
...
...
1
1
Pod stałym ciśnieniem i w stałej temperaturze:
k
k
i
i
dn
dn
dn
dG
µ
µ
µ
+
+
+
+
=
...
...
1
1
A wtedy, praca nieobjętościowa może być spowodowana zmianą
składu chemicznego układu. Np. w ogniwie galwanicznym zmiana
jego składu chemicznego (od substratów do produktów) prowadzi
do uzyskania pracy elektrycznej.
Chem. Fiz. TCH II/05
11
Gazy
Zagadnienia omawiane w tej części, przynajmniej w
pewnym zakresie, traktuję jako powtórkę.
Niektóre przeźrocza w tej serii są zatem jedynie
materiałem pomocniczym!
Większość omawianych tutaj zagadnień można
(i należy) powtórzyć sobie z dowolnego podręcznika
chemii fizycznej lub fizyki.
Chem. Fiz. TCH II/05
12
Gaz doskonały(1).
Prawa gazowe
Boyle’a
izoterma
const
T
const
PV
=
=
dla
;
Charlesa
izobara
const
P
T
V
=
∝
dla
;
Gay-Lussaca
izochora
const
V
T
P
=
∝
dla
;
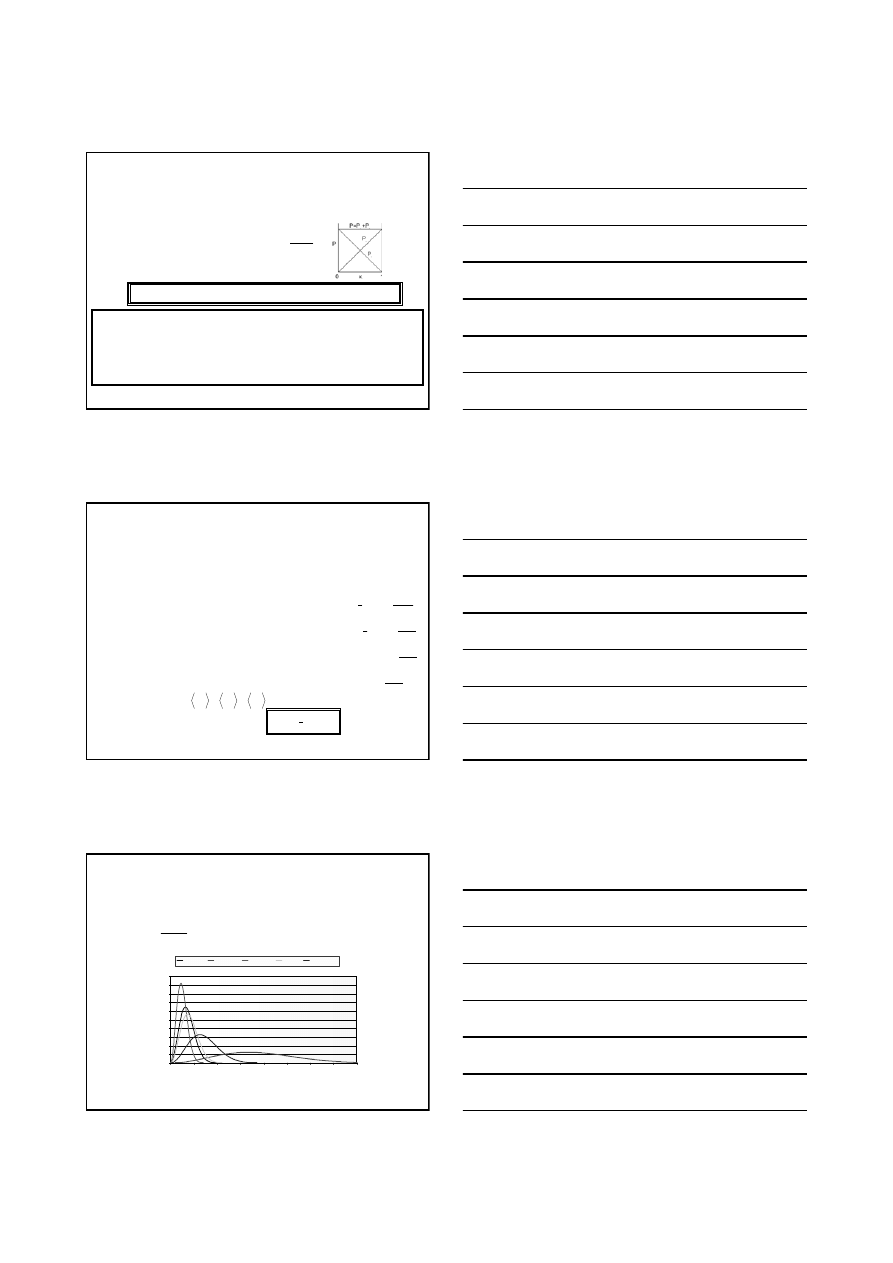
Równanie Clapeyrona
nRT
PV
=
T
PV
R
m
P 0
lim
→
=
•5
Chem. Fiz. TCH II/05
13
Gaz doskonały(2).
Prawa gazowe, c.d.
Avogadro
n
const
V
⋅
=
Daltona
;
;
1
i
i
k
i
i
x
P
P
P
P
⋅
=
=
∑
=
∑
=
=
k
i
i
i
i
n
n
x
1
Założenia kinetycznego modelu gazu doskonałego:
•
Cząsteczki gazu, o masie m znajdują się w ciągłym, chaotycznym
ruchu.
•
Jedyne oddziaływanie pomiędzy cząsteczkami, jak również
między nimi a ścianką zbiornika to zderzenia idealnie sprężyste.
•
Rozmiary cząsteczek są pomijalnie małe w porównaniu ze średnią
drogą pomiędzy zderzeniami (średnią drogą swobodną).
Chem. Fiz. TCH II/05
14
Gaz doskonały(3).
Ciśnienie wg modelu kinetycznego
1
2
mv
mv
t
F
−
=
∆
Druga zasada dynamiki Newtona:
V
nN
t
Av
l
A
x
∆
=
2
1
Liczba cząsteczek uderzających w ściankę o pow.
A
w czasie
∆
t
(zmiana szybkości z mv
x
na – mv
x
):
Ich masa (masa jednej cząsteczki to M/N
A
) :
Ich całkowita zmiana pędu :
V
nM
t
Av
lm
x
∆
=
2
1
V
nM
t
Av
t
F
x
∆
=
∆
2
Wywierane ciśnienie (F/A) :
V
nM
v
P
x
2
=
Ponieważ :
2
2
2
2
z
y
x
v
v
v
c
+
+
=
To ostatecznie :
nM
c
PV
2
3
1
=
Chem. Fiz. TCH II/05
15
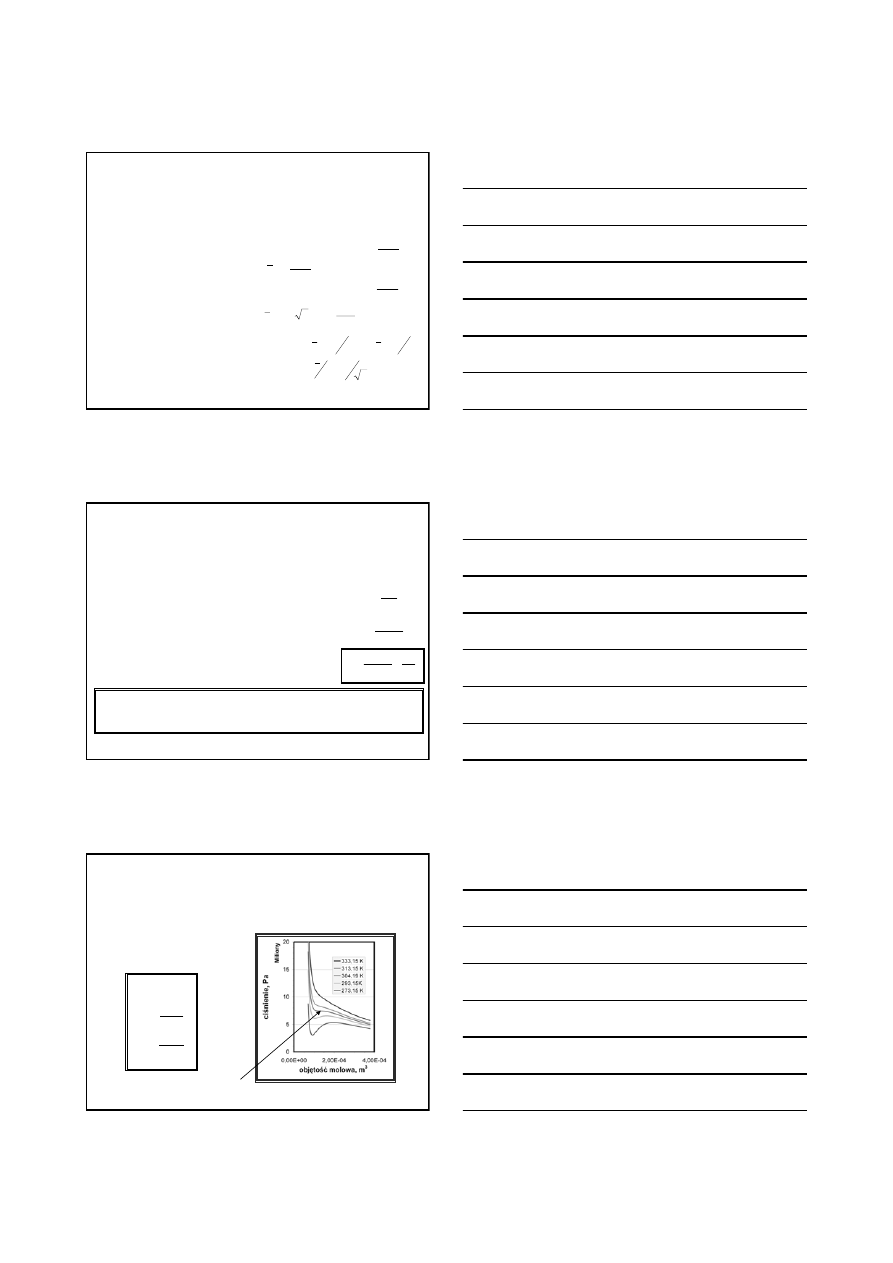
Rozkład Maxwella (1)
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
0
10
20
30
40
50
60
70
80
s zybkoś ć , m/s
wz
g
lę
dna
l
ic
z
b
a
c
z
ą
st
e
c
z
e
k
He ; 10 K
He ; 273 K
Rn; 273 K
Kr; 273
Azot;273 K
)
2
/(
2
2
/
3
2
2
4
)
(
RT
Mv
e
v
RT
M
v
f
−
=
π
π
bez wyprowadzania
•6
Chem. Fiz. TCH II/05
16
Rozkład Maxwella (2)
2
/
1
3
=
M
RT
c
Wykorzystując rozkład Maxwella można także wyznaczyć
takie własności cząsteczek gazu jak:
szybkość średnia kwadratowa :
szybkość średnia :
szybkość najbardziej prawdopodobna:
średnia szybkość względna:
częstość zderzeń:
średnia droga swobodna:
2
/
1
8
=
M
RT
c
π
2
/
1
2
*
=
M
RT
c
2
/
1
8
2
=
⋅
=
πµ
kT
c
c
wzgl
kT
P
c
V
N
c
z
wzgl
wzgl
⋅
=
⋅
=
σ
σ
P
kT
z
c
σ
λ
2
=
=
Chem. Fiz. TCH II/05
17
Gazy rzeczywiste. Równanie
stanu Van der Waalsa
nRT
PV
=
Udoskonalenie (urealnienie) równania Clapeyrona:
na 1 mol gazu:
Poprawka na objętość mola cząsteczek gazu, b
m
V
RT
P
=
b
V
RT
P
m
−
=
Poprawka na oddziaływania międzycząsteczkowe, a
2
m
m
V
a
b
V
RT
P
−
−
=
• same cząsteczki gazu zajmują pewną objętość, b
• atraktywne (przyciągające) oddziaływania międzycząsteczkowe zmniejszają pęd
(zatem siłę) cząsteczek zmierzających ku ściance, a także częstość ich zderzeń ze
ścianką, dodatkowo zmniejszają więc ciśnienie (do kwadratu stężenia cząsteczek).
Chem. Fiz. TCH II/05
18
Izotermy Van der Waalsa
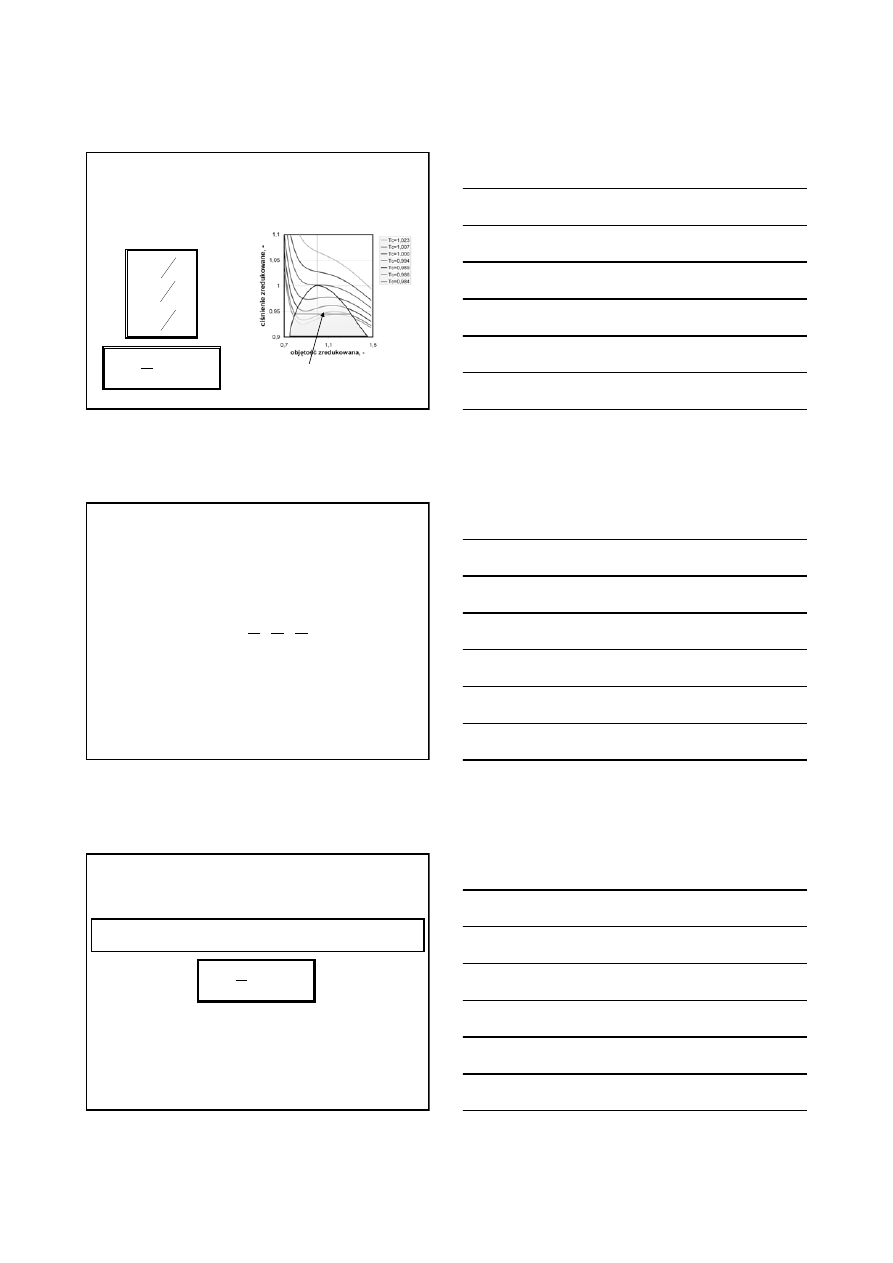
(1)
punkt krytyczny
b
V
c
3
=
2
27b
a
P
c
=
bR
a
T
c
27
8
=
Związki między
współczynnikami r-nia Van
der Waalsa a parametrami
krytycznymi:
•7
Chem. Fiz. TCH II/05
19
Izotermy Van der Waalsa
(2)
c
P
P
=
π
c
V
V
=
φ
c
T
T
=
θ
rzeczywisty przebieg
izotermy podczas skraplania
zredukowane parametry stanu
i równanie Van der Waalsa
(
)
θ
φ
φ
π
8
1
3
3
2
=
−
+
Chem. Fiz. TCH II/05
20
Wirialne równania stanu
...)
'
'
'
1
(
3
2
+
+
+
+
=
P
D
P
C
P
B
RT
PV
m
+
+
+
+
=
...
1
3
2
m
m
m
m
V
D
V
C
V
B
RT
PV
Zaproponował je Kamerlingh Onnes (dwie formy):
Współczynniki B, C, D (B’, C’, D’) noszą nazwę
współczynników wirialnych (zależą od temperatury).
Często stosuje się jedynie drugi współczynnik wirialny:
P
B
RT
PV
m
"
+
=
Chem. Fiz. TCH II/05
21
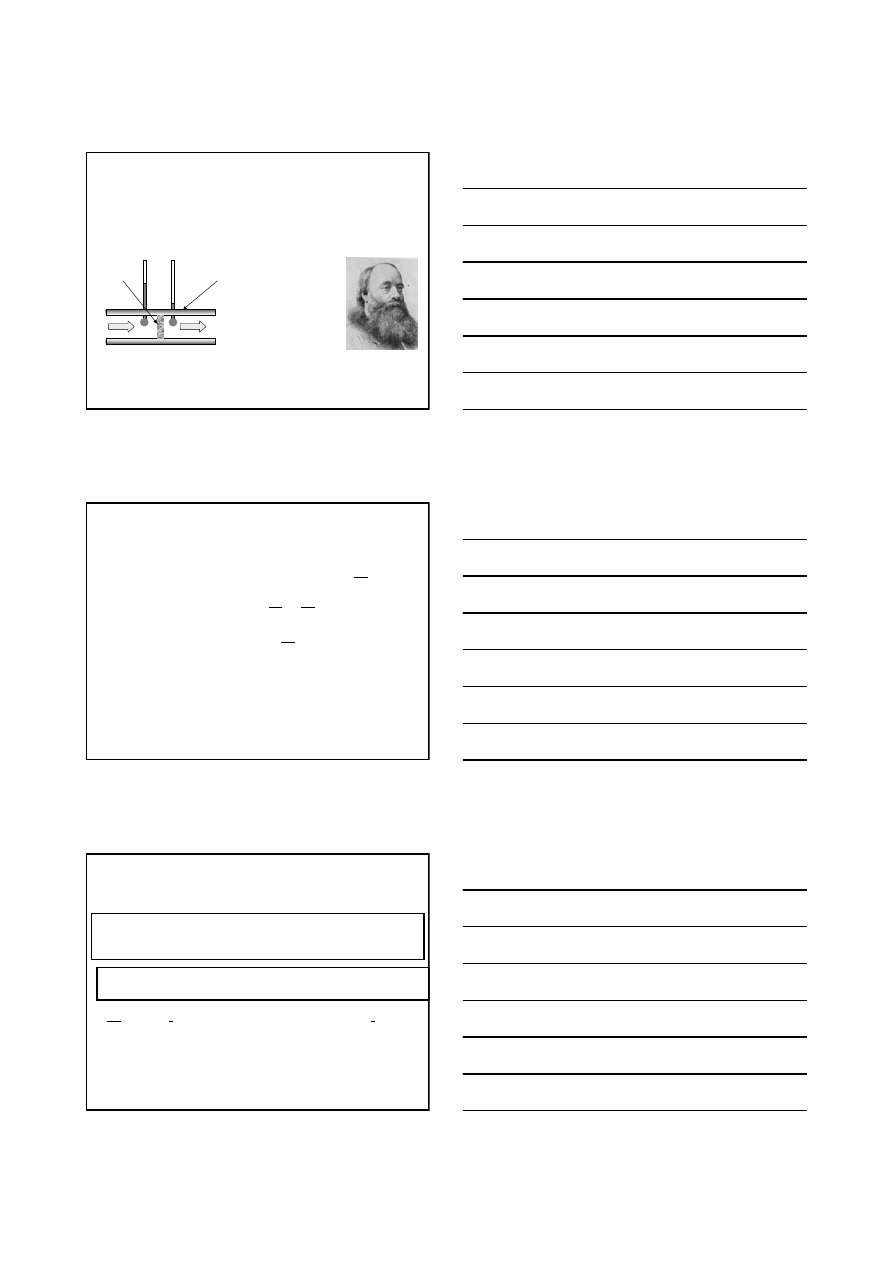
Gazy rzeczywiste w tych samych warunkach zredukowanej objętości
i temperatury wywierają takie samo ciśnienie zredukowane.
Zasada stanów
odpowiadających sobie
Wynika ze zredukowanego r-nia Van der Waalsa (znikają w nim
bowiem charakterystyczne dla poszczególnych gazów współczynniki
a
i b). Inne równania stanu też ją zawierają.
Zasada ta zawodzi, gdy cząsteczki gazu są nie sferyczne lub polarne.
(
)
θ
φ
φ
π
8
1
3
3
2
=
−
+
•8
Chem. Fiz. TCH II/05
22
Skraplanie gazów. Efekt
Joule’a-Thomsona (1).
Klasyczne metody sprężania poniżej w temperaturze T<T
c
często nie
wystarczały do skroplenia gazów o coraz niższych T
c
.
q = 0
adiabatyczne
rozprężanie
-w = P
2
V
2
– P
1
V
1
James Joule
rura izolowana
termicznie
przegroda
porowata
P
1
,T
1
P
2
,T
2
U
2
– U
1
= P
2
V
2
– P
1
V
1
U
2
+ P
2
V
2
= U
1
+ P
1
V
1
H
2
= H
1
proces izoentalpowy
Chem. Fiz. TCH II/05
23
Skraplanie gazów. Efekt
Joule’a-Thomsona (2).
Współczynnik Joule’a – Thomsona:
H
JT
P
T
∂
∂
=
µ
Można dowieść, że :
−
∂
=
V
dT
V
T
C
P
P
JT
1
µ
Dla gazu doskonałego (V=RT/P):
0
;
0
=
=
−
∂
JT
P
V
dT
V
T
µ
Dla każdego gazu rzeczywistego istnieje tzw. temperatura inwersji, T
inw
.
Gdy T < T
inw
,
µ
JT
>0
, gdy T > T
inw
,
µ
JT
<0
, gdy T = T
inw
,
µ
JT
=0
.
T
inw
zależy od ciśnienia (mogą być dwie T
inw
– dolna i górna).
Chem. Fiz. TCH II/05
24
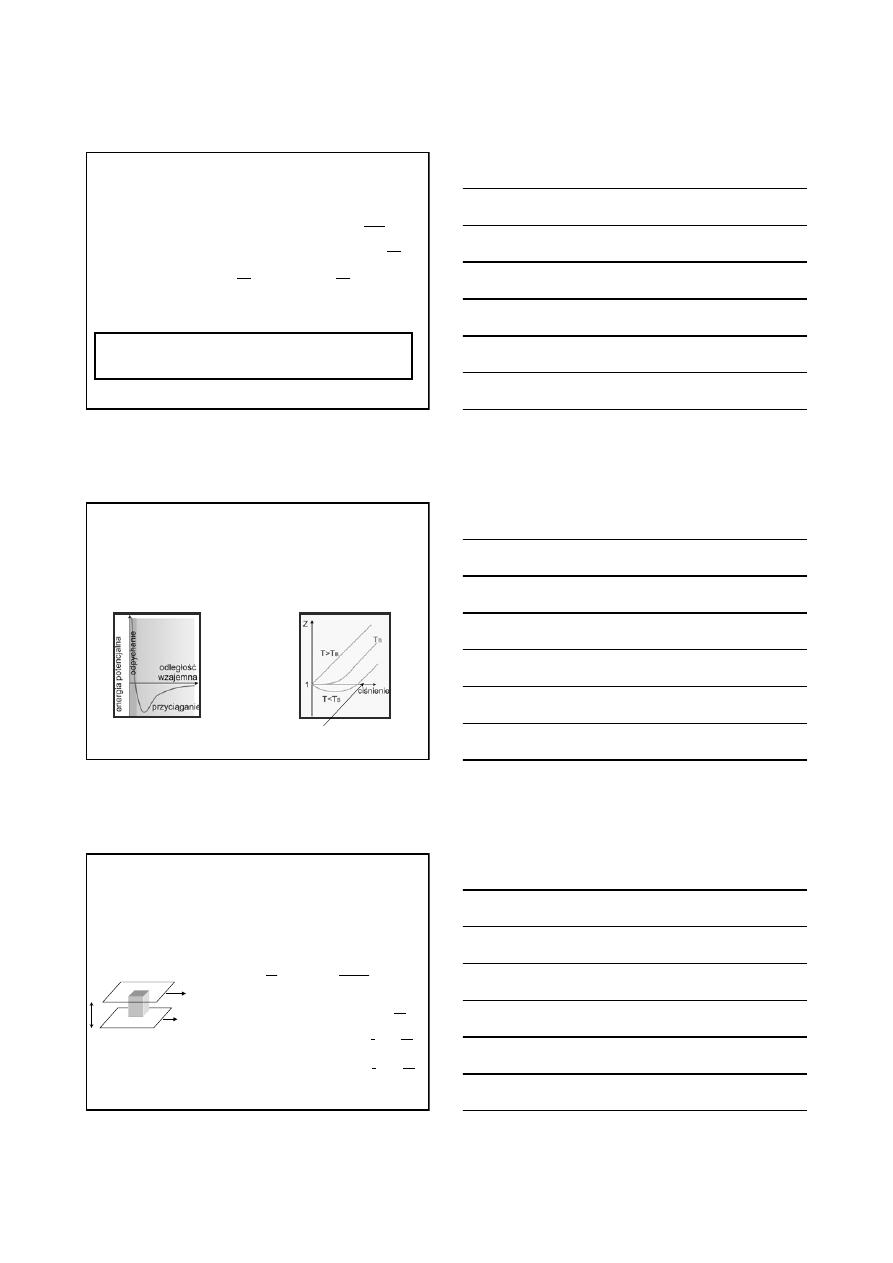
Na każdy stopień swobody ruchu translacyjnego (na każdy człon
kwadratowy energii kinetycznej) cząsteczki przypada identyczna
energia równa ½kT
Zasada ekwipartycji energii
Prawidłowość ta jest spełniona tylko dla helu (i innych gazów
jednoatomowych).
Najwyraźniej, cząsteczki gazu posiadają jeszcze inną energię.
Gdyby, jak wynika z modelu kinetycznego, energia translacyjna była
jedyną energią cząsteczek gazu, to:
R
C
dT
du
V
V
2
3
=
=
a ze znanych względów
R
C
P
2
5
=
•9
Chem. Fiz. TCH II/05
25
Cząsteczki mogą także wykonywać ruch rotacyjny (obrót wokół osi
symetrii). Dla cząsteczek dwuatomowych lub o budowie liniowej (2
osie, moment bezwładności wokół trzeciej – głównej osi cząsteczki
liniowej – jest pomijalny), istnieją dwa stopnie swobody rotacji, zatem
Pojemności cieplne gazów
(1)
gdzie J jest momentem bezwładności.
Dla cząsteczek przestrzennych, o trzech momentach bezwładności
RT
J
E
rot
=
=
2
2
1
ω
RT
E
rot
2
3
=
Chem. Fiz. TCH II/05
26
Zatem, dla cząsteczek liniowych:
Pojemności cieplne gazów
(2)
Stwierdzono jednak, że w wysokich temperaturach, krzywa
ogrzewania gazów wieloatomowych wykazuje jeszcze większe
pojemności cieplne. Dochodzi wtedy do wzbudzenia oscylacyjnego.
a dla nieliniowych (przestrzennych):
R
C
V
2
5
=
R
C
P
2
7
=
R
C
V
3
=
R
C
P
4
=
Chem. Fiz. TCH II/05
27
a na każde drganie przypadają dwa stopnie swobody (energia
potencjalna i kinetyczna). Zatem w wysokich temperaturach, dla
gazów dwuatomowych
Liczba drgań normalnych wynosi, dla cząsteczek :
nieliniowych liniowych
Pojemności cieplne gazów
(3)
Ogólnie, energia wewnętrzna gazów dana jest równaniem:
5
3
−
N
6
3
−
N
R
C
V
2
7
=
R
C
P
2
9
=
const
RT
RT
RT
E
E
E
E
U
nukl
el
osc
rot
tr
+
+
+
=
+
+
+
=
2
3
.
.,
•10
Chem. Fiz. TCH II/05
28
Ściśliwość gazów (1)
RT
PV
Z
m
=
0
=
dP
dZ
Współczynnik ściśliwości gazów dany jest wzorem:
Dla gazu doskonałego wynosi on zawsze 1 zaś pochodna:
'
lim
...
'
2
'
0
B
dP
dZ
C
B
dP
dZ
P
=
+
+
=
→
Dla gazów rzeczywistych:
Jednak B nie musi być równe zeru, ponadto zależy od temperatury.
Istnieje temperatura, zwana temperaturą Boyle’a, w której B = 0
dla P
→ 0, czyli gazy rzeczywiste zachowują się w niej naprawdę
jak gaz doskonały (w niskich ciśnieniach).
Chem. Fiz. TCH II/05
29
Powodem są oddziaływania międzycząsteczkowe odpychające
(bliskiego zasięgu) i przyciągające (dalszego zasięgu).
Ściśliwość gazów (2)
gaz doskonały
Chem. Fiz. TCH II/05
30
Lepkość gazów (1)
(1)
;
A
dx
dv
F
η
=
(2)
;
8
4
τ
η
π
l
∆P
r
v
=
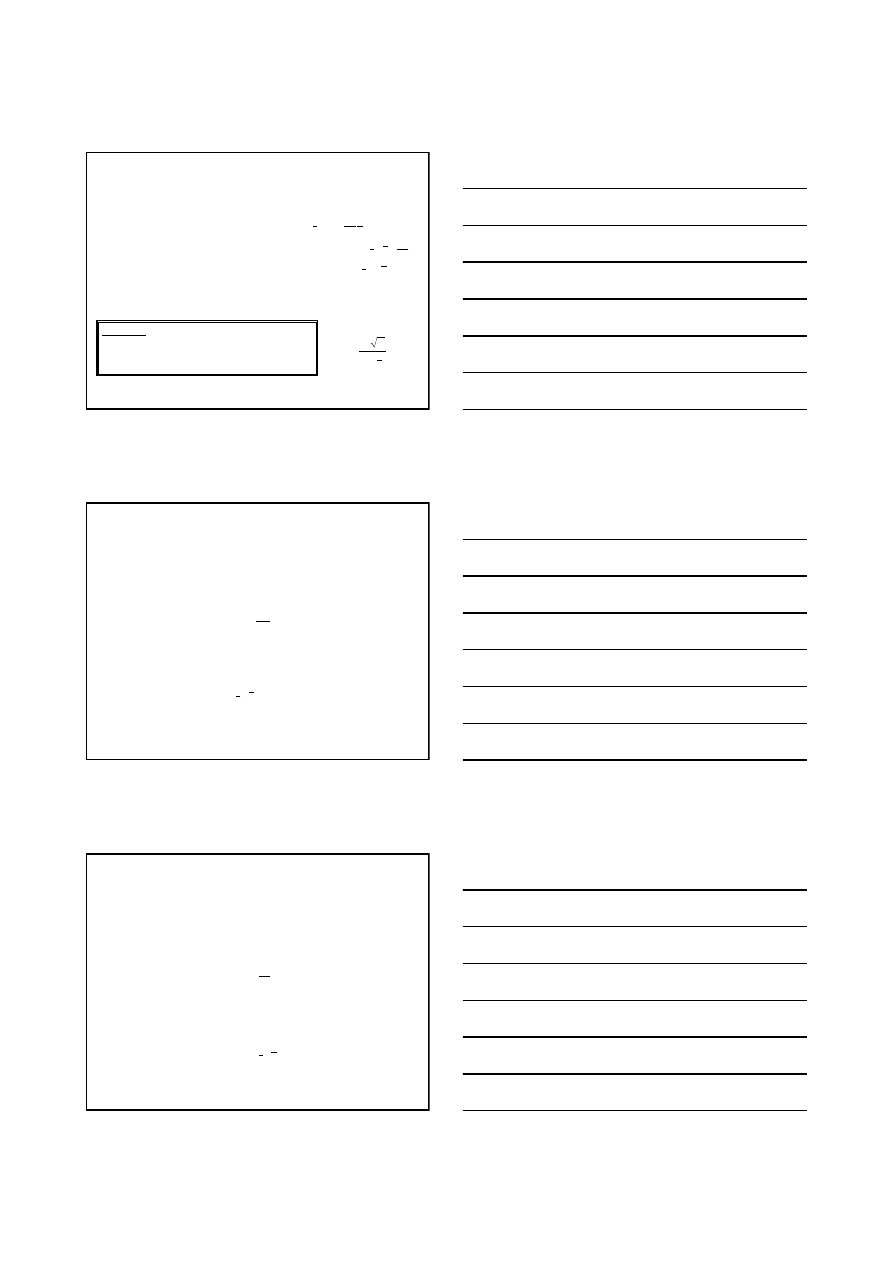
Współczynnik tarcia wewnętrznego,
η
, czyli lepkość, możemy
rozpatrywać w kategoriach teorii kinetycznej gazów, jako wymianę pędu
przez cząsteczki sąsiadujących warstw poruszającego się gazu.
(tylko 1/3 wymienia pęd wzdłuż osi x); N=N
A
·
λ
A/V
m
λ
v
v+
λ
·dv/dx
Jedna cząsteczka przenosi pęd:
dx
dv
m
p
λ
=
1
Jeżeli w V=
λ
A
znajduje się N cząsteczek:
dx
dv
Nm
p
t
λ
3
1
=
dx
dv
A
p
t
2
3
1
ρλ
=
•11
Chem. Fiz. TCH II/05
31
Lepkość gazów (2)
Porównując ostatnie równanie z r-niem Poiseuille’a:
Jeżeli wszystko dzieje się w czasie
τ
=1/z
:
τ
ρλ
1
2
3
1
dx
dv
A
F
=
a ponieważ
λ
/
τ
=ĉ
:
A
dx
dv
c
F
λ
ρ
3
1
=
c
ρλ
η
3
1
=
który to wynik możemy dowolnie komplikować podstawiając doń
wielkości uzyskane z rozkładu Maxwella.
Wnioski:
(sprawdzające się doświadczalnie)
• lepkość nie zależy od ciśnienia,
• lepkość zależy od pierwiastka kwadrato-
wego temperatury
Wzór Sutherlanda:
T
c
T
+
=
1
0
η
η
Chem. Fiz. TCH II/05
32
Przewodnictwo cieplne
gazów
τ
χ
Ad
dx
dT
dq
=
V
V
c
c
c
η
λ
ρ
χ
=
=
3
1
Przepływ ciepła zależy od gradientu temperatury dT/dx. Ilość ciepła
przechodząca przez prostopadłą do gradientu temperatury
powierzchnię A, w czasie d
τ
wynosi:
Przewodnictwo cieplne gazu,
χ
, polega na przenoszeniu energii
kinetycznej przez cząsteczki pomiędzy sąsiadującymi warstwami.
Rozumując analogicznie jak w przypadku lepkości, otrzymujemy:
Przewodnictwo cieplne gazów jest niezmiernie ważne w
niektórych metodach detekcji gazów i par (np. w GC).
Chem. Fiz. TCH II/05
33
Dyfuzja
τ
Ad
dx
dc
D
dm
=
c
D
λ
3
1
=
Dyfuzja zależy od gradientu stężeń dc/dx. Masa substancji
przechodząca przez prostopadłą do gradientu stężenia powierzchnię
A
, w czasie d
τ
wynosi (II prawo Ficka):
Współczynnik dyfuzji, D, jest charakterystyczny dla substancji i tem-
peratury. Rozumując analogicznie jak w poprzednich przypadkach, gdy
autodyfuzja polega na ruchu termicznym cząsteczek gazu, otrzymujemy:
•12
Chem. Fiz. TCH II/05
34
Efuzja
π
ρ
π
ρ
µ
2
2
4
1
P
RT
M
P
c
=
=
=
πρ
2
P
v
=
Efuzja polega na wypływie gazu z naczynia pod ciśnieniem przez otwór
(lub otwory) o wielkości mniejszej od średniej drogi swobodnej.
Strumień masy gazu przechodzący przez takie otwory
(masa na 1 cm
2
na 1 sekundę) wynosi:
Efuzja ma ogromne znaczenie praktyczne. Opisuje przepływ przez
przegrody porowate (np. separacja izotopów).
Objętościowo zaś (cm
3
/(cm
2
s)):
Chem. Fiz. TCH II/05
35
Entalpia swobodna a
lotność gazów (1)
V
dP
G
T
=
∂
Z części termodynamicznej wykładu pamiętamy, że:
Dla izotermy zatem:
VdP
dG
=
oraz:
∫
=
−
2
1
1
2
P
P
VdP
G
G
1
2
1
2
ln
2
1
P
P
RT
P
dP
RT
G
G
P
P
=
=
−
∫
Dla gazu doskonałego:
Jeżeli P
1
= P
0
, a odpowiadającą mu G oznaczymy G
0
, to dla innego
ciśnienia P:
0
0
ln
P
P
RT
G
G
+
=
Równanie to spełnione jest dla gazów rzeczywistych jedynie
ze wszystkimi znanymi ograniczeniami.
Chem. Fiz. TCH II/05
36
Entalpia swobodna a
lotność gazów (2)
Definicja lotności (ciśnienia efektywnego):
0
0
ln
P
f
RT
G
G
+
=
P
f
φ
=
gdzie
φ
jest współczynnikiem lotności.
Stan standardowy gazu rzeczywistego jest hipotetycznym stanem, w
którym gaz znajdujący się pod ciśnieniem P
0
zachowuje się jak gaz
doskonały.
Ogólnie zatem:
φ
ln
ln
0
0
RT
P
P
RT
G
G
+
+
=
Można dowieść, że:
(
)
∫
−
=
P
P
dP
Z
0
1
ln
φ
a także:
...
'
'
ln
2
2
1
+
+
=
P
C
P
B
φ
•13
Chem. Fiz. TCH II/05
37
Potencjał chemiczny
mieszanin gazowych (1)
i
n
T
i
V
dP
i
=
∂
,
µ
Dla mieszanin obowiązuje analogicznie:
Jeżeli mieszanina spełnia prawo Daltona:
i
i
dP
V
d
=
µ
i
i
i
P
RT
n
V
=
Dla mieszaniny gazów doskonałych:
a zatem:
i
n
T
P
i
i
i
P
RT
n
V
V
i
j
=
∂
∂
=
≠
,
,
oraz
i
i
i
P
dP
RT
d
=
µ
Ostatecznie:
0
0
ln
P
P
RT
i
i
i
+
=
µ
µ
Gdzie
µ
i
0
jest standardowym potencjałem chemicznym składnika i
(odpowiadającemu jego ciśnieniu równemu standardowemu).
Chem. Fiz. TCH II/05
38
Potencjał chemiczny
mieszanin gazowych (2)
Uprzednie równania spełniane są przez gazy rzeczywiste ze
wszystkimi znanymi ograniczeniami.
Musimy więc stosować lotności.
'
"
'
"
ln
i
i
i
i
f
f
RT
µ
µ
−
=
Oznaczając przez
µ
i
0
potencjał chemiczny składnika i
w stanie standardowym
0
0
ln
P
f
RT
i
i
i
+
=
µ
µ
Wyszukiwarka
Podobne podstrony:
chf tch I wykl 010Ac
chf tch I wykl 004c
chf tch I wykl 010a
chf tch I wykl 007c
chf tch I wykl 001c
chf tch I wykl 012c
chf tch I wykl 001c
chf tch I wykl 004b
chf tch I wykl 006b
chf tch I wykl 008c
chf tch I wykl 009c
chf tch I wykl 003c
chf tch I wykl 002c
chf tch I wykl 005c
chf tch I wykl 010c
chf tch I wykl 006c
chf tch I wykl 011c
chf tch I wykl 010Ac
więcej podobnych podstron