3582327823
Chemia fizyczna - termodynamika molekularna 2010/2011 31
Wykład 9 3.12.2010
1. Symulacje komputerowe - metody dynamiki molekularnej i Monte Cado.
Rozwój technik komputerowych sprawił, że właściwości makroskopowe mogą być bezpośrednio obliczone na podstawie właściwości cząsteczek. Można to zrobić dla skończonej liczby cząsteczek, stosunkowo niewielkiej (do paru tysięcy, w przypadku dużych, rozbudowanych cząsteczek, liczba ta jest mniejsza), ale w większości przypadkach wystarczającej, żeby w ten sposób można było uzyskać parametry termodynamiczne. Symulacje komputerowe dzielą się na 2 grupy:
- metody dynamiki molekularnej (MD), w których bezpośrednio rozwiązuje się równania ruchu, obserwując w ten sposób ewolucję wirtualnego układu;
- metody Monte Carlo (MC), w których generuje się łańcuch konfiguracji zgodnie z prawdopodobieństwem termodynamicznym wynikającym z określonego zespołu.
Symulacje komputerowe stanowią swoisty eksperyment obliczeniowy. Istotna różnica polega na tym, że otrzymany wynik nie jest obserwacją rzeczywistości, a wirtualnego układu, zdefiniowanego poprzez przyjęte potencjały oddziaływań międzycząsteczkowych.
2. Podsumowanie termodynamiki statystycznej
a. Właściwości makroskopowe mogą być wyprowadzone z właściwości cząsteczek.
b. Istnieją dwa sposoby takiego wyprowadzenia
- uśrednianie statystyczne (N —» °°),
- bezpośrednie obliczenia dla skończonej liczby cząsteczek (symulacje komputerowe).
c. Uśrednianie statystyczne - reguły;
- zakłada się zasadę równych prawdopodobieństw i hipotezę ergodyczną; ergodyczność układu można przyjąć dla układów kwazyergodycznych,
- łącznikiem pomiędzy światem mikro- i makro- jest funkcja podziału (suma stanów), której definicja zależy od zespołu statystycznego; stanowi ona czynnik normalizujący w wyrażeniu na prawdopodobieństwo zaistnienia stanu kwantowego; dla zespołu kanonicznego wyraża się wzorem:
n
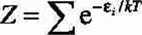
d. Znąjomość funkcji podziału umożliwia wyznaczenie wszystkich funkcji termodynamicznych.
e. Podstawowa metoda obliczania funkcji podziału dla układów niedoskonałych opiera się na przybliżeniu pseudoklasycznym
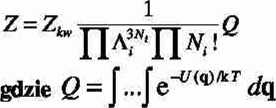
jest konfiguracyjną funkcją podziału.
Wyszukiwarka
Podobne podstrony:
Chemia fizyczna - termodynamika molekularna 2010/2011 1 Wykład 1.8.10.2010 1. Plan
Chemia fizyczna - termodynamika molekularna 2010/2011 36 Wykład 1010.12.2010 1. Wyprowadzenie równan
Chemia fizyczna - termodynamika molekularna 2010/2011 43 Wykład 12 7.01.2011 1. Parametry struktural
Chemia fizyczna - termodynamika molekularna 2010/2011 8 Wykład 322.10.2010 1. Jaki
Chemia fizyczna - termodynamika molekularna 2010/2011 12 Wykład 4 29.10.2010 1. Trudności w bezpośre
Chemia fizyczna - termodynamika molekularna 2010/2011 20 Wykład 6 (skrócony)12.11.2010 1. Kontynuuje
Chemia fizyczna - termodynamika molekularna 2010/2011 50 nadmiarowa entropia nie może znikać. Ściśle
Chemia fizyczna - termodynamika molekularna 2010/2011 9 dziedzin ludzkiej aktywności. Warto zaznaczy
Chemia fizyczna - termodynamika molekularna 2009/2010 37 Wykład 10 11.12.2009 1. Równania stanu w te
Chemia fizyczna - termodynamika molekularna 2009/2010 52 Wykład 13 15.01.2010 1. O
Chemia fizyczna - termodynamika molekularna 2009/2010 32 Wykład 9 4.12.2009 1. Uogólniona funkcja po
Chemia fizyczna - termodynamika molekularna 2009/2010 1 Wykład 1. 2.10.2009 1. Pla
Chemia fizyczna - termodynamika molekularna 2009/2010 42 Wykładll 18.12.2008 1. Roztwór doskonały w
Chemia fizyczna - termodynamika molekularna 2009/2010 11 Wykład 4 23.10.2009 1.
Chemia fizyczna - termodynamika molekularna 2009/2010 15 Wykład 5 30.10.2009 1. Wa
Chemia fizyczna - termodynamika molekularna 2009/2010 19 Wykład 6 5.11.2009 1. Wątpliwość (wyrażona
Chemia fizyczna - termodynamika molekularna 2009/2010 7 6. Przykład z życia. Chcemy znaleźć temperat
Chemia fizyczna - termodynamika molekularna 2009/2010 38 Chemia fizyczna - termodynamika molekularna
Chemia fizyczna - termodynamika molekularna 2009/2010 43 M(xlel roztworu doskonałego jest stosowany
więcej podobnych podstron